
ABSTRACT
Purpose Common LOXL1 protein-altering variants are significant genetic risk factors for exfoliation syndrome (XFS) and the related secondary glaucoma (XFG). A rare LOXL1 missense allele has been associated with protective effects in a Japanese cohort, suggesting that other rare alleles may also exhibit protective effects. The goal of this study was to assess the contributions of rare LOXL1 variants to XFS/XFG risk in cases and controls from the United States.
Methods LOXL1 rare (minor allele frequency less than 1%) variants were identified from Humanexome BeadArray (Illumina) data for 1118 XFS/XFG cases and 3661 controls. Distribution of rare variants, haplotypes (defined using IMPUTE2) and diplotypes were examined using the Fisher’s exact test. Rare variant allele distribution was confirmed in an independent set of primary open angle glaucoma (POAG) controls and multi-ethnic datasets. Correlation of LOXL1 common allele homozygosity with disease risk used data from gnomAD (gnomad.broadinstitute.org/) and an existing multi-ethnic meta-analysis.
Results Four rare LOXL1 missense alleles were identified, and all were more common in controls (combined P= 7.6E-4), with two of these located in a LOXL1 intrinsic disordered region (IDR) known to be involved in LOXL1 aggregation. Haplotypes that included the rare or minor variants were more common in controls compared to cases (OR= 0.33, P=1.7E-8). Heterozygous diplotypes were significantly associated with reduced risk overall (OR= 0.45 P= 1.7 E-89) with the largest effects observed for diplotypes with more than one heterozygous genotype (OR= 0.05, P= 1.0E-39). A homozygous diplotype was associated with increased disease risk (OR= 6.8, P= 4.7E-157) and homozygosity was correlated with disease risk for common LOXL1 variants across multi-ethnic populations (Pearson= 0.92, P<0.001).
Conclusions Using exome array data from XFS/XFG cases and controls from the United States, we identify 4 rare protective LOXL1 missense variants and show that the distribution of the corresponding haplotypes and diplotypes are associated with reduced risk of XFS/XFG. The diplotype results also demonstrate that LOXL1 allelic heterozygosity is protective while homozygosity is associated with increased disease risk. These results suggest that LOXL1 minor allele frequency variation among populations, with corresponding variation in genotype heterozgyosity and homozygosity, determines the XFS/XFG association effects and that genotypic effects may also impact protein aggregation involving intrinsic disordered regions.
INTRODUCTION
Exfoliation syndrome (XFS) is a systemic disorder characterized by progressive accumulation of abnormal fibrillar protein aggregates that can obstruct drainage of fluid from the eye, raising intraocular pressure (IOP) and causing exfoliation-related secondary glaucoma (XFG). XFG is the most common secondary glaucoma worldwide and is a major cause of irreversible blindness (Nazarali, 2018). XFS is also associated with pre-mature cataract formation and complications during cataract surgery. Systemic conditions have also been associated with XFS, including cardiovascular disease (Chung, 2018), sleep apnea (Shumway, 2021), and obstructive pulmonary disease (Taylor, 2019).
LOXL1 (lysyl oxidase-like 1) is a major genetic risk factor for XFS/XFG in all populations examined (Aung, 2017), with LOXL1 risk variants occurring in up to 98% of patients (Fan, 2011). Aggregated LOXL1 protein has been shown to be one component of the exfoliation fibrillar material (Zenkel, 2014; Sharma, 2009) that has been identified in extracellular spaces throughout the body but in particular in the ocular anterior segment (Schlötzer-Schrehardt, 2009). The LOXL1 protein includes several intrinsic disordered regions (IDRs) that could be sites of initiation of protein aggregates, and recent studies suggest that deletion of the most disordered regions reduces protein aggregation (Bernstein, 2019).
Two of the major LOXL1 risk variants, (G153D and R141L) are common missence alleles located within the IDR with high likelihood of aggregation (Bernstein, 2019). In particular, the G153D variant falls within the largest protein segment with the highest disorder probability (Figure 1). Of interest, in most populations studied, the common allele ‘G’ at 153 and the common ‘R’ allele at 141 are associated with increased risk; however, in East Asian populations the ‘L’ allele at 141 is associated with increased risk (Hayashi 2008), and in South Africans, the ‘D’ allele at 153 is associated with increased risk (Williams 2010). Interestingly, both R141 ‘L’ and G153 ‘D’ are the ‘common’ alleles in East Asian and South African populations, respectively. A number of studies have investigated the role of LOXL1 genetic variants including effects on protein function (Sharma 2016), gene expression (Pasutto, 2017) and dysregulation related to a long noncoding RNA also located within the LOXL1 genomic region (Hauser 2015). However, none of these studies have observed a consistent effect for the associated risk allele in all populations, which limits the determination of the underlying disease causing mechanism(s).

LOXL1 variants are shown in blue font. The deaminase domain is indicated by the blue line. The region that includes G153D, S159A and A160P is the largest protein region with highest disordered probability indicating greatest likelihood of aggregation. Disordered probably greater than 50% is considered to be significant. The regions resulting in reduced aggregation when deleted were residues 28-95 and residues 125-185. This figure was modified from Bernstein et al. (Adv Protein Chem Struct Biol. 2019;118:273-288).
Recently, a rare LOXL1 protein altering variant was associated with significantly reduced risk in a population of Japanese cases and controls (Aung, 2017). This finding, as well as the observation that the less frequent allele at G153D and R141L is protective in all populations studied so far, suggests that other rare or low frequency alleles may also be protective. To investigate this hypothesis, we examined the effects on risk of rare LOXL1 coding variants in a cohort of XFS cases and controls with European descent from the United States.
METHODS
Study participants
This study adhered to the tenets of the Declaration of Helsinki and has been reviewed and approved by the Institutional Review Boards of the Massachusetts Eye and Ear Infirmary, Harvard School of Public Health, and the Brigham and Women’s Hospital. Informed consent was obtained from the participants after explanation of the nature and possible consequences of the study.
Exfoliation cases were recruited from the Glaucoma service of the Massachusetts Eye and Ear Infirmary (MEEI, Harvard), Duke University School of Medicine Ophthalmology Department, the Glaucoma service of the Mayo Clinic, the Ophthalmology department from the University of Iowa School of Medicine, the Bascom Palmer Eye Institute (University of Miami), and the Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS). The controls were previously genotyped samples from the NHS and HPFS and were over the age of 40 and self-reported Caucasians with European ancestry and are representative of the United States population (Kang 2016). Controls have no evidence of XFS by clinical exam or medical record or repeatedly denied a self-report of glaucoma on biennial questionnaires administered over two decades. We also accessed genotype data from the MEEI and NEIGHBOR controls. Detailed information on this dataset has been previously described (Wiggs, 2012; Margeta 2020).
All XFS cases are self-reported European ancestry Caucasians and are over the age of 40 and have documentation of characteristic ocular exfoliation material at the pupil margin or surface of the ocular lens, either though clinical exam or medical records. Clinical examination included measurement of visual acuity and intraocular pressure, slit lamp biomicroscopy, and fundoscopy. Cases also had visual field assessment primarily using the Humphrey automated visual fields. Individuals were excluded if other types of glaucoma (pigment dispersion, steroid-induced, uveitis) were evident on exam. All cases provided blood samples for DNA extraction.
Genotyping and quality control
Cases were genotyped at the Center for Inherited Disease Research (CIDR; http://www.cidr.jhmi.edu/) using the Illumina OmniExpress+Exome platform that includes 700,000 common SNPs (MAF >0.3) and 250,000 rare or low frequency functional exonic SNPs collected from 12,000 exomes (http://genome.sph.umich.edu/wiki/Exome_Chip). The XFS/XFG controls were previously genotyped using the Omni Express platform. The NEIGHBOR control data set was also genotyped using the Illumina HumanExome BeadArray (Illumina, Inc., San Diego, CA) at CIDR.
Quality control for human exome array genotype data (Igo 2016) for all cases and controls was carried out as follows. The Illumina Genome Studio (Illumina) and PLINK (Purcell 2007) were used for all quality controls (QC) steps except where noted. Basic QC for samples included screens for call rate (≥98.5%) and high (≥95%) concordance with a previous Illumina 660K panel run on the same sample where available (about 80% of samples). We verified recorded sex in the clinical records with genotyped sex by two criteria: mean fluorescence intensity on the X and Y chromosomes, plus genotype heterozygosity on the X chromosome and call rate on the Y, allowing male and female samples to have heterozygous X-linked and successful Y-linked genotypes, respectively. We tested samples for pairwise relationships and unexpected duplication using KING (Manichaikul, 2010).
We verified European ancestry from the first two principal components derived from genotypes at 9000 ancestry-informative markers by means of the SNP weights program (Chen, 2013), including representative HapMap CEU, YRI, CHB, and JPT samples as reference populations. Moreover, we conducted a principal components analysis over 52,040 independent (pairwise r2 < 0.1), common (MAF ≥0.005) SNPs using the smartpca program in EIGENSOFT (Price, 2006) to detect finer population structure.
Initial QC screens for markers included call rate (≥98%) and consistency with Hardy-Weinberg proportions (P > 10−6 by Fisher’s exact test). We screened markers for differences in allele frequency between whole-genome-amplified DNA samples and all other samples by the Fisher’s exact test and removed from analysis all markers with P < 0.0001. All pseudoautosomal, Y-linked, and mitochondrial SNPs were subject to review in Illumina Genome Studio (Illumina, Inc.), and if necessary, were manually re-clustered. We also confirmed genotype clustering for rare (MAF <0.02) variants from fluorescence intensity data by means of zCall, run with a stringent z score threshold of z = 21 for calling heterozygous genotypes. Every rare SNP with two or more additional heterozygous calls by zCall than by GenCall was reviewed in Genome Studio, and if necessary, cluster locations were adjusted manually. Data for four rare coding variants (G120S, S159A, A160P, S427F) included on the genotyping platform were extracted for analyses.
Analyses
Distribution of the four rare LOXL1 variants in cases compared to controls was assessed using the Fisher’s exact test. Haplotypes were constructed using IMPUTE2 (Howie 2009) and haplotype distribution between cases and controls was also assessed using the Fisher’s exact test. Diplotypes were identified using the haplotype data for each study participant. Pearson’s correlation was used to assess percent homozygosity with disease risk for common LOXL1 risk alleles.
RESULTS
Cases and controls
After sample and genotyping quality control, data was available for 1,118 cases and 3,661 XFS/XFG controls. Additionally, genotype data was available for 2,606 primary open angle glaucoma (POAG) subjects without XFS/G from the NEIGHBOR study (Wiggs 2013). Cases and controls have similar proportions of females (Table 1). The cases are on average older than both control cohorts; however, the overall difference was not significant (P= 0.26).
Distribution of rare missense alleles in XFS cases and controls suggest protective effects
We identified four LOXL1 missense alleles with minor allele frequencies (MAFs) less than 1%: G120S, S159A, A160P and S427F (Table 2). Three of these have Combined Annotation Dependent Depletion (CADD) (Kircher 2014) scores >20, indicating potential deleterious effects on protein structure/function. Two missense alleles (S159A and A160P) are located within the LOXL1 intrinsic disordered region (IDR) with high propensity to protein aggregation (Figure 1). All four missense variants were observed more often in controls compared to cases (Table 2) for both the XFS/XFG (combined P= 7.6E-4) and NEIGHBOR control datasets (combined P= 2.2E-3). Using data from an international XFS meta-analysis (Aung 2017), we assessed the allelic distributions for these variants in other populations. While these rare variants are not present in many populations, we were able to observe consistent protective effects in an Icelandic case control study for A160P and S427F and in a South African cohort for S159A (Table 3). G120S was not associated (P> 0.05) with XFS in any other population (1 of 458 cases from Italy and 1 of 2827 cases from Japan and not in any controls from either population (267 and 3013 respectively).
LOXL1 rare haplotypes are more common in controls than in cases
To further explore the protective effects of the rare missense alleles, we investigated the distribution of haplotypes defined by the 4 missense alleles as well as the two common LOXL1 variants, G153D and R141L. All haplotypes that include a rare (minor) missense allele are found more often in controls compared to cases (OR = 0.33, P=1.7E-173) (Table 4). In addition, only the haplotype that includes the common allele at all 6 missense variants was adversely associated with disease risk (OR = 4.76, P= 1.7E-173).
LOXL1 heterozygous diplotypes are more common in controls than in cases
As rare alleles are more likely to be present as heterozygous genotypes, we examined the distribution of the 6-variant diplotypes (derived from the haplotypes in Table 4) among XFS/XFG cases and controls. Similar to the haplotype distribution, only the homozygous diplotype comprised of the common alleles (Diplotype A, Table 5) is associated with increased disease risk (OR= 6.78, P=4.72E-157). All diplotypes that include heterozygous genotypes are protective with the exception of diplotype F, which was associated with an OR of 1.64 (Table 5) but was also very rare and the distribution between cases and controls was not statistically significant (P= 0.55). Overall heterozygous diplotypes are significantly protective (OR= 0.45, P=4.2E-89) (Table 5).
Protective effects of diplotypes are correlated with the number of heterozygous variants
Among the diplotypes identified, the number of heterozygous variants varies from 0 to 3. All diplotypes with heterozygous variants exhibit protective effects, however the observed ORs are lowest for diplotypes with more than one heterozygous variant (OR= 0.05, P= 1.0E-39) (Table 5, Figure 2). In fact, diplotypes with 2 or 3 heterozygous variants are only observed in controls while diplotypes with 1 heterozygous variant are found in both cases and controls, but more commonly in controls (Table 5). Among the single heterozygous diplotypes, the G153D exhibits the most protective effects (Diplotype D, OR= 0.06) followed by S427F (Diplotype E, OR=0.47) and R141L (Diplotype C, OR= 0.70). The distribution of diplotypes among cases and controls does not vary substantially with age in this population (P>0.05, Figure 3).

ORs for diplotypes with at least nominal evidence of association (P<0.05) are displayed on the x-axis for each diplotype labeled on the y-axis. The diplotype letter corresponds to the diplotype from Table 5. Only diplotype A that includes all homozygous genotypes is associated with increased XFS/XFG risk. All others have ORs less than 1 indicating protective effects.
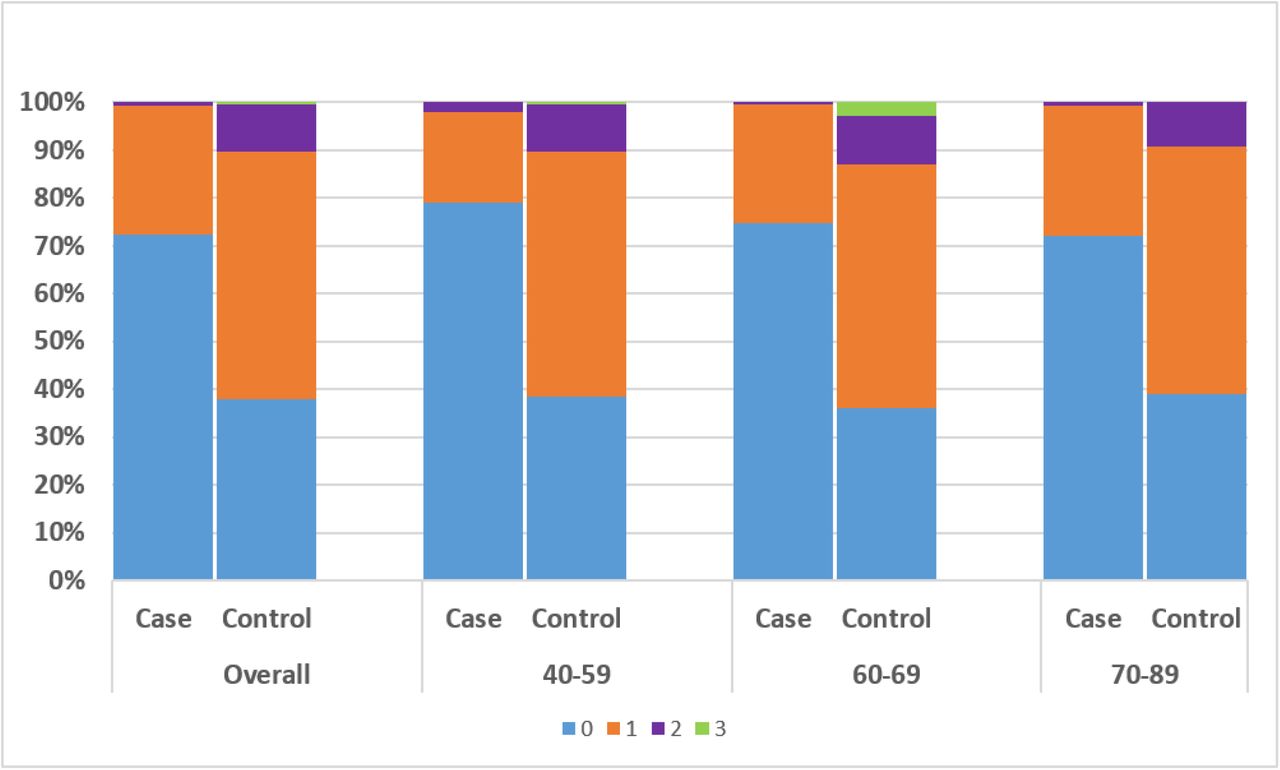
Total diplotypes with 0 heterozygous genotypes (ie homozygous) (blue), 1 heterozygous genotype (orange), 2 heterozygous genotypes (purple) and 3 heterozygous genotypes (green) were summed for cases and controls according to age groups: 40-59, 60-69, 70-89 and compared with the distribution among cases and controls overall. Similar distribution of diplotypes was observed for all age groups (P>0.05) with the exception of the rare diplotypes with 3 heterozygous variants which were found only in controls primarily in the 60-69 age range.
Homozygosity for common G153D and R141L alleles is correlated with disease risk
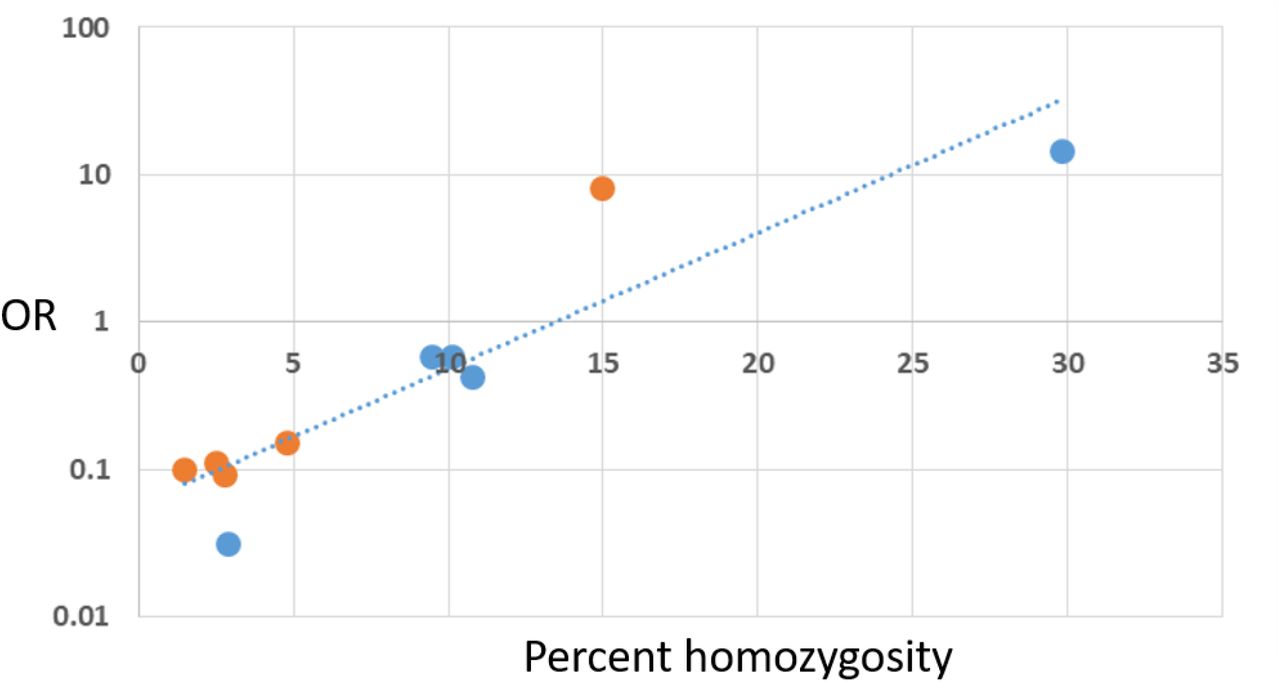
Considering the consistent protective effects for heterozygous variants, we sought to examine the effects of homozygosity on disease risk. As homozygous genotypes are not found for the rare variants this analysis was limited to the alternate alleles for the common variants: allele ‘D’ for G153D and allele ‘L’ for R141L. We used population specific data for the alternate alleles from gnomAD (gnomad.broadinstitute.org/) and association results for similar populations from the XFS international meta-analysis (Aung 2017). Figure 4 shows that the percent homozygosity for the alternate allele for G153D and also for R141L is strongly correlated with the risk effects for the same allele (Pearson = 0.92, P<0.001). The correlation is evident for populations where the alternate allele is the minor allele, and is also evident for populations where the ‘alternate’ allele is actually the common allele (i.e., the L allele at R141L is the ‘common’ allele in East Asians). Indeed this result suggests that the population minor allele frequency is a key factor in determining the observed associations due to the percent homozygosity with the most common alleles having highest percent homozgyosity and highest disease risk.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The percent homozygosity for 5 populations for the ‘alternate allele’ for common missense variants G153D (allele D) (orange dots) and R141L (allele L) (blue dots) are displayed on the x-axis (linear scale) and the corresponding OR for XFS/XFG for that allele in the same population is shown on the y-axis (logarithmic scale). The regression line indicates excellent correlation (Pearson coefficient= 0.9233, P<0.001). The values for each population are shown in Table 6. Percent homozygosity was obtained from gnomAD (gnomad.broadinstitute.org/) and the ORs for XFS/XFG risk were obtained from Khor et al. (Nat Genet. 2017 Jul;49(7):993-1004.).
DISCUSSION
Common LOXL1 coding variants are major XFS/XFG genetic risk factors, but despite the strong association with disease risk, the underlying pathogenic mechanism(s) are not known. The discovery of a protective rare LOXL1 variant in a Japanese case control study provided the first insight that LOXL1 protein-altering variants could reduce disease risk. In this study, we have expanded this initial observation to include 4 other rare LOXL1 missense alleles that are all more commonly found in unaffected controls compared to cases in a U.S. population with European ancestry. Of these variants, some are also found in other populations where they also exhibit protective effects. We also show that haplotypes and diplotypes that include these rare coding variants are more common in controls and that overall heterozygosity is protective and homozygosity is associated with increasing disease risk.
Only the diplotype that includes common homozygous variants at all 6 variants (diplotype A) is associated with increased disease risk. Interestingly, only one other homozygous diplotype was observed (diplotype B), and this diplotype, while rare, is found more often in controls. Diplotype B includes the homozygous ‘alternate’ R141 allele L that is frequently found as a heterozygous variant in the protective diplotypes that also include heterozygous rare variants. Homozygosity of R141L alone would be expected to be associated with increased risk, and it is possible that other, as yet undetected rare variants, are present on this diplotype that are underlying the protective effects. One diplotype (diplotype F) that includes a heterozygous rare variant is found more commonly in cases, however this association was not statistically significant. In this population, diplotype C that includes the single heterozygous genotype for R141L is less protective than diplotype D that includes the single heterozygous genotype for G153D. Accordingly, the cumulative protective effects of rare variants is more evident in diplotypes that include the ‘L’ allele for R141 and do not include the ‘D’ allele for G153.
Two of the rare variants as well as the common variant G153D are located within a LOXL1 intrinsic disordered region that has been shown to be correlated with protein aggregation (Bernstein 2019). This observation, coupled with the protective effects of heterozygosity, including the profound protective effect of heterozygous G153D, suggests that protein variation within this region could reduce protein aggregation. IDRs can contribute to pathogenic aggregation for several neurodegenerative diseases including Alzheimer’s, Parkinson’s, Huntington’s, and prion-related diseases, among others (Eftekharzadeh 2016). Understanding the nature of aggregation induced by IDRs is an area of intense investigation. Protein variants may increase or decrease aggregation (Candelise 2021). A missense allele protective for Alzheimer disease has been shown to reduce abeta aggregation (Das, 2017), supporting a hypothesis that heterozygous LOXL1 variants could potentially reduce protein aggregation through a similar mechanism.
In some populations, the risk allele at the common LOXL1 variants (G153D and R141L) are reversed making it difficult to evoke a specific biological mechanism that explains disease risk. For example, a molecular interaction with the allele associated with risk or allele specific gene expression influencing risk would have to be population dependent. While possible, it seems unlikely that a gene variant could underly a drastically different function in one population compared to another. The results from this study, showing a strong correlation between homozygosity and disease risk combined with overall protective effects observed for heterozygous diplotypes supports a novel hypothesis that the observed risk allele reversal in some populations is simply the result of variation in minor allele frequency leading to variation in relative homozygosity and heterozygosity of risk variants. For example, R141 allele ‘L’ is the minor allele in most populations and is also associated with protective effects in most populations. However, in East Asians, where ‘L’ is the risk allele (Hayashi 2008), the ‘L’ allele is the more frequent allele and therefore is more likely to be homozygous and associated with disease risk. A similar argument can be made for the reversed risk observed for the G153D variant in South Africans (Williams 2010). This hypothesis also provides an explanation for the protective effects of rare LOXL1 protein coding variants as these are likely to be heterozygous due to their low minor allele frequencies. Further work will be required to confirm this hypothesis including testing heterozygous versus homozygous variants for protective functional effects.
There are several limitations of our study. First, our analyses are based on exome array data which does not comprehensively sample all LOXL1 variants. Only the variants present on the array could be examined in this study. In future work, comprehensive sequence data will be necessary to more completely examine the effects of LOXL1 protein altering variants on disease risk. Second, the differences in average ages for cases and controls may influence these results; however, the overall difference in age was not significant, and the distribution of diplotypes among case and control age groups was not significantly different. Finally, because only one haplotype including a rare variant did not also include the protective allele for the common variants, we could not examine the effects of rare variants independent of the common risk alleles. However, the observation that the increased number of risk variants correlates with increased protective effects supports an effect of rare variants independent of the common risk alleles.
In summary, we identify 4 rare protective LOXL1 protein coding variants and show that the distribution of the haplotypes and diplotypes that include these variants suggest an association with reduced risk of XFS/XFG. We also show that only a homozygous diplotype comprised of all common alleles is significantly associated with disease risk and that homozygosity of the common risk variants G153D and R141L is correlated with disease risk across multi-ethnic populations. These results support a novel hypothesis that the reversal of LOXL1 risk alleles observed in some populations is dependent on variation of minor allele frequencies among populations and that LOXL1 allelic homozygosity is associated with increased disease risk potentially due to effects on protein aggregation involving intrinsic disordered regions. Further work identifying additional protective variants and testing effects on protein aggregation will be necessary to confirm this hypothesis.
Data Availability
Exom array data for this study has been deposited in dbGaP.
ACKNOWLEDGEMENTS
Supported by NIH/NEI R01 EY020928, EY015473, and an unrestricted challenge grant from Research to Prevent Blindness (NYC to Icahn School of Medicine at Mount Sinai)
REFERENCES




