
Summary
Background A SARS-CoV-2 protein-based heterodimer vaccine, PHH-1V, has been shown to be safe and well-tolerated in healthy young adults in a first-in-human, Phase I/IIa study dose-escalation trial. Here, we report the interim results of the Phase IIb HH-2, where the immunogenicity and safety of a heterologous booster with PHH-1V is assessed versus a homologous booster with BNT162b2 at 14 and 98 days after vaccine administration.
Methods The HH-2 study is an ongoing multicentre, randomised, active-controlled, double-blind, non-inferiority Phase IIb trial, where participants 18 years or older who had received two doses of BNT162b2 were randomly assigned in a 2:1 ratio to receive a booster dose of vaccine – either heterologous (PHH-1V group) or homologous (BNT162b2 group) – in 10 centres in Spain. Eligible subjects were allocated to treatment stratified by age group (18-64 versus ≥65 years) with approximately 10% of the sample enrolled in the older age group. The endpoints were humoral immunogenicity measured by changes in levels of neutralizing antibodies against the ancestral Wuhan-Hu-1 strain and different variants of SARS-CoV-2 after the PHH-1V or the BNT162b2 boost, the T-cell responses towards the SARS-CoV-2 spike glycoprotein peptides and the safety and tolerability of PHH-1V as a boost. This study is ongoing and is registered with ClinicalTrials.gov, NCT05142553.
Findings From 15 November 2021, 782 adults were randomly assigned to PHH-1V (n=522) or BNT162b2 (n=260) boost vaccine groups. The geometric mean titre (GMT) ratio of neutralizing antibodies on days 14 and 98, shown as BNT162b2 active control versus PHH-1V, was, respectively, 1·68 (p<0·0001) and 0·87 (p=0·43) for the ancestral Wuhan-Hu-1 strain; 0·61 (p<0·0001) and 0·57 (p=0·0064) for the beta variant; 1·01 (p=0·89) and 0·52 (p=0·0003) for the delta variant; and 0·59 (p=<0·0001) and 0·56 (p=0·0026) for the omicron variant. Additionally, PHH-1V as a booster dose induced a significant increase of CD4+ and CD8+ T-cells expressing IFN-γ on day 14. There were 458 participants who experienced at least one adverse event (89·3%) in the PHH-1V and 238 (94·4%) in the BNT162b2 group. The most frequent adverse events were injection site pain (79·7% and 89·3%), fatigue (27·5% and 42·1%) and headache (31·2 and 40·1%) for the PHH-1V and the BNT162b2 groups, respectively. A total of 52 COVID-19 cases occurred from day 14 post-vaccination (10·14%) for the PHH-1V group and 30 (11·90%) for the BNT162b2 group (p=0·45), and none of the subjects developed severe COVID-19.
Interpretation Our interim results from the Phase IIb HH-2 trial show that PHH-1V as a heterologous booster vaccine, when compared to BNT162b2, elicits a strong and sustained neutralizing antibody response against Wuhan-Hu-1 strain, and a superior one concerning the previous circulating beta and delta SARS-CoV-2 variants, as well as the currently circulating omicron. Moreover, the PHH-1V boost also induces a strong and balanced T-cell response. Concerning the safety profile, subjects in the PHH-1V group report significantly fewer adverse events than those in the BNT162b2 group, most of mild intensity, and both vaccine groups present comparable COVID-19 breakthrough cases, none of them severe.
Funding HIPRA SCIENTIFIC, S.L.U.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) identified in Wuhan, China, in December 2019, has brought an urgency to prophylactic measures globally. Consequently, the world has witnessed a striking change in the speed of vaccine development targeting the SARS-CoV-2’s membrane spike protein1, particularly its receptor binding domain (RBD), which interacts with the angiotensin converting enzyme 2 (ACE2) receptor in the host cells and generates a neutralizing antibody response 2.
RBD-specific neutralizing antibodies prevent the interaction between the SARS-CoV-2 spike protein and ACE2 receptor, impeding virus entry to lung cells and further transmission of the virus3. Indeed, there is a positive correlation between the levels of SARS-CoV-2 neutralizing antibodies induced by vaccination and the conferred level of protection4. Antibody levels have been previously used as endpoints for many viral vaccines, such as measles, polio virus and hepatitis A vaccines5. Additionally, the time-dependent decrease in antibody levels after vaccination called for the need of booster administrations during the COVID-19 pandemic6.
Clinical trials for several approved SARS-CoV-2 vaccines7-9 also revealed the induction of a rapid and specific T-cell-mediated response in most participants noted from day 14 post-vaccination, asserting its value as a basis for immunological memory and its role in the rapid response to pathogen re-exposure, conferring protection from severe disease even with lower levels of antibodies10.
To date, two main types of vaccines have been developed for SARS-CoV-211; the first type consists of those that carry the genetic information required for the biosynthesis of the spike glycoprotein, such as mRNA (BNT162b2 [Pfizer/BioNTech] and mRNA-1273 [Moderna]) and adenoviral vector vaccines (Ad26.COV2.S [Janssen] and ChAdOx [Oxford-AstraZeneca]), and the second are protein-based vaccines that contain the spike protein itself in different forms and combinations with adjuvants. Vaccines based on the mRNA technology have inherent limitations: the liability of the mRNA molecule requires that mRNA-based vaccines are encapsulated in lipid nanoparticles and kept at very low temperatures (between –20 °C and –80 °C)12. Regarding protein-based vaccines, only NVX-CoV2373 (Novavax) has been approved by the WHO and the EMA. This subunit vaccine contains the full-length spike protein with the Matrix M1 adjuvant8,13. In contrast to mRNA, protein-based vaccines can be stored under refrigerated conditions14.
Although various SARS-CoV-2 vaccines have been approved, there is still the utmost need for options able to meet global demands, since only 65% of the world population, and, more starkly, only 15·2% of people in low-income countries, have received at least one dose of a COVID-19 vaccine15. Vaccination campaigns protect against severe disease, reduce viral load and therefore lower transmission risk, which implies a public health benefit16. Booster vaccination has become necessary because immunity diminishes with time and new variants emerge. Here, there is increasing evidence that heterologous vaccination is safe, demanding more available vaccine platforms17,18. New vaccine presentations, preferentially stable at 2-8°C and ready-to-use, will facilitate global distribution and be valuable options in the current pandemic setting.
Recombinant protein-based vaccines have emerged as competitive vaccine candidates. They possess a proper safety profile, no risk of genome integration, no live components, and are suitable for people with compromised immune systems19, showing high productivity yields and good stability profiles 20. PHH-1V is a protein-based vaccine intended for the prevention of COVID-19 caused by SARS-CoV-2. This vaccine is based on a fusion heterodimer consisting of the spike RBD sequence from the SARS-CoV-2 beta (B.1.351) and alpha (B.1.1.7) variants21, formulated with an oil-in-water based adjuvant produced by HIPRA known as SQBA, and prepared as an emulsion for intramuscular administration. PHH-1V comprises three key mutations of high relevance for previously and currently circulating SARS-CoV-2 variants: K417N, E484K and N501Y22. Previous studies revealed that the E484K and N501Y mutations are present in the RBD of the spike protein of beta, gamma, and mu variants; and both the K417N and N501Y mutations present in the PHH-1V antigen are common to the beta and to the currently predominant omicron variants23.
A first-in-human Phase I/IIa study dose-escalation, randomized, double-blinded, active-comparator controlled clinical trial in 30 healthy adults demonstrated that the PHH-1V vaccine is safe and well-tolerated in healthy young adults, with even fewer reported solicited adverse events compared to the control (BNT162b2; publication under review).
The HH-2 study is an ongoing multicentre, randomised, active-controlled, double-blind, non-inferiority Phase IIb trial that aims to assess the immunogenicity and safety of PHH-1V as a heterologous boost versus a homologous boost in individuals after receiving a primary vaccination series of BNT162b2. Here we show interim data up to 98 days after administration of the booster for the Phase IIb HH-2 study.
Methods
Study design and participants
This multicentre, randomised, active-controlled, double-blind, non-inferiority Phase IIb trial where immunogenicity and safety of the PHH-1V vaccine were assessed, was carried out in 10 centres in Spain (Supplementary Information).
Eligibility criteria were individuals aged 18 years or older, who had received two doses of the BNT162b2 vaccine at least 182 days and less than 365 days after their second dose; Body Mass Index (BMI) between 18 and 40 kg/m2; negative SARS-CoV-2 PCR test at the time of enrolment; willingness to avoid all other vaccines within 4 weeks before and after vaccination in this study (seasonal influenza vaccination was allowed if it was received at least 14 days before or after the study booster). Key exclusion criteria included pregnancy or breastfeeding; an ongoing serious psychiatric condition; history of respiratory disease requiring daily medications; history of significant cardiovascular disease; history of neurological or neurodevelopmental conditions; ongoing malignancy or recent diagnosis of malignancy in the last five years excluding basal cell and squamous cell carcinoma of the skin; any confirmed or suspected autoimmune, immunosuppressive or immunodeficiency disease/condition (iatrogenic or congenital) including human immunodeficiency virus infection; use of immunosuppressants; coagulation or bleeding disorder; chronic liver disease; history of SARS-CoV-2 infection; close contact with anyone positive for SARS-CoV-2 infection within 15 days before screening and life expectancy of less than 12 months.
The trial was conducted in accordance with the Declaration of Helsinki, the Good Clinical Practice guidelines, and national regulations. The study protocol was reviewed and approved by the Spanish Agency of Medicines and Medical Devices (AEMPS) as well as Independent Ethics Committee from the Hospital Clínic de Barcelona. Written informed consent was obtained from all participants before enrolment. The biologic biosafety committee of the Research Institute Germans Trias i Pujol approved the execution of SARS-CoV-2 experiments at the BSL3 laboratory of the Centre for Bioimaging and Comparative Medicine (CSB-20-015-M8).
Randomisation and masking
Subjects were randomly assigned in a 2:1 ratio to receive a booster dose of vaccine (third immunization) either with the HIPRA PHH-1V vaccine (PHH-1V group) or with the Pfizer/BioNTech BNT162b2 vaccine (BNT162b2 group). Subjects were allocated to treatment using an Interactive Response Technology (IRT). The allocation sequence was stratified by age group: approximately 90% of the total recruited participants were in the 18 to 64 years group, and 10% in the group 65 years or older. Participants, site staff, including clinical staff involved in study drug preparation and administration, the sponsor, and the Clinical Research Organisation (CRO) were blinded to treatment assignment/allocation. Unblinded hospital pharmacists or other qualified personnel prepared the booster dose, and unblinded site staff members, who were not otherwise involved with study procedures (except for blood extraction), administered treatment to subjects. This article refers to the data obtained on days 14 and 98 and the analysis could be carried out after the unblinding.
Procedures
The study visits were scheduled on day 0, day 14, day 28 and at 3, 6 and 12 months. This article reports data up to day 98. Participants who, at that moment, met eligibility criteria were vaccinated in the visit on day 0. The BNT162b2 vaccine was given as a 0·3 mL (30 μg) and the PHH-1V as a 0·5 mL (40 μg) by intramuscular injection into the deltoid muscle. The first 30 participants were observed for 60 minutes and monitored during the following 72 hours by phone. Other participants were observed for 30 minutes and contacted again on day 7. During the day-0 visit, participants were given a hard copy diary to record local and systemic solicited reactions within the 7 days after vaccination.
The neutralization titres of antibodies were determined by the inhibitory concentration 50 (IC50, reported as reciprocal dilution) using a pseudovirion-based neutralisation assay (PBNA) as described previously24. The immunogenicity against the SARS-CoV-2 spike glycoprotein was assessed by the percentage of subjects having a ≥4-fold increase in the binding antibodies titre 14 and 98 days after boosting using the Elecsys Anti-SARS-CoV-2 S immunoassay (Roche Diagnostics). The titre of neutralizing antibodies was also analysed in a subset of subjects by infectious SARS-CoV-2 neutralisation test (VNA) and measured as inhibitory dilution 50 (ID50), also reported as reciprocal dilution. The geometric mean titre (GMT) and the geometrical mean fold rise (GMFR) was calculated for each parameter.
The T-cell mediated immune response against the SARS-CoV-2 spike glycoprotein was assessed after the in vitro peptide stimulation of peripheral blood mononuclear cells (PBMC) followed by IFN-γ enzyme-linked immune absorbent spot (IFN-γ ELISpot) and intracellular cytokine staining (ICS). Six peptide pools of overlapping SARS-CoV-2 peptides, each encompassing the SARS-CoV-2 regions S (two pools) and RBD (four peptides’ pools covering Wuhan-Hu-1, alpha, beta, and delta variants) were used (details of procedures are described in the Supplementary methods).
The safety assessment included the incidence and description of solicited local and systemic reactions, unsolicited local and systemic adverse events, serious adverse events, and adverse events of special interest. This is an ongoing study, and both solicited and unsolicited local and systemic adverse events were assessed through days 7 and 28, respectively, and safety laboratory parameters together with medically attended adverse events through the end of the study. Additionally, the incidence of SARS-CoV-2 infections, including severe infections, the requirement of hospital and intensive care unit (ICU) admissions, and deaths associated with COVID-19, if any, through the end of the study will be assessed. In the safety analysis, adverse events were coded using the MedDRA Version 24.1 coding system.
Outcomes
The endpoints were humoral immunogenicity measured by changes in levels of neutralizing antibodies by PBNA and VNA, binding antibodies by ELISA, T-cell responses by ELISpot, and the Th-1/Th-2 T-cell mediated response towards the SARS-CoV-2 spike glycoprotein peptides, all of them assessed at least in the sample of 14 days after the boost with PHH-1V or BNT162b2. Exploratory endpoints included the incidence of COVID-19 and the number and percentage of severe COVID-19 cases, both happening from day 14 after administration of the booster and through the end of the study.
Statistical analysis
The sample size calculation is detailed in Supplementary methods. The following analyses of populations were included in this study: Intention-to-treat (ITT) population, including all subjects who were randomly assigned to treatment, regardless of the subject’s treatment status in the study; modified intention-to-treat (mITT) population, consisting of all subjects in the ITT who met the screening criteria and received one vaccine dose; Immunogenicity (IGP) population, including all subjects in the mITT who had a valid immunogenicity test result before receiving study drug and at least one valid result after dosing; per-protocol (PP), all subjects in the mITT who received one vaccine dose and had no major protocol deviations; safety (SP), comprised of all randomised subjects who received one vaccine dose, and were analysed according to the treatment received. The efficacy analysis was done in the mITT population and the T-cell response in the IGP population. The safety analysis was conducted in the safety population and the assessment of COVID-19 cases in the PP population.
The efficacy analyses tested the following hypotheses to show non-inferiority of PHH-1V when compared to the BNT162b2 booster vaccine: i) Null hypothesis, H0: the ratio of the GMTs (BNT162b2: PHH-1V) exceeds the non-inferiority margin (NIm); equivalently, the difference in log (GMT) exceeds log (NIm); ii) Alternative hypothesis, H1: the ratio of GMTs is below NIm; equivalently the difference in log (GMT) is less than log (NIm). The NIm established for this study was 1·4, whereby the upper bound of the 95% confidence interval (95% CI) had to be lower than this value to accept the null hypothesis and was defined for each endpoint separately.
To analyse the antibody neutralisation titre against SARS-CoV-2 (Wuhan-Hu-1, beta, delta, and omicron) as measured by IC50 with PBNA or ID50 with VNA, a mixed effects model for repeated measures (MMRM) was carried out on log transformed data. Summary statistics for the log10 transformations for each individual sample were calculated based on the log10-transformed titres at baseline, on day 14 and day 98, and are presented for the mITT population.
The GMFR analysis was performed for participants who, after a booster dose, had a ≥4-fold change in binding antibodies titre from baseline to day 14 and day 98 (responders), summarised by the number and proportion of responders along with an exact 95% Clopper-Pearson CI for the proportion.
For the cellular immunogenicity analysis, a MMRM of T-cell data (interferon gamma [IFN-γ] Spot-Forming Units [SFU]/106 PBMCs) was employed, using the angular-transformed proportions as the response variable.
For the incidence of COVID-19 data are shown as the number of events and percentage of participants affected in the PP population. An exact 95% Clopper-Pearson CI for the proportion of each endpoint was also presented.
All statistical tests were performed using a two-tailed 5% overall significance level, unless otherwise stated, using SAS (Version 9.4) or R (Version 4.0.5).
This trial is registered at ClinicalTrials.gov, NCT05142553.
Role of the funding source
This study was sponsored by HIPRA SCIENTIFIC, S.L.U (HIPRA). HIPRA was involved in the study design; in the collection, analysis, and interpretation of data; in writing of the report; and in the decision to submit the paper for publication.
Results
Baseline characteristics
From 15 November 2021, 862 participants were screened, of whom, 782 adults were randomly assigned to PHH-1V (n=522) or BNT162b2 (n=260) booster vaccine groups (Figure 1). A total of 504 participants in the PHH-1V group and 248 participants in the BNT162b2 group were included in the mITT population. For the SP population, 513 and 252 participants were included in the PHH-1V and BNT162b2 groups, respectively. Five hundred and three subjects were included in the PHH-1V group and 246 participants in the BNT162b2 group (IGP population). Since this trial is ongoing, the PP population has not yet been defined as protocol deviations are still being assessed. Baseline characteristics were well balanced between the two groups, where the median age of participants was 42 years (range 19–76 years; p=0·65), 484 (63·3%) were female (p=1·00) and 755 (98·7%) identified as White (p=1·00; Table 1).
Baseline characteristics of participants included in the analysis.

PHH-1V=PHH-1V vaccine, HIPRA. BNT162b2= BNT162b2 vaccine, Pfizer–BioNTech.
Neutralizing antibodies response
The GMT and GMFR of neutralizing antibodies determined by PBNA on day 14 and 98 post-booster are shown in Table 2 and Figure 2A for the mITT population. GMT on day 14 for SARS-CoV-2 Wuhan-Hu-1 was 1998·95 (95% CI: [1760·56, 2269·61]) for the PHH-1V group and 3357·50 (95% CI [2882·12, 3911·28]) for the BNT162b2 group, with a GMT ratio of 1·68 (95% CI [1·44, 1·96]; p<0·0001), which could not demonstrate non-inferiority of the PHH-1V vaccine to the BNT162b2 vaccine in immune response. However, on day 98, the GMT ratio was 0·87 (95% CI [0·60, 1·24]; p=0·43), which shows non-inferiority of the PHH-1V vaccine to the BNT162b2 vaccine immune response. The GMT for the beta variant on day 14 was 4328·93 (95% CI [3747·80, 5000·16]) in the PHH-1V group and 2658·04 (95% CI [2234·60, 3161·73]) in the BNT162b2 group, with a GMT ratio of 0·61 (95% CI [0·51, 0·73]; p<0·0001), indicating a superiority of the PHH-1V vaccine. The GMT ratio of 0·57 (95% CI [0·38, 0·85]; p=0·0064) on day 98 confirms the superiority of the PHH-1V for the beta variant. Regarding the delta variant, the GMT on day 14 was 1471·60 (95% CI [1281·71, 1689·62]) and 1487·13 (95% CI [1269·58, 1748·85]) for the PHH-1V and BNT162b2 groups, respectively, with a GMT ratio of 1·01 (95% CI [0·87, 1·18]; p=0·89), demonstrating non-inferiority of the PHH-1V vaccine to BNT162b2. On day 98, the GMT ratio of 0·52 (95% CI [0·36, 0·74]; p=0·0003) indicates a superiority of the PHH-1V vaccine for the delta variant. For the omicron variant, the PHH-1V and the BNT162b2 boosted groups presented a GMT of 2053·73 (95% CI [1770·39, 2382·41]) and 1219·08 (95% CI [1025·61, 1449·03]), respectively, and a GMT ratio of 0·59 (95% CI [0·50, 0·70]; p<0·0001), thus demonstrating superiority of the PHH-1V vaccine to the BNT162b2 vaccine against the omicron SARS-CoV-2 variant. This result is further confirmed on day 98, with a GMT ratio of 0·56 (95% CI [0·38, 0·82]; p=0·0026).
Analysis of neutralizing and binding antibodies against SARS-CoV-2 variants on days 14 and 98 post-vaccination boost.

Analysis of the antibody response to PHH-1V vaccination. (A) Neutralizing antibody responses against multiple SARS-CoV-2 variants by PBNA in the mITT population. Comparison of neutralizing antibody titres at baseline, day 14 and day 98, between the PHH-1V and BNT162b2-vaccinated groups. (B) SARS-CoV-2 binding antibodies titre. Comparison of total binding antibody titres at baseline, day 14 and day 98, between the PHH-1V and BNT162b2-vaccinated groups. The analysis has been performed considering only responder subjects (≥4 fold-change in binding antibodies titre from baseline to Day 14 or Day 98). (C) SARS-CoV-2 neutralisation assay (VNA) for the omicron variant (B.1.1.529). VNA assay was performed with serial dilutions of heat-inactivated serum samples from individuals receiving a PHH-1V or BNT162b2 boosting vaccine on day 14. VNA titres are plotted as ID50 (the reciprocal dilution inhibiting 50% of the cytopathic effect). *p-value for ratio=1, BNT162b2 active control vs PHH-1V; p=0·0001. Adjusted treatment mean is shown for VNA analysis for BNT162b2 active control (n=24) vs PHH-1V (n=34) as LS mean (95% CI). GMT is shown for adjusted treatment mean (95% CI). GMT ratio is the GMT for treatment ratio for BNT162b2 active control vs PHH-1V (95% CI) followed by p-value for ratio=1.
Boxplots with grey dots and blue dots refer to PHH-1V and BNT162b2, respectively. Statistically significant differences are shown as * for p ≤ 0·05; ** for p ≤ 0·01; *** for p ≤ 0·001. Non-significant comparisons have been indicated with “ns”.
The response of neutralizing antibodies assessed by PBNA is also shown as GMFR in Table 2, confirming the results observed for GMT on day 14. A booster vaccination with PHH-1V could not demonstrate non-inferiority for Wuhan-Hu-1 (ratio 1·76, 95% CI [1·46, 2·13]; p<0·0001) on day 14, but provided non-inferiority for the delta variant (ratio 1·12, 95% CI [0·93, 1·34]; p=0·24), and elicited statistically significant higher levels of neutralizing antibodies for the beta (ratio 0·69, 95% CI [0·56, 0·84]; p=0·0003) and omicron (ratio 0·68, 95% CI [0·56-0·83]; p=0·0001) variants, thus, demonstrating superiority of the PHH-1V booster against beta and omicron variants. On day 98 post-boost, the GMFR ratio for Wuhan-Hu-1 indicates that PHH-1V administration reaches non-inferiority to BNT162b2 (ratio 0·85, 95% CI [0·60, 1·19]; p=0·33), and superiority for the beta (ratio 0·66, 95% CI [0·44, 1·00]; p=0·05), delta (ratio 0·59, 95% CI [0·41, 0·87]; p=0·0069), and omicron variants (ratio 0·61, 95% CI [0·43, 0·89]; p=0·0091).
The percentage of participants with a ≥4-fold change in binding antibodies in the PHH-1V and BNT162b2 vaccine groups were similar, 98·4% (n=490) and 98·8% (n=238) respectively, on day 14 post-boost (Table 2). Accordingly, there were non-significant differences between the odds of the two groups, with an odds ratio of 1·29 (95% CI [0·34, 4·91]; p=0·71). The data concerning day 98 after booster administration reinforce this result, with non-significant differences in the odds between treatment groups (odds ratio 1·12, 95% CI [0·21, 5·97]; p= 0·90). Nevertheless, the quantitative comparison of the total binding antibody titres between treatments reflects that vaccination with PHH-1V induced higher antibody titres on days 14 and 98 compared with the BNT162b2 booster dose (p<0·0001; Figure 2B). These results indicate that a similar percentage of subjects respond to both vaccines with a ≥4-fold change in binding antibodies, although the subjects vaccinated with PHH-1V reach higher titres compared with those immunized with BNT162b2. Interestingly, quantitative results on day 98 show that the decrease in the titre in the PHH-1V arm is not as pronounced as in the BNT162b2 arm, with geometric mean fold reduction ratio from day 14 to day 98 of 1·87 (95% CI [1·35, 2·60]; p<0·0001), suggesting a greater maintenance of the long-term antibody titre (Figure 2B).
The results for the SARS-CoV-2 neutralisation test (VNA) for the omicron variant on day 14 show a GMT for treatment ratio for BNT162b2 active control vs PHH-1V of 0·44 (95% CI [0·30, 0·65]) followed by p value for ratio=1 (p=0·0001; Figure 2C and Table 2).
T-cell-mediated response
Participants in the PHH-1V group showed a significant increase of IFN-γ producing lymphocytes upon in vitro re-stimulation with SARS-CoV-2 spike peptide pools SA (Figure 3E, p=0·0016) and RBD (Figure 3A-3D, Wuhan-Hu-1, alpha, beta, and delta variants; all p<0·0001) at 2 weeks post-boost in comparison with the levels observed at baseline. Similarly, the administration of the booster dose of BNT162b2 increased the proportion of IFN-γ producing cells in response to the stimulation with spike peptide pool SA (Figure 3E, p<0·0001), pool SB (Figure 3F, p=0·0105) and RBD (Figure 3A-3D, Wuhan-Hu-1, and variants; all p< 0·0001) at 2 weeks post-boost compared to baseline (pre-boost). After the booster dose, the two groups showed similar increase rates in the IFN-γ response when PBMC were stimulated in vitro with RBD antigens. Nevertheless, the BNT162b2 group showed a significantly higher increase in the number of IFN-γ producing cells after in vitro stimulation with the Spike SA (p=0·0043) and Spike SB (p=0·0173) antigens at 2 weeks post-boost compared with the PHH-1V group (Figure 3E and 3F).

Cellular SARS-CoV-2 specific immune response. PBMCs from participants receiving either PHH-1V (in grey) or BNT162b2 (in blue) were isolated before (Baseline) and two weeks after the boost immunization (Day 14). Results of IFN-γ ELISpot assay stimulating PBMCs with RBD and variants peptide pools [RBD (A); RBD B.1.1.7 (B); RBD B.1.351 (C) and RBD B.1.1617.2 (D)] and Spike [SA (E) and SB (F)] peptide pools are shown. Boxes depict the median (solid line) and the interquartile range (IQR), and whiskers expand each box edge 1.5 times the IQR. Interaction contrasts have been displayed in the plots, comparing the increase rates over time between the two vaccination groups. Non-significant differences in the increase rates between groups have been reported with “ns”, while p-values lower than 0·05 indicate that the BNT162b2-vaccinated group has experienced a stronger boost compared to the PHH-1V arm.
IQR=interquartile range; RDB; receptor binding domain for the SARS-CoV-2 spike protein (ancestor Wuhan-Hu-1 strain); RDB B.1.1.7 (alpha variant); RDB B.1.351 (beta variant); RDB B.1.1617.2 (delta variant); Spike SA corresponds to 194 spike protein peptide pools overlapping the S1-2016 to S1-2196 region of the Spike protein; Spike SB corresponds to 168 spike protein peptide pools overlapping the S1-2197 to S2-2377 region of the Spike protein. Statistically significant differences are shown as * for p ≤ 0·05; ** for p ≤ 0·01. Non-significant comparisons have been indicated with “ns.
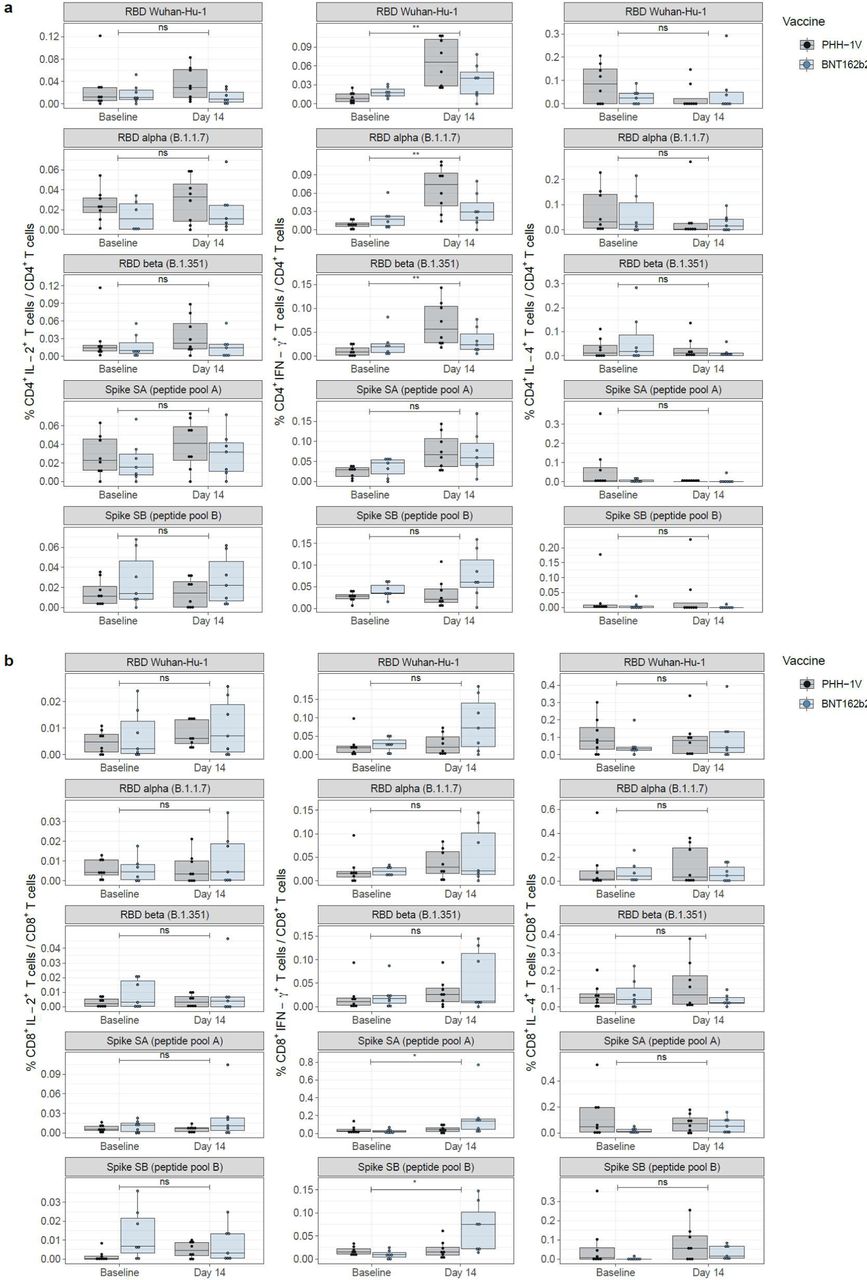
A significant activation of CD4+ T-cells expressing IFN-γ was observed for the PHH-1V group at 2 weeks compared with baseline upon the stimulation with the RBD (Wuhan-Hu-1, alpha, and beta variants) and Spike SA peptide pool (Figure 4A; p<0·0001). For these variants, the increase rate in the percentage of CD4+ IFN-γ+ T-cells from baseline to 2 weeks was higher in the PHH-1V group compared to the BNT162b2 group (Wuhan-Hu-1, p=0·0097; alpha, p=0·0030; beta, p=0·0069). Regarding the CD8+ T-cells, the boost with the PHH-1V vaccine significantly increased the percentage of IFN-γ-expressing cells upon re-stimulation with RBD from SARS-CoV-2 beta variant at week 2 compared to baseline (Figure 4B; p=0·0372). The BNT162b2 vaccine boost increased the percentage of CD8+ IFN-γ+ T-cells after re-stimulation with a spike peptide pool SA (p=0·0089) and SB (p=0·0018) at week 2 compared with baseline, showing higher values in the increase rate than the PHH-1V boost (p=0·0147 and p=0·0248, respectively). Moreover, the BNT162b2 group increased the percentage of CD8+ IL-4+ T-cells after re-stimulation with the SB spike peptide pool at week 2 compared with baseline (p=0·0123).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Characterization of T-cell responses in PBMCs from groups receiving a heterologous boosting vaccination with PHH-1V (in grey) or BNT162b2 (in blue). CD4+ T (A) and CD8+ T (B) cells were characterized by intracellular cytokine staining (ICS) of interleukin-2 (IL-2+; left panel), interferon gamma (IFN-γ+; middle panel) and interleukin-4 (IL-4+, right panel). Boxes depict the median (solid line) and the interquartile range (IQR), and whiskers expand each box edge 1·5 times the IQR. Interaction contrasts have been displayed in the plots, comparing the increase rates over time between the two vaccination groups. Statistically significant differences are shown as * for p ≤ 0·05; ** for p ≤ 0·01; *** for p ≤ 0·001. Non-significant comparisons have been indicated with “ns”. P-values lower than 0·05 indicate that one of the vaccinated groups has experienced a stronger boost compared to the other.
Safety
The most frequent solicited local reactions on day 1 were pain (51·1% and 69·8% for the PHH-1V and the BNT162b2 group, respectively) and tenderness (48·5% for the PHH-1V and 63·5% for the BNT162b2 group) (Table 3). These local reactions remained the most frequent up to day 7. The most frequent post-vaccination solicited systemic adverse events, for the PHH-1V and the BNT162b2 groups on day 1, respectively, were fatigue (16·0% and 35·3%), headache (14·2% and 27·8%), muscle pain (11·7% and 29·4%), and fever (0·6% and 7·1%), and these were the main adverse events observed up to day 7 post-boost. In this period, 257 subjects reported headache, where 157 (30·6%) were in the PHH-1V group reporting a mean (SD) duration of 2·0 (1·63) days, and 100 (39·7%) in the BNT162b2 group, reporting a duration of 1·8 (1·19) days. Additionally, in the same period 32 subjects reported fever, with 9 of them (1·75%) belonging to the PHH-1V group and reporting a mean (SD) duration of 1·7 (1·00) days, and 23 (9·12%) belonging to the BNT162b2 group, reporting 1·5 (0·90) days of fever. The complete data for the vaccination diary from day 0 (12 hours) until day 7 are shown in the Supplementary Table 1.
Solicited local reactions and systemic adverse events.
Overall, 458 (89·3%) subjects in the PHH-1V group and 238 (94·4%) subjects in the BNT162b2 group experienced at least one adverse event, of which 66·7% and 57·9% were of mild intensity in the PHH-1V and BNT162b2 groups, respectively. The most frequent adverse events were injection site pain (79.7% and 89·3%), fatigue (27·5% and 42·1%) and headache (31·2 and 40·1%) for the PHH-1V and the BNT162b2 groups, respectively (Table 4). Treatment-related adverse events were reported in 434 (84·6%) in the PHH-1V group and 231 (91·7%) in the BNT162b2 group. At the data cut-off date of the report, the percentage of patients who reported experiencing COVID-19 (n=82) ≥14 days post-booster administration remained similar in the PHH-1V (10·14%) and BNT162b2 (11·90%) groups (OR 0·83, 95% CI [0·51, 1·36]; p= 0·46). None of the subjects developed severe COVID-19 and, consequently, no hospital or intensive care unit (ICU) admission were required.
Summary of Adverse Events by Treatment Group (Safety Population).
Discussion
Our interim results from the Phase IIb HH-2 trial show that PHH-1V as a heterologous boost elicits a non-inferior neutralizing antibody response to SARS-CoV-2 delta variant, a superior neutralizing antibody response against beta variant and, most importantly, a superior neutralizing antibody response against the omicron variant of concern (sublineage BA.1, predominant during the development of the trial) at 14 days post-vaccination when compared to the BNT162b2 boost. Although the PHH-1V as a booster could not demonstrate non-inferiority to BNT162b2 against the initial Wuhan-Hu-1 SARS-CoV-2 strain on day 14, it provided non-inferiority 98 days after booster administration. Moreover, on day 98, PHH-1V vaccine showed superiority for the delta variant. These results suggest a lower decrease in the neutralizing antibody titres after the PHH-1V boost compared to the BNT162b2 boost. Accordingly, given the very high levels of community transmission observed for the currently circulating delta and omicron variants, neutralisation antibody response against these variants is of paramount importance in contrast to ancestral strains. No significant differences were observed between the groups for those participants with ≥4-fold change in binding antibodies up to 98 days after boost administration, although the PHH-1V induced higher titres compared with the BNT162b2 boost. Our results have also shown that PHH-1V as a booster dose induces a significant increase of CD4+ and CD8+ T-cells expressing IFN-γ on day 14. Concerning the safety profile, PHH-1V group had less percentage of adverse events compared with BNT162b2 group, with most of mild intensity, and similar non-severe COVID-19 cases.
SARS-CoV-2 delta, and mostly omicron (BA.1), were the most concerning variants circulating at the time this trial was started. PHH-1V has consistently showed good response against each new variant in many animal models 21, a result which has been confirmed in the data presented here. These data indicate that PHH-1V is superior to BNT162b2 in terms of neutralizing antibodies against SARS-CoV-2 for the beta and omicron variants, and non-inferior against delta variant (or even superior on day 98) and Wuhan-Hu-1 (on day 98). Remarkably, PHH-1V was able to sustain a good neutralising ability against the omicron SARS-CoV-2 variant despite the heavily mutated spike protein of this variant, many of them located in the RBD25. This fact strongly supports the high efficacy of the PHH-1V RBD-based candidate against a wide range of potential new mutations since the PHH-1V antigen comprises key mutations that are also present in the omicron variant, as well as in many other SARS-CoV-2 variants.
Results of the T-cell-mediated response determined by ELISpot in the present study indicate that the PHH-1V boost, after a primary vaccination with BNT162b2, increases the cellular immune response after the in vitro re-stimulation. The ICS data for T-cell response characterization demonstrates that the booster immunization with the PHH-1V vaccine induces the activation of CD4+ T-cells expressing IFN-γ upon re-stimulation with pools of RBD peptides from different variants. Interestingly, this response is stronger compared to those subjects boosted with BNT162b2 vaccine, indicating that the PHH-1V heterologous boost is more efficient than the homologous boost in the activation of the CD4+ T-cell memory previously induced by the primary vaccination protocol. As no IL-4 expression was detected in the activated CD4+ T-cells after the in vitro re-stimulation, the ICS results suggest that the PHH-1V booster induces a Th1-biased T-cell response. The detection of the IFN-γ expression by the ELISpot assay confirms the Th1-biassed T-cell response upon boost immunization with the PHH-1V vaccine. Avoiding a Th2-biased immune response is important as it has been related with ineffective vaccines that induce vaccine-associated enhanced respiratory disease (VAERD) after subsequent infection26. Moreover, the heterologous boost with the PHH-1V vaccine has proven to induce the activation of CD8+ T-cells expressing IFN-γ. The T-cell response is crucial to confer protection from COVID-19 severe disease10, and these results show the potential ability of PHH-1V not only to fight against the virus with neutralizing antibodies but also with cellular immunity specifically to destroy infected cells.
The percentage of subjects with local reactions (injection site pain, induration, erythema), systemic reactions (fatigue, headache, muscle pain and fever), total adverse effects and treatment-related adverse events were lower in the PHH-1V group compared to the BNT162b2 group. No deaths were reported in the study in either of the PHH-1V or BNT162b2 group and one SAE was reported, which was resolved and found to be nonrelated to the vaccination. A total of 82 SARS-CoV-2 cases, all of them mild or asymptomatic, occurred between day 14 and the data cut-off date post-booster administration with no statistically significant differences between both groups, suggesting that both vaccine boosters provide protection to moderate and severe forms of SARS-CoV-2 infections and that PHHV-1V efficacy is comparable to that of BNT162b2. Noteworthy, this study temporarily took place during a high COVID-19 incidence period, namely the currently circulating delta and omicron (BA.1 variant) waves in Spain. Moreover, most of participants in the study were healthcare workers, thus with a higher infection exposition compared to the general population27.
The main limitations of this study are related to the changing epidemiological and social situation during the pandemics. Although it was initially proposed that neutralising antibodies by means of PBNA would have to be determined for Wuhan-Hu-1, alpha, beta and delta SARS-CoV-2 variants, the situation of the pandemics during the study made us change the proposal and so alpha was substituted for omicron which was the relevant circulating variant at that time, adding a very important value to the study that has demonstrated the effective potential of the PHH-1V vaccine as a heterologous booster.
In conclusion, the HH-2 study demonstrates that the PHH-1V vaccine is safe and well tolerated, eliciting a strong neutralizing antibody response against all tested SARS-CoV-2 variants and Wuhan-Hu-1 strain. The study specifically shows superiority against the beta and omicron SARS-CoV-2 variants when compared to BNT162b2 homologous vaccine boost and non-inferiority against delta on Day 14.
Importantly, the PHH-1V vaccine also allows for better sustained levels of antibodies over time (Day 98). This demonstrates that PHH-1V can elicit protection against current circulating variants of concern and, most importantly, can anticipate protection against potentially new emerging variants. The PHH-1V vaccine also induces a strong and balanced T-cell response against SARS-CoV-2. At present, a phase III trial is ongoing where the inclusion criteria have been opened further and studies on the vaccine are continued. The interim results presented here provide data on the balance observed between safety and efficacy elicited by the PHH-1V boost. These data support PHH-1V vaccine as a valuable tool to the current authorized COVID-19 vaccines and as a booster dose in the vaccination campaigns.
Data Availability
All data relevant to the study are included in the article or uploaded as supplementary information. Further data are available from the authors upon reasonable request and with permission of HIPRA S.A.
Contributors
Veristat was responsible for managing the data and performing the statistical analyses. Authors contributed to the acquisition, analysis, and/or interpretation of data. All authors had full access to all the data, revised the manuscript critically for important intellectual content, approved the version to be published, and accepted responsibility for publication.
Declaration of interests
The authors of this manuscript declare Julià Blanco has received institutional grants from HIPRA, Grifols, Nesapor Europe and MSD. Outside of this work she is the CEO and founder of AlbaJuna Therapeutics, S.L. Alex Soriano has received grants from Pfizer and Gilead Sciences and honoraria for lectures for Pfizer, MSD, Gilead Sciences, Shionogi, Angelini, Roche and Menarini. Nuria Izquierdo-Useros declares institutional grants from HIPRA, Pharma Mar, Grifols, Dentaid, Palobiofarma and Amassence. Jose R Arribas has received honoraria for lectures and advisory boards from Janssen, Gilead, MSD, Lilly, Roche and Pfizer. Susana Otero-Romero has received speaking and consulting honoraria from Genzyme, Biogen-Idec, Novartis, Roche, Excemed and MSD. Julia G Prado declares institutional grants from HIPRA and Grifols.
Some authors are employees of HIPRA, as indicated in the affiliation section, a private pharmaceutical company that develops and manufactures vaccines.
Several patent applications have been filed by HIPRA SCIENTIFIC S.L.U. and Laboratorios HIPRA, S.A. on different SARS-CoV-2 vaccine candidates and SARS-CoV-2 subunit vaccines, including the novel recombinant RBD fusion heterodimer PHH-1V. These patent applications are not yet public. Antonio Barreiro, Antoni Prenafeta, Luis González, Laura Ferrer, Ester Puigvert, Jordi Palmada, Maria Teresa Prat and Carmen Garriga are the inventors of these patent applications.
Data sharing
All data relevant to the study are included in the article or uploaded as supplementary information. Further data are available from the authors upon reasonable request and with permission of HIPRA S.A.
Funding
This work was supported by HIPRA SCIENTIFIC, S.L.U (HIPRA) and partially funded by the Centre for the Development of Industrial Technology (CDTI, IDI-20211192), a public organisation answering to the Spanish Ministry of Science and Innovation.
Acknowledgements
Medical writing support was provided by Vanessa Chigancas at Dynamic Science S.L.U. (Evidenze Clinical Research, Madrid, Spain) during the preparation of this paper, and funded by HIPRA SCIENTIFIC, S.L.U.
We especially acknowledge the following members of Veristat, who contributed in the success of this trial. The following were responsible for study management, biostatistics, medical monitoring, data management, and database programming of the study: Lubia Álvarez, MD, Robin Bliss, PhD, Judith Oribe, Emma Albacar, MPH, Nancy Hsieh, MPH, Marcela Cancino, MSc, Rachel Smith, Montse Barcelo, MD, Mariska van der Heijden, MSc, Amy Booth, Edmund Chiu and Rodney Sleith, MS, Avani Patel, Atalah Haun, MD and Cesar Wong, MD.
We are grateful to Daniel Perez-Zsolt and Jordana Muñoz-Basagoiti for their outstanding contribution to VNA.
The authors would like to thank Silvia Marfil, Raquel Ortiz and Carla Rovirosa for technical assistance.
The authors thank Ruth Peña, Esther Jimenez-Moyano for technical assistance with sample management and ELISpot and Gabriel Felipe Rodriguez-Lozano for ELISpot database generation.
We would like to express our gratitude to Marina Machado, Ana Álvarez-Uría, Sara Rodríguez, Ma Jesús Pérez Granda, Juan Carlos López Bernaldo de Quirós, Ma Teresa Aldamiz, Francisco Tejerina, Rocío Fernández, Martha Kestler, Cristina Díez, Iván Adán, Ana Mur, Patricia Gómez, Félix García and Víctor Fernández for their tireless effort and contribution to this important public health clinical trial.
We would like to thank Glòria Pujol and Eduard Fossas for their assistance in the revision of the manuscript; Fiorella Gallo, Núria Fuentes and Miriam Oria for the ELISA analysis; Clara Panosa, Thais Pentinat and Ester Puigvert for their assistance in the production of the vaccine and Jordi Palmada and Eva Pol for carrying out manufacturing controls. And of course, we would like to especially thank all the HIPRA workers who in one way or another have contributed to making this project a reality.
The authors thank the members of the DSMB for their expertise and recommendations.
We are indebted to the HCB-IDIBAPS Biobank, integrated in the Spanish National Biobanks Network, for the biological human samples and data procurement. We want to express our gratitude to all the volunteers for their time and effort. With their contribution they have enabled the generation of medical and scientific knowledge that will enable us to draw closer to the end of this pandemic.
Footnotes
Please, we've amended the name of the vaccine "PHH-IV" to "PHH-1V" in the title of the manuscript, since it was a typographic error.
References




