
Abstract
As increasing numbers of people recover from and are vaccinated against COVID-19, tests are needed to measure levels of protective, neutralizing antibodies longitudinally to help determine duration of immunity. We developed a lateral flow assay (LFA) that measures levels of neutralizing antibodies in plasma, serum or whole blood. The LFA is based on the principle that neutralizing antibodies inhibit binding of the spike protein receptor-binding domain (RBD) to angiotensin-converting enzyme 2 (ACE2). The LFA compares favorably with authentic SARS-CoV-2 and pseudotype neutralization assays with an accuracy of 98%. Sera obtained from patients with seasonal coronaviruses did not prevent RBD from binding to ACE2. To demonstrate the usefulness of the LFA for measuring antibodies in convalescent plasma used for therapy, we measured conversion of non-immune plasma into strongly neutralizing plasma. This is the first report of a neutralizing antibody test that is rapid, highly portable and relatively inexpensive that might be useful in assessing COVID-19 vaccine-induced immunity.
Introduction
The pandemic virus, Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) continues to be transmitted by person to person spread from its origin in Wuhan, China in December 2019(1–3). Vaccine trials are ongoing, and preliminary results from at least 2 vaccines show that the vaccines elicit protective immunity, but durability of vaccine responses is not known(4).
Molecular tests such as PCR detect SARS-CoV-2 nucleic acid in nasopharyngeal secretions and saliva and are diagnostic of infection(5). These molecular tests can determine the rate of infection and potential re-infection. When negative they indicate that patients have cleared the virus. In response to infection, all immunocompetent hosts generate antibodies against the virus. Patients may have a positive serologic test result as well as a PCR-positive test result if they are early in convalescence(6). For SARS-CoV-2, antibodies to spike protein do not always predict recovery from COVID-19(7).
Although immunocompetent individuals infected with SARS-CoV-2 generate antibodies against the virus, it is essential to know which individuals generate high levels of neutralizing antibodies (NAbs) so that they can resume normal activities without fear of being re-infected and spreading the virus to others(8–10). The goal of COVID-19 vaccines is to induce NAbs and T cell responses that prevent infection/re-infection. Additionally, development of NAbs indicates which individuals might be optimal donors for convalescent plasma protocols. Although NAbs are important for elimination of the virus and protection from subsequent infection, it has been reported by several groups that up to one-third of convalescent plasma samples from individuals who have recovered from COVID-19 do not neutralize SARS-CoV-2 or spike pseudotype virus infection(11–13).
Viral neutralization assays measure levels of antibodies that block infection of host cells. Two main types of viral neutralization assays are utilized for SARS-CoV-2. Authentic microneutralization assays measure reduction of viral plaques or infectious foci in microneutralization assays in susceptible host cells using SARS-CoV-2 under BSL3 conditions. These assays are slow, laborious, require highly trained personnel and require a BSL3 facility. Pseudotype virus neutralization assays have been developed in which SARS-CoV-2 spike protein is expressed in a virus such as vesicular stomatitis virus or lentivirus(14, 15). These assays are faster and less dangerous than authentic microneutralization assays, but still require BSL2 conditions and 24-48 hours for results. Another challenge is that both authentic and pseudovirus virus neutralization assays depend on host cells for infection which adds variability to the assay.
It is known that SARS-CoV-2 uses receptor binding domain (RBD) on spike protein to bind angiotensin converting enzyme 2 (ACE2) on host cells; this appears to be the principal neutralizing domain of SARS-CoV-2(16, 17). Using this knowledge, we developed a lateral flow assay (LFA) that measures levels of (neutralizing) antibodies which block RBD from binding to ACE2. Other groups have developed RBD-ACE2-based competition ELISAs(18)-(19) but none have developed a rapid, highly portable semi-quantitative point-of-care (POC) test.
Methods
Human Subjects and Samples
Peripheral blood, serum and plasma were collected for this study under an Arizona State University institutional review board (IRB) approved protocol #0601000548 and Mayo Clinic IRB protocol #20-004544. Plasma was obtained by ficoll gradient separation of peripheral blood and serum was obtained by centrifugation 30 minutes after drawing blood. Plasma and serum samples obtained from excess clinical samples at Mayo Clinic were pre-existing, de-identified and leftover from normal workflow. COVID-19 samples ranged from 3 to 84 days post PCR positive result. Residual clinical samples were stored at 2-8°C for up to 7 days, and frozen at −80°C thereafter.
Serum samples from patients with non-COVID-19 respiratory illnesses were also tested. These specimens were collected from patients from 2/14/17 – 4/6/20 and were tested by the FilmArray Respiratory Panel 2 (RP2) (Biofire Diagnostics, Salt Lake City, UT) respiratory virus panel as part of routine clinical workflow. Specimens were stored at −80°C until analysis.
Pseudotype Virus Neutralization Assays
Titers were obtained from 60 COVID-19 patient sera using a vesicular stomatitis virus (VSV) spike protein pseudotype assay as previously reported with modifications (14). Patient sera had been stored at −80°C prior to testing. Briefly, SARS-CoV-2 pseudotypes were created by replacing VSV G glycoprotein with SARS-CoV-2 spike protein resulting in a pseudotyped virus with the C-terminal 19 amino acids removed from spike. The pseudotyped virus is called VSV-SARS-CoV-2-S-Δ19CT and was produced by BHK-21 cells. It induces syncytium formation when incubated with vero cells. Two different Vero cells encoding split product luciferase (DSP1 and DSP2) were cultured at a 1:1 ratio as a monolayer. Virus-induced fusion leads to luciferase complementation and in the presence of luciferase substrate EnduRen™ (Promega, Madison, WI) generates luminescence. The higher luminescence, the more infection events by the virus. A human monoclonal antibody (Regeneron, Tarrytown, NY) targeting the SARS-CoV-2 spike protein was diluted to set a cut-off at 50% neutralizing activity and utilized as a single point calibrator. Patient sera with serial dilutions (1:80-1:2560) and the calibrator were pre-incubated with virus for 30 minutes (neutralization) prior to addition to DSP1 and DSP2 Vero cells. The virus-cell mixture was incubated at 37°C in a 5% C02 incubator for a total of 24 hours before luminescence reading; the substrate EnduRen™ was added at 18 hours of incubation. The raw luminescence signal of each sera dilution was compared against the raw luminescence signal of the calibrator. The last consecutive dilution with luminescence signal below the calibrator was considered the end-point titer. Samples were tested in duplicate. The same serum samples, 68 coded sera (10 serum samples at titers of 1:80, 1:160, 1:320, 1:640, 1:1280, 1:2560 and 8 serum samples from individuals never infected with COVID-19) were frozen at −20°C and shipped by Dr. Mills from Mayo Clinic Rochester to Mayo Clinic Arizona and the Lake Lab for LFA testing.
Authentic SARS-CoV-2 Microneutralization Assay
The authentic microneutralizing assay was performed using a recombinant SARS-CoV-2 that expresses mNeonGreen (SARS-CoV-2ng) during replication in permissive cells, as previously described (20). Inhibitory concentrations for which 50% of virus is neutralized by serum antibodies (IC50 values) were obtained on a unique set of 38 COVID-19 sera. Sixty µl aliquots of SARS-CoV-2ng were pre-incubated for 1 h in 5% CO2 at 37°C with 60µl serial 2-fold serum dilutions in cell culture media, and 100µl were inoculated into Vero-E6 monolayers in black polystyrene 96-well plates with clear bottoms (Corning, Tewksbury, MA). Each serum was tested in duplicates. The final amount of the virus was 200 PFU/well, and the starting serum dilution was 1:20 and the end dilution was 1280 unless an IC50 was not reached in which case serum was diluted to 1:10240. Cells were maintained in Minimal Essential Medium (ThermoFisher Scientific, Waltham, MA) supplemented by 2% FBS (HyClone, Logan, UT) and 0.1% gentamycin in 5% CO2 at 37°C. After 2 days of incubation, fluorescence intensity of infected cells was measured at a 488 nm wavelength using a Synergy 2 Cell Imaging Reader (Biotek, Winooski, VT). The signal readout was normalized to virus control aliquots with no serum added and was presented as the percentage of neutralization. IC50 was calculated with GraphPadPrism 6.0 software. Work with infectious SARS-CoV-2ng was performed in a BSL-3 biocontainment laboratory of the University of Texas Medical Branch, Galveston, Tx, Galveston National Laboratory.
Serologic Antibody Assay
The Ortho Vitros Anti-SARS-CoV-2 IgG test was performed on the Ortho Clinical Diagnostics Vitros 3600 Immunodiagnostics System (Ortho, Raritan, NJ) following manufacturer’s instructions at the Mayo Clinic. This assay is approved for use in clinical testing under FDA Emergency Use Authorization to qualitatively detect antibody to the S1 subunit of SARS-CoV-2 spike protein. Results are reported as reactive (S/CO ≥ 1.0) or Nonreactive (S/CO <1.0). Quantitative results are not reported for clinical workflows but are measured and recorded in this assay. Specimens were tested within 7 days of collection and were stored at 2-8°C per manufacturer’s instructions. This assay was performed after the VSV assay was performed on the set of 60 serum samples, then the samples were frozen at −80°C before running them on the LFA. A separate set of 38 samples was run concurrently in the SARS-CoV-2 authentic microneutralizing antibody assay, the LFA, and the Ortho Vitros Anti-SARS-CoV-2 IgG assay.
Lateral Flow Neutralizing Antibody Assay
The Lateral Flow Neutralizing Antibody assay was developed to detect antibodies that compete with ACE2 for binding to RBD. The LFA cassette contains a test strip composed of a sample pad and blood filter, conjugate pad, nitrocellulose membrane striped with test and control lines, and an absorbent pad to wick excess moisture (Axim Biotechnologies Inc, San Diego, CA). The blood filter allows serum to pass through but prevents red blood cells and leukocytes from interfering with the assay. Test strips are secured in a cassette that contains a single sample port (Empowered Diagnostics, Pompano Beach, FL). For procedural control purposes, the LFA also contains a control mouse antibody conjugated to red gold nanospheres and corresponding anti-mouse control line.
LFAs were performed at ambient temperature and humidity on a dry, flat surface and left to run undisturbed for 10 minutes prior to reading results. First, 10µl of plasma, serum or whole blood were transferred to the cassette sample port and immediately followed by two drops (∼50µl) of chase buffer. After 10 minutes, each test was placed in an iDetekt RDS-2500 LFA reader (Austin, TX) and the densities of both test and control lines were recorded electronically. This assay was performed on the VSV subset (n = 60) as well as the authentic SARS-CoV-2 microneutralizing antibody assay subset (n = 38).
Serum samples from COVID-19 patients as well as sera from patients with PCR-confirmed seasonal respiratory viruses including PCR-confirmed seasonal coronaviruses were tested in the LFA as controls. Twenty-seven sera from patients with seasonal respiratory viruses were tested in the LFA in a manner identical to COVID-19 sera.
The test leverages the interaction between RBD-conjugated green-gold nanoshells (Nanocomposix, San Diego, CA) that bind ACE2 at the test line when RBD-binding/neutralizing antibodies (RBD-NAbs) are absent or low. As indicated by the competitive nature of this assay, test line density is inversely proportional to RBD-NAbs present within the sample. Thus, an absent or faint test line indicates high levels of RBD-NAbs, whereas a dark or strong test line suggests lack of RBD-NAbs within a given plasma sample (Figure 1A).


(A) Schematic of Neutralization LFA. Below each graphic is a representative image of a lateral flow strip demonstrating actual line density. Addition of non-COVID19-immune serum or plasma (top) does not block binding of RBD-beads (green particles) to ACE2 resulting in the RBD-bead–ACE2 complex creating a visible line. Addition of patient serum with moderate titer NAbs to the sample pad creates a weak line (middle). Addition of patient serum with high titer NAbs (> 1:640) blocks binding of RBD-beads to ACE2 such that no line is observed at the test location on the strip (bottom). Red control line represents capture of a mouse monoclonal antibody coupled to red beads. (B) Scorecard for measuring levels of NAbs. Red control line across from the “C” on the cassette indicates that the test ran properly and the green test line across from the “T” can be used to measure the ability of plasma or serum to block RBD on gold nanoshells from binding to ACE2. (0) represents patient serum producing a visually non-existent line with density units of 10,095 and an IC50>500 (IC50=1151); (1) represents patient serum with a line density of 132,503 and an IC50 of 396; (2) represents patient serum with a line density of 239,987 and an IC50 of 243; (3) represents patient serum with a line density of 485,665 and an IC50 of 96.
As a semi-quantitative test, the results of the LFA can be interpreted using a scorecard or a densitometer. Figure 1B demonstrates the scorecard. Red control line across from the “C” on the cassette is composed of a mouse monoclonal antibody coupled to red-gold beads binding to an anti-mouse IgG. A red line indicates that the test ran properly and the green test line across from the “T” can be used to semi-quantitatively measure the ability of plasma, serum or whole blood to block RBD on green-gold nanoshells from binding to ACE2.
To demonstrate the ability of the test to measure NAbs in whole blood, 10µg of a neutralizing monoclonal antibody (mAb) based on the sequence of B-38(20)(Axim Biotechnologies, Inc, San Diego, CA) was mixed with 10µl of normal donor whole blood collected in a heparinized blood collection tube. Two-fold dilutions were made in whole blood to a final concentration of 0.625µg/ml neutralizing mAb. Then, 10µl of each dilution were transferred to LFA cassettes, chased with 50µl running buffer, and read with an LFA reader after 10 minutes.
Conversion of Non-immune Normal Human Plasma (NIHP) into Neutralizing Plasma
To convert NIHP into strongly neutralizing plasma (SNP), plasma from a convalescent donor who demonstrated the ability to block RBD from binding to ACE2 (M21) was mixed with NIHP collected prior to December 2019. For example, for a 1% mixture, 1µl of SNP was mixed with 99µl NIHP; for a 5% mixture 5µl of SNP was admixed with 95µl NIHP, etc. We performed the test with 10µl of 1%, 5%, 10%, and 20% SNP admixed into NIHP.
Data Analysis
Pearson’s correlation (r) was conducted to assess the strength and significance of associations between the LFA, the Ortho Vitros Anti-SARS-CoV-2 IgG test, VSV titers and IC50 values. Regression analysis using IC50 values was performed to evaluate consistency(18) while Bland-Altman plots were constructed to assess agreement and bias(21, 22). Correlation analysis was conducted using IBM SPSS (Armonk, NY). Regression analysis was performed using Microsoft Excel (Redmond, WA); Bland-Altman plots were visualized using IBM SPSS. For two-group analysis, IC50 values corresponding to >240 were categorized as titer of ≥1:320 (neutralizing), whereas IC50 values ≤240 were categorized as ≤1:160 (non-neutralizing). Receiver operating characteristic (ROC) analysis was performed to assess classification accuracy, sensitivity, and specificity of the LFA and Ortho Vitros Anti-SARS-CoV-2 IgG test methods in assessing neutralizing capacity; optimal cutoffs for each method were established to maximize area under curve (AUC)(23, 24). ROC analysis was conducted using R language in the RStudio environment (version 3.6.2; RStudio PBC, Boston, MA). All analyses were conducted using raw values; data were not normalized, transformed, or scaled.
Results
The lateral flow assay reported here is a rapid 10-minute POC test. As shown in the schematic in Figure 1A, this test utilizes serum, plasma or whole blood to semi-quantitatively measure levels of NAbs. An example of strong, moderate and non-neutralizing sera is shown in Figure 1B. As diagrammed in Figure 1A, levels of NAbs in serum or plasma are reflected by the intensity of the test line which can be read on a hand-held densitometer or compared visually to a scorecard (Figure 1B). Strong neutralization results in a weak line because neutralizing antibodies are binding RBD on green gold beads and preventing RBD-beads from binding to the ACE2 cellular receptor at the test line.
We tested 60 serum samples with known neutralization titers obtained in a VSV-based spike pseudotype assay in our LFA. De-identified serum samples were sent from Mayo Clinic Rochester to our laboratories at Mayo Clinic Arizona and run in a blinded manner. The LFA result compared favorably with serum titers, especially at higher titers and correctly distinguished all eight non-neutralizing serum samples (Figure 2A).

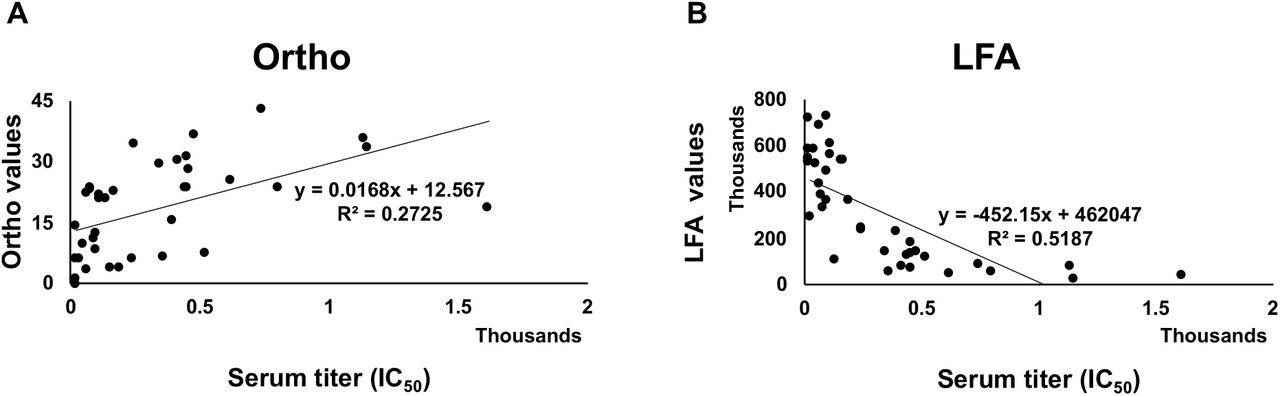
(A) Comparison of RBD-ACE2 competition LFA Density Units with VSV-Spike pseudotype virus assay titers using 60 convalescent serum samples. Titers from the pseudotype assay are shown on the X-axis. Scatter plots with bar graph including standard deviation of the mean for 10 serum samples at each titer except for negative donor serum which is eight samples. (B) Comparison of RBD-ACE2 competition LFA with IC50 values determined in a SARS-CoV-2 microneutralization assay on 38 samples (collected 3 to 90 days after PCR positive result). Ranges of IC50 values are shown on the X-axis plotted against LFA line density units on the Y-axis.
To further support the application of our lateral flow test to measure levels of antibodies that neutralize SARS-CoV-2, we tested a different set of 38 serum samples that were assigned IC50 values in an authentic SARS-CoV-2 microneutralization assay(25). Again, the experiment was performed in a blinded manner such that personnel running either the LFA or the microneutralization assay did not know the results of the comparator test. When line densities from the LFA were plotted against IC50 values determined in the SARS-CoV-2 microneutralization assay, serum samples with strong neutralization activity demonstrated low line densities; this indicates that neutralizing antibodies inhibited RBD from binding to ACE2 (Figure 2B).
Next, we determined if our test detected any neutralization activity in serum samples collected from patients with other PCR-confirmed respiratory viruses including seasonal coronaviruses (Figure 3A) and for serum samples collected prior to December 2019 (Figure 3B). None of the seasonal respiratory virus plasma samples, including pre-December 2019 samples showed neutralizing activity.

A) Serum samples collected with PCR-confirmed diagnosis of seasonal respiratory viruses (Coronavirus OC43, blue; Coronavirus HKU-1, green; Coronavirus NL-63, pink; influenza A, orange, influenza B, red ; parainfluenza, purple ; rhinovirus, teal ; respiratory syncycial virus, yellow ; and adenovirus, black were run on the LFA as described in Methods. A positive control serum from a convalescent COVID-19 patient is shown on the far right of the bar graph in white. B) Serum samples collected pre-December 2019. Cutoff value of 263,000 density units was calculated based on receiver operating characteristic curves (see Figure 6).
We evaluated how the Ortho Vitros Anti-SARS-CoV-2 IgG test and our LFA compared to IC50 values determined in an authentic SARS-CoV-2 microneutralization assay using 38 COVID-19 sera. FDA guidance indicates that an Ortho Vitros Anti-SARS-CoV-2 IgG test of ≥12 meets a threshold for convalescent plasma use in patients with COVID-19. To determine the agreement between our lateral flow assay and the Ortho Vitros Anti-SARS-CoV-2 IgG test, density units from our LFA and values from the Ortho test were regressed onto IC50 values (Figure 4).

Regression analysis between (A) LFA and serum titer, and (B) Ortho Vitros SARS-CoV-2 IgG test and titer. Regression plots show explained variance (R2) between compared methods. Thirty-eight samples were tested.
LFA values accounted for roughly 52% of observed variance in IC50 values, while Ortho Vitros Anti-SARS-CoV-2 IgG test accounted for approximately 27% of IC50 variance. Since the format of the LFA reports decreasing density values as neutralization increases, LFA was significantly inversely correlated with IC50 values (r = −0.720, p < 0.001), while Ortho Vitros Anti-SARS-CoV-2 IgG test values showed a significant positive correlation to IC50 values (r = 0.522, p = 0.001). Additionally, LFA and Ortho Vitros Anti-SARS-CoV-2 IgG test values showed a significant relationship with each other (r = −0.572, p < 0.001).
To evaluate bias between the assays, mean differences and 95% confidence intervals (CIs) were calculated and plotted alongside limits of agreement (Figure 5). Both LFA and Ortho Vitros Anti-SARS-CoV-2 IgG test values showed high agreement with titer, although Ortho Vitros Anti-SARS-CoV-2 IgG test showed a tendency to underestimate neutralizing capacity while the LFA method showed no bias.

Bland-Altman plots showing bias (mean difference and 95% CI) and computed limits of agreement (mean difference ± 2SD) between (A) Ortho Vitros Anti-SARS-CoV-2 IgG test and IC50 values and (B) our LFA and IC50 values. Thirty-eight samples were tested.
Univariate ROC analysis was performed to assess the performance of the newly developed LFA and Ortho assay to classify non-neutralizing (Neg—1:160), and neutralizing groups (≥1:320) (Figure 6). As can be seen in Figure 6B and 6D, the LFA misclassified one non-neutralizing sample (Neg—1:160) as neutralizing (≥1:320). In contrast, Ortho Vitros Anti-SARS-CoV-2 IgG test misclassified that same non-neutralizing sample as neutralizing, in addition to incorrectly classifying five additional neutralizing samples as non-neutralizing. Importantly, our LFA method exhibited a greater dynamic range of differential values as compared to Ortho’s test. (Figure 5), translating to superior classification accuracy of the LFA method in detecting neutralizing samples (titer ≥ 1:320) compared to Ortho Vitros Anti-SARS-CoV-2 IgG test (Figures 6A and 6C).

(A) Univariate ROC analysis of Ortho Vitros Anti-SARS-CoV-2 IgG test for discrimination of neutralizing samples (≥1:320) [AUC: 0.856, 95% CI: 0.697—0.953, sensitivity = 0.7, specificity = 0.9]. (B) Box plot of Ortho Vitros Anti-SARS-CoV-2 IgG test values between neutralizing (≥1:320) and non-neutralizing (Neg—1:160) groups. (C) Univariate ROC analysis of LFA for discrimination of neutralizing samples (≥1:320) [AUC: 0.978, 95% CI: 0.908—1.0, sensitivity = 1.0, specificity = 0.9]. (D) Box plot of LFA values between neutralizing (≥1:320) and non-neutralizing (Neg—1:160) groups.
Our LFA showed high accuracy for classification of neutralizing samples (AUC = 0.978), while the Ortho Vitros Anti-SARS-CoV-2 IgG test showed modest accuracy for classification of neutralizing samples (AUC = 0.856). Furthermore, while the LFA method showed a narrow confidence interval (95% CI: 0.908—1.00), ROC analysis of Ortho Vitros Anti-SARS-CoV-2 IgG test values showed a wider range (95% CI: 0.697—0.953), indicating less certainty. Notably, while both methods showed roughly 90% specificity, Ortho Vitros Anti-SARS-CoV-2 IgG test Vitros Anti-SARS-CoV-2 IgG test showed only 70% sensitivity. In contrast, the novel LFA method showed perfect sensitivity (100%) in this analysis of 38 samples.
Optimal cutoffs were computed to maximize AUC. For the LFA, density unit values below 263,000 classify samples as neutralizing and correspond to titers ≥1:320. Density unit values above this LFA cutoff classify samples in the non-neutralizing group. For the Ortho Vitros Anti-SARS-CoV-2 IgG test, values between 0 and 23.3 were representative of non-neutralizing capacity, whereas values above 23.3 were reflective of the neutralizing group. If we used the FDA cutoff of 12 instead of the 23.3 value calculated using our results, AUC would be reduced to 0.69 (sensitivity = 0.55, specificity = 0.83).
Polyclonal antisera was used in this study, not an individual Mab, so the limit of detection (LoD) does not exactly apply). However, we can calculate the LoD by line density based on the method of Armbruster and Pry(26) as the highest line density from samples still containing neutralizing antibodies and distinguishable from blank except that the operand sign was changed because due to the competitive format of the LFA: LoD= limit of blank (LoB) – (1.65* SDlow conc sample): LoD=1,047,382-(1.65 * 63,769)= 942,481 Test Line Density Units. The average line density observed for the top 10 donors who demonstrated the strongest ability to prevent RBD from binding to ACE2 was 20,706.
The principle of infusing convalescent plasma from recovered individuals into patients fighting COVID-19 is to transfer NAbs from the donor to the recipient, as has been done for several other diseases(27–29). The rapid test described here, could quickly and efficiently classify COVID-19 convalescent plasma (CCP) units so that the sickest patients might receive the most potently neutralizing plasma. Although levels of neutralizing antibody to achieve in patients receiving CCP remains undefined, our point-of-care test might be useful at the bedside to monitor a patient receiving highly neutralizing CCP as he/she begins to demonstrate NAbs in circulation, as shown in Figure 7A. It could help in deciding whether to administer another unit of highly neutralizing CCP to a patient fighting COVID19. The same scenario might also be applied to hyperimmune gammaglobulin or neutralizing monoclonal antibody infusion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Highly neutralizing plasma from an individual recovered from COVID-19 (M21) converts non-immune human plasma (ND82) into highly neutralizing plasma; 10µl of plasma was added to each lateral flow cassette. Text below each lateral flow cassette indicates the percent M21 and percent ND82 plasma used to run the lateral flow test. (B) Titration of anti-RBD neutralizing mAb into 10µl of heparinized whole blood. For both types of cassettes, plasma or blood sample was immediately chased with 50µl sample buffer. Densities were read and cassettes were imaged after 10 minutes development.
To demonstrate the utility of our LFA to measure NAbs in whole blood, we used a lateral flow strip with a blood filter and performed an experiment in which a neutralizing monoclonal antibody based on the B-38(20) sequence was titrated into whole blood as shown in Figure 7B. Density units were not obtained in these experiments but both Figures 7A and 7B demonstrate the semi-quantitative nature of this test to visually distinguish different levels of NAbs in plasma and whole blood.
Discussion
Serologic tests that detect responses to infection are an important population surveillance tool during pandemics because they provide data on pathogen exposure, especially when a subset of the population is asymptomatic and would not have been diagnosed by molecular methods(30, 31). While over 90% of individuals diagnosed with COVID-19 generate antibody responses(30) we are only beginning to learn about the prevalence and duration of NAbs induced by a natural infection. This lack of knowledge is mainly due to the fact that virus-based neutralization assays require i) BSL2 or BSL3 facilities, ii) highly skilled personnel, iii) permissive cells and quantified virus, and iv) take longer than 24 hours to obtain results. Since every viral vaccine, including COVID-19 vaccines, administered to humans is designed to elicit antibodies that neutralize virus by blocking host cell infection, development of NAbs is a hallmark of protection from disease. Therefore, there is a need for rapid, accurate tests that measure levels of NAbs. Furthermore, in a post-vaccine world, longitudinal measurement of protective, neutralizing antibodies is crucial. Measurement of anti-viral T cells also indicates that infected individuals could destroy infected host cells if virus escapes neutralization.
We developed a rapid, 10-minute lateral flow test that measures levels of NAbs in serum and plasma. As shown in Figure 2, the lateral flow test correlates well with serologic titers determined using a VSV-based pseudotype assay, and IC50 values in an authentic SARS-CoV-2 microneutralization assay, especially when serum sample titers are ≥640 and IC50 values are >250. Samples with less potent NAbs in both viral assays correlated with decreased ability to block RBD from binding to ACE2 in the LFAs.
The LFA and Ortho Vitros Anti-SARS-CoV-2 IgG test methods showed a strong, significant correlation with each other (r = −0.572, p < 0.001), displaying an appreciable degree of linear relation (r = −0.720, p < 0.001)(32). Importantly, LFA accounts for 52% of observed IC50 variance (R2 = 0.5187) while, in comparison, Ortho Vitros Anti-SARS-CoV-2 IgG test accounts for 27% (R2 = 0.2725). Although absolute quantitation of a construct demands an excellent coefficient of determination (R2 ≥ 0.99)(33), variables with R2 ≥ 0.5 are highly predictive in univariate regression models while measures with R2 < 0.5 are recommended for use in multivariate models in combination with complementary measures to increase predictive accuracy(34, 35). Additionally, Bland-Altman analysis (Figure 5) showed the Ortho Vitros Anti-SARS-CoV-2 IgG test to be prone to underestimation of IC50 values. In contrast, the LFA method did not exhibit any over- or underestimation bias. Furthermore, across mean values for both methods, LFA showed discrete differential values while Ortho Vitros Anti-SARS-CoV-2 IgG test struggled to differentiate samples with high neutralizing capacity (IC50 values).
Since the Ortho Vitros Anti-SARS-CoV-2 IgG test uses S1, which includes RBD, it is likely that antibodies reactive with other parts of S1–even some that may neutralize via N-terminal domain–are responsible for increased reactivity to S1 when they are not neutralizing in our RBD-ACE2 competition assay. Advantages of our POC test is that it can be inexpensively and rapidly deployed in studies to determine levels of NAbs that protect against re-infection and limit transmission of the virus. Moreover, rapid inexpensive tests can be used longitudinally to evaluate duration of protective immunity in both naturally infected and many more vaccinated individuals than could ever be evaluated using BSL2 or BSL3-based neutralization assays.
In the setting of much debated CCP, a rapid test could be used to measure levels of NAbs in a CCP product prior to infusion, or potential donors who recovered from COVID-19. During infusion of CCP, a rapid test could be used to help decide if a patient fighting COVID-19 requires another unit of highly neutralizing plasma. If a particular donor’s CCP has very high levels of CCP, patients might need only one unit. On the other hand, if donor CCP contains moderate levels of NAbs, the patient may require multiple units to achieve therapeutic levels of NAbs. However, since we do not know what an adequate therapeutic dose of NAbs is, this rapid test could be a valuable tool in trials to determine under what conditions CCP, hyperimmune globulin, and neutralizing mAbs are effective therapeutic agents for COVID19 patients.
Limitations of the LFA are that it uses only the RBD portion of SARS-CoV-2 spike protein. Although the vast majority of reports indicate that the principle neutralizing domain is RBD portion of spike protein, mAbs have been reported that neutralize SARS-CoV-2 by binding to the N-terminal domain of spike protein(36, 37). Also, since the spike protein assumes several conformations during viral binding and entry(38), neutralizing epitopes exist on the quaternary structure of spike(37). Although RBDs on the nanoparticles may associate, it is unlikely they assume a native quaternary conformation.
Other limitations are the binary nature of the data analysis (neutralizing and non-neutralizing) of a continuous assay. Clearly, line densities demonstrate moderate levels of neutralization. Since blood draws and subsequent assays are a “snapshot in time” of neutralizing antibody activity, levels were undoubtedly increasing in some patients and decreasing in others. LFAs are generally inexpensive and highly portable compared to other laboratory-based tests, so neutralizing antibody levels could be measured using the LFA to longitudinally assess protective neutralizing antibody immunity. Another limitation is that the LFA does not differentiate high affinity anti-RBD NAbs from an abundance of lower affinity anti-RBD NAbs. In related experiments, we have observed patient sera that bind strongly to RBD, but do not demonstrate neutralizing activity (data not shown).
This test may prove very useful in monitoring COVID-19 vaccine recipients. Although vaccines have now been approved for distribution and administration, durability of protective immunity elicited by any COVID-19 vaccine is unknown. It is the goal of all COVID-19 vaccines to induce protective NAbs. However, since clinical trials of vaccines have enrolled 30,000 to 60,000 participants, it is not logistically possible to draw a tube of blood from each vaccine recipient longitudinally to determine duration of protective NAbs. Application of our test in vaccine recipients using a drop of blood obtained from a finger-stick as shown in Figure 7B might lead to more comprehensive longitudinal monitoring of increases and decreases in protective humoral immunity.
Data Availability
All data in the manuscript is available.
Data availability
The authors declare that data supporting the findings of this study are available within the paper. Additional data mentioned but not shown are available from the corresponding author upon reasonable request.
Footnotes
↵* Co-first authors
References




