
Abstract
Deer tick virus (DTV) is an emerging pathogen in North America. This virus can cause nervous system complications such as encephalitis in humans. Further, no data has been surmounted around long-term effects of infection from DTV patients across variable age groups. Diagnostic tools of DTV used by government laboratories are based on RT-PCR using patient serum or ticks. This paper explores the feasibility of a colorimetric loop-mediated isothermal amplification (LAMP) assay to create a point-of-care diagnostic methodology for use in field and in primary care. LAMP consists of six primers that bind to target DNA and amplifies variable length nucleotide strands that can be visualized through side reactions or via electrophoresis. First, a viable LAMP primer set, and a primer set that dimerizes and amplifies DNA regardless of compatibility were created in silico and validated in vitro. Then, a specific LAMP assay was developed. Our findings showed this method can be performed within 30 minutes and can measure with limits of detection comparable to PCR.
Introduction
Deer tick virus (DTV) has a single-stranded RNA genome and belongs to the genus Flavivirus in the family Flaviviridae. Other members of this virus genus and family are dengue virus, Zika virus and West Nile virus. The genome length of DTV is roughly 10,800 bases and it known to cause tick-bourne encephalitis (Artsob et al., 2001).
The increasing presence of DTV throughout North America is coupled with various tick species (CDC, 2019). DTV is also referred to as the second lineage Powassan virus (POW2), deviating from a native strain in Europe and Russia. POW1, or Powassan virus lineage I, has a variable genetic diversity along the theoretical divergence cutoff at 84% nucleotide identity to DTV, 94% amino acid identity, and is estimated to have diverged around 200 years ago (Pesko et al., 2010). This cutoff provided the difference between the two Powassan viruses on the cladogram of Flaviviridae (Telford III, 1997). Patient testing for DTV is usually performed in response to severe cases where symptoms arrive suddenly. Most people tested for DTV have symptoms of acute encephalitis or overactive inflammation of the brain, which leads to death in some cases without proper medical treatment (Solomon et al., 2018; Tavakoli et al., 2009). Remaining cases go unbeknownst because testing measures are not approached from typical or asymptomatic standpoints as other tick-borne infections. Current diagnostic testing is limited to the Center of Disease Control and its partnering laboratories using either reverse transcription-polymerase chain reaction (RT-PCR) or immunohistochemical techniques (CDC, 2019; Morozova et al., 2002). A sample of serum or cerebral spinal fluid is collected and sent to one of the offsite diagnostic centers, further complicating the testing process and taking up to a few days for results to become available.
A more time effective detection method than RT-PCR would be beneficial for point-of-care testing (POCT), as well as for field surveys of tick samples that are performed by state game commissions. Three different species of ticks, i.e., Ixodes cookie, Ixodes marxi, and Ixodes scapularis, have the potential to transmit DTV from their main vertebrate hosts, i.e., woodchucks, squirrels, and white-footed mice, to humans (Brackney et al., 2010). White-tailed deer is also a known host to Ixodes scapularis. DTV has no known pathology on these intermediate mammal hosts, but concern is comprehensible for humans (CDC, 2019). The main tick vector that raises human concern is Ixodes scapularis. This tick species has a parasitic relationship with white-tailed deer, Odocoileus virginianus, and other species of deer that humans encounter frequently in rural areas (Brackney et al., 2010). This host relationship is unique to DTV and unlike the previously mentioned POW1 strain. The prevalence of outdoor recreation and leisure activities give exposure to the tick vectors and potential pathogens to be transmitted.
Monitoring the presence of DTV provides the first steps to surveying disease spread and to developing mitigation efforts. A rapid DTV detection assay like colorimetric loop-mediated isothermal amplification (LAMP) would prove useful for field surveys and POCT. LAMP is an alternative nucleic acid amplification technique that has gained popularity in the past decade (Anupama et al., 2019; Chen et al., 2014; Lamb et al., 2018; Lau et al., 2015; Luo et al., 2011; Shin et al., 2018; Tomlinson, 2008). LAMP is a nucleotide amplification process that occurs through the binding of specially designed primers at approximately 60°C and will generate variable ladder structures that can be visualized in two manners. Colorimetric LAMP does not require expensive and sophisticated equipment or visualization techniques other than the naked eye. In addition, it is inexpensive and quick. For example, the Colorimetric Warmstart® MasterMix (New England Biolabs, Cat# M1800S) LAMP technique, utilized in this paper, is up to five times quicker than PCR and changes color for visual assessment of the results (Nagamine et al., 2002). The colorimetric test starts out as pink and turns a bright yellow color if the result is positive. Color change is dependent on the emission of protons through nucleotide assimilation on target DNA (Mitra et al., 2019). The amplified DNA can then be confirmed through electrophoresis on an agarose gel followed by staining with ethidium bromide or Coomassie Blue® to display banding, validating the presence of DTV in the tick sample tested. In this paper we present our preliminary data on the development of a colorimetric LAMP detection of DTV and discuss perspectives of our research for POCT.
Materials and Methods
Nucleotide Sequence Analysis and Homology Analysis for DTV Strains
The greatest amount of genetic variance between strains of DTV were estimated through a maximum composite likelihood analysis method. The greatest amount of conservation was determined to be at the RNA-dependent RNA Polymerase (RdRp) coding site using the codon based Z-test. Two bioinformatics methods, i.e., the codon-based Z-test, and the UPGMA phylogenetic tree method, were used to analyze the RNA-dependent RNA polymerase (RdRp) coding region of the DTV genome of four strains and determine nucleotide sequence conservation. These tests were performed using the software suite MEGA7: Molecular Evolutionary Genetic Analysis version 7.0 (Kumar et al., 2015) using DTV accession numbers MG196295.1, EU338403.1, NC003687.1, and HM991145.1 retrieved from GenBank, an open-source genetic sequence database operated by the National Institute of Health (Soloman et al., 2018; Tavakoli et at., 2008, Unpublished Accession, 2010).
Codon Based Z-Test
Alignment tools were utilized to investigate the homology between the conserved sequences of DTV. Positive selection for the resistance to mutation was analyzed through the codon-based Z-test as defined by Equation 1. When analyzing the homology, the null hypothesis is regarded such that the sequences are not similar and only by rejecting the null hypothesis when p < 0.05, can we assume that the sequences are considered homologous.
 Equation 1. The Z-test for codon homology testing across multiple DTV genomes. Variables denoted in the equation are defined as the number of nonsynonymous substitutions per nonsynonymous site (dN), number of synonymous substitutions per synonymous site (dS), and the respective variances of these variables (Var(dN) and Var(dS)).
Equation 1. The Z-test for codon homology testing across multiple DTV genomes. Variables denoted in the equation are defined as the number of nonsynonymous substitutions per nonsynonymous site (dN), number of synonymous substitutions per synonymous site (dS), and the respective variances of these variables (Var(dN) and Var(dS)).
Primer Design
Primers for the LAMP assay were designed through the PrimerExplorerV5 LAMP Primer Designer using the following criteria: (1) genomic regions with a GC ratio of 50-60%, (2) no more than four successive G’s or C’s to prevent primers from binding too tightly to the target DNA strand, and (3) stability of F3 and B3 primer ends be lower than −4.00 to create the most stable 5’ and 3’ loop structures for LAMP proliferation (Wang et al., 2015). The region of the viral genome that was selected for primer design was NS5a that codes the non-structural polymerase.
Primer Constructs
Below are the two primer constructs evaluated in this paper (Table 1 and Table 2).
Primer candidate 1 sequences generated using the PrimerExplorerV5 software with respective loop primers (LF and LB).
Primer candidate 2 sequences generated using the PrimerExplorerV5 software with respective loop primers (LF and LB).
Reagents added to reaction vessels to perform the colorimetric LAMP assay based on New England Biolabs® protocol.
The probability of rejecting the null hypothesis of strict-neutrality (dN = dS) (below diagonal) is shown. Values of P less than 0.05 are considered significant at the 5% level and are highlighted. The test statistic (dN - dS) is shown above the diagonal. dS and dN are the numbers of synonymous and nonsynonymous substitutions per site, respectively. The variance of the difference was computed using the analytical method. Analyses were conducted using the Nei-Gojobori method [17]. This analysis involved 4 nucleotide sequences. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There was a total of 3556 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [16]
LAMP primers were determined using the PrimerExplorerV5 software and the AF311056.1 accession (Kuno et al., 2001). The following primers have been designed using the PrimerExplorerV5 software and have been experimentally tested in the laboratory. Primer candidates 1 and 2 are displayed with length and position on accession sequence in Tables 1 and 2, respectively. Four primers are designed within the first iteration of software for binding to a target sequence. Figure 1 depicts the amplification process that occurs to create the loop products generated by Bst polymerase. The inner primers denoted Forward Inner Primer (FIP) and Backward Inner Primer (BIP) are the central component of amplification and loop structure formation. Two additional primers F3 and B3, complementary to F3c and B3c respectively, can bind to the target DNA and displace the amplification product. This allows a repetitive, rapid amplification process. The final two primers, Loop Front (LF) and Loop Back (LB) are not pictured in Figure 1 but are able to further amplify and accelerate the loop creation process by providing templates. The reaction is still possible without LF and LB, but a greater limit of detection can be achieved using loop primers. The constructs for LF and LB were generated by inserting the initial primer file into the PrimerExplorerV5 software and following the same protocol.

Each arrow indicates a new step of the process and arrows indicate polymerase writing genetic code. Three prime and five prime ends are indicated on each side of the strands of DNA as well as the LAMP products. Horizontally positioned arrows indicate polymerase activity and amplification. The initial double stranded DNA is representative of the target cDNA that was cloned into a plasmid. F1, F2, and F3 represent the forward primer sequences and are contained within the FIP, F3, and LF primers. F1c, F2c, and F3c represent coding sequences of target DNA. This same principle applies to the B1, B2, B3, B1c, B2c, B3c, BIP and LB sequences in a similar manner to create the end loop product of LAMP.
Loop-Mediated Isothermal Amplification
For the purposes of this experimentation, the optimal temperature for primer candidate 1 was determined to be 59.3°C and the optimal time was twenty minutes. The Colorimetric Warmstart® MasterMix contains nucleotides, Bst polymerase, 16.0 mM Magnesium Sulfate, and pH sensitive indicator phenol red. The general reaction scheme for colorimetric based LAMP assays is seen in Figure 2. After the addition of genetic material and molecular grade water, the reaction will proceed in either a hot water bath or a thermocycler held at constant temperature. Results are visualized by the naked eye to be pink (negative) or yellow (positive).
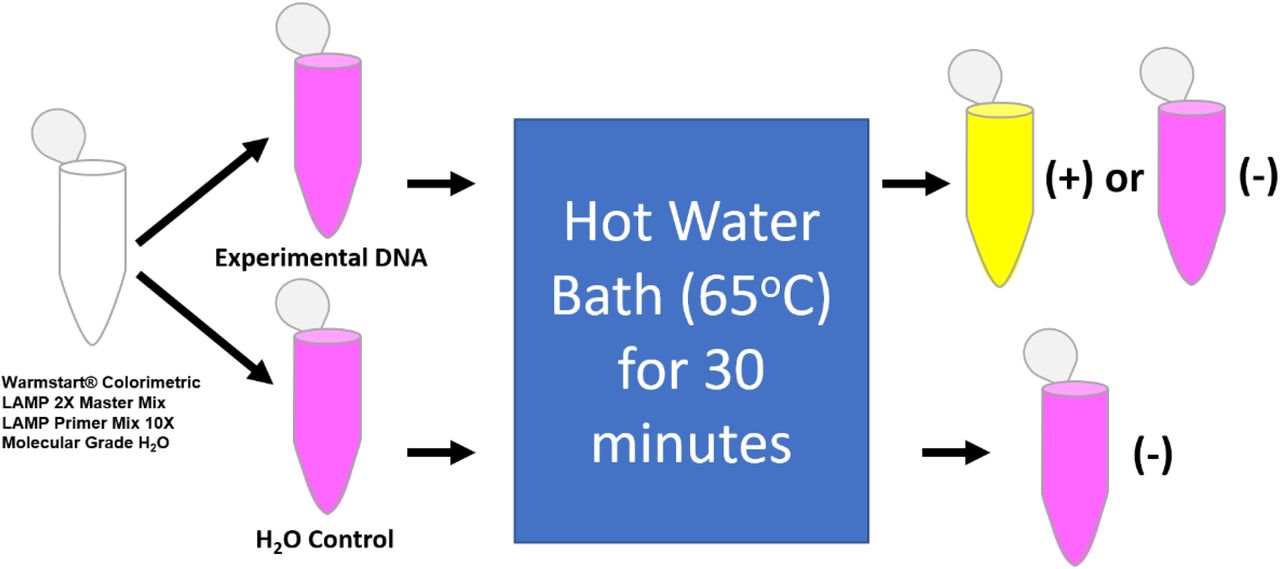
Reagents are listed below first reaction vessel on left and diverge based on using either 1 μL of sample or water control. The results from this procedure include the stagnant pink color for the negative control, and either a color change to yellow, indicating a positive result, or remain pink indicating a negative result for the experimental DNA.
Transformation and Purification of Plasmid cDNA
A plasmid was created (GenScript) containing a 500 bp cDNA of DTV’s RdRp, with flanking EcoRI and XhoI restriction enzyme sites. The plasmid was transformed into DH5α chemically competent E. coli for 2 minutes at 37°C and plated on nutrient agar plates containing ampicillin. DNA stocks were harvested from single clones by plasmid miniprep kits (Qiagen). The desired cDNA was digested from the vector by EcoRI and XhoI, run on an agarose gel, and purified from the gel with a gel extraction kit (Qiagen). The cDNA from this purification process was used for the experimentation as the DTV RdRp coding strand sample.
Discussion
From the analysis methods, we concluded that these sequences are statistically significant (p<0.05) to move forward with the RdRp targeted primer design. A 2,000 base-pair sequence was chosen from the DTV NS5a coding region GenBank accession NC003687.1, which includes the coding region for the RdRp. The codon-based test resulted in rejecting the null hypothesis for comparing the four different accessions, with p<0.05 for each entry. An evolutionary analysis showed the branching of four accessions from GenBank including one complete genome and three RdRp sequences and is visualized in Figure 3. The maximum divergence was 19.22% across all accessions, and primer development should represent the ideal areas of conservation. Alignment was performed against the full genome to match the RdRp sequences.

The evolutionary history was inferred using the UPGMA method [14]. The optimal tree with the sum of branch length = 0.19223306 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method [15] and are in the units of the number of base substitutions per site. This analysis involved 4 nucleotide sequences accessed through GenBank. Codon positions included were 1st+2nd+3rd+Noncoding. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There was a total of 10840 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [16
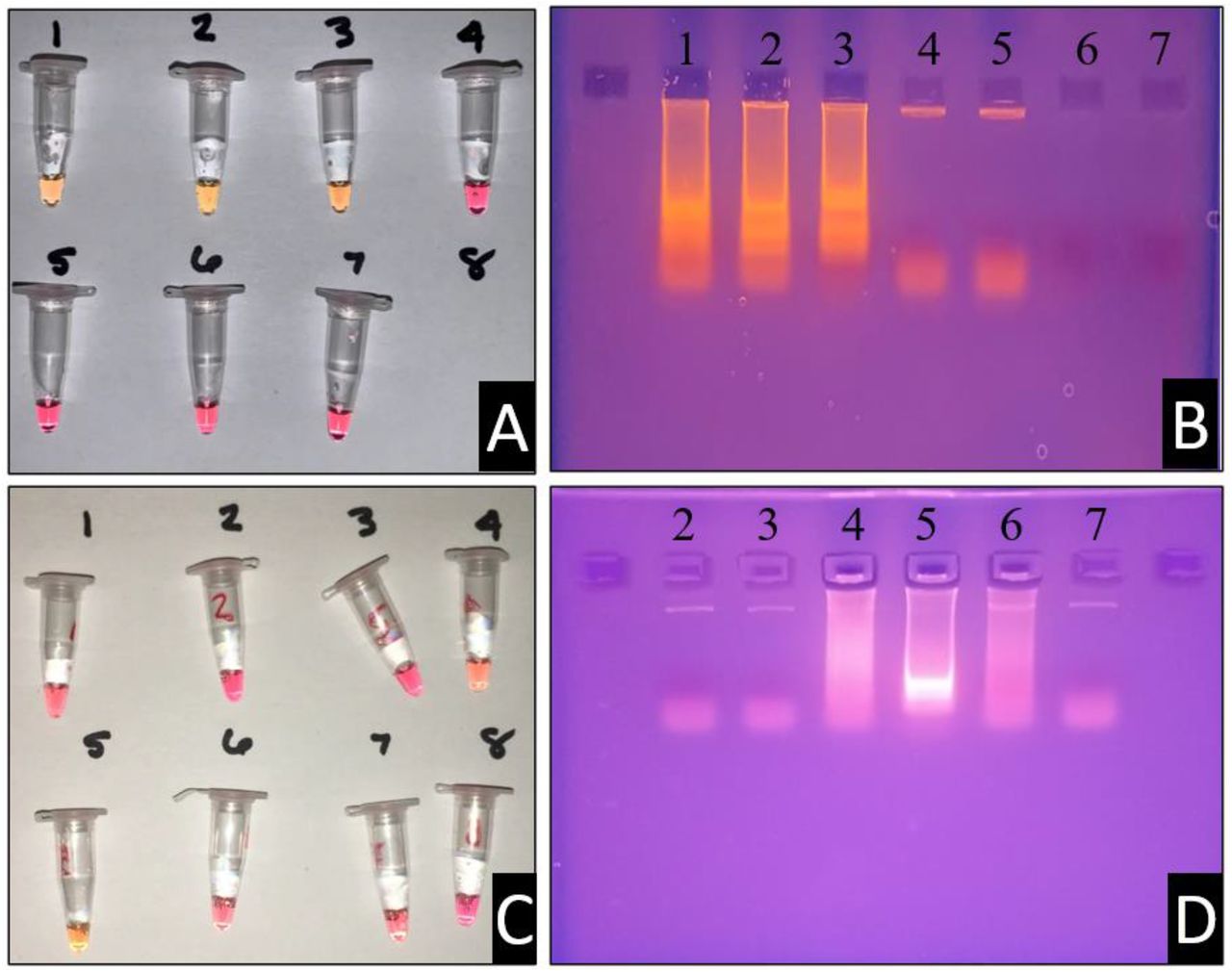
Primer candidate 1 displayed potential and consistency within LAMP assays. Dimerization events were unlikely to occur and provided consistent results. In Figure 4 typical results of this primer set are displayed. When LAMP is run with these primers, a colorimetric change is visualized, and the corresponding products are visualized following electrophoresis on an agarose gel. Controls of nonspecific DNA and water provide negative results as expected. These are desirable traits with LAMP primers to avoid false positives or false negatives. Additional primer sets were designed to confirm our design strategies. The optimal temperature at which this primer candidate function was 59.3°C (Figure 4.D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1 – Primer w/DNA, 2 – Primer w/DNA, 3 – Primer w/DNA, 4 – Primer w/o DNA, 5 – Primer w/o DNA, 6 – No Primer/DNA, 7 – No Primer/DNA. (B) Ethidium bromide electrophoresis gel with samples of panel (A). The containments of each lane correspond with the reaction vessels as indicated. (C) Temperature gradient assay for LAMP primer set 1.0. Each of the reaction vessels (left) were also run with negative controls, not pictured. Reaction vessel numbers correspond with the ethidium bromide gel pictured in box (D), with the exception of vessel (1) and (8). From vessel (1) to (8), the temperature gradient follows the order (in °C): 70.8, 69.3, 67.8, 64.8, 59.3, 57.8, 56.3, and 54.8.
The second primer set displayed dimerization concerns after looking closely at the dimerization rate of the 5’ end of the F3 primer. This observation was denoted after consistent false positives were obtained with primer candidate 2. The instability of these primers was directly correlated to the instability of the F3 primer being more positive than dG = −4.00 (Table 2). This observation can be regarded as a learning guide for future primer design experimentation and allowed for a consistency check with the first iteration generated.
Future primer sets will be compared to these initial two primer candidates and put through a comparative analysis for accuracy and limit of detection. An additional primer candidate has already been synthesized and experimentation shows additional promise. The efforts of this research are to bring attention to this pathosystem and the lack of robust diagnostic testing for downstream health effects.
DTV is a recent pathogen of interest and needs additional detection methods to monitor the long-term effects and spread. In the creation of a LAMP detection system point-of-care and field diagnostic, additional tools can be enacted to understand the spread of this virus. Primer 1.0 was an excellent candidate for the detection of in vitro genetic material of the virus. Our second primer candidate was faulty but further improved primer design hypotheses and tactics. Further validation is currently being investigated for newer primer sets. Additional experimentation will be performed with RNA expression samples, tick samples spiked with DNA and RNA, and additional collaborations with state environmental agencies for tick testing. Through these efforts we hope to further promote mitigation tactics and reduce the number of infections in North America.
Data Availability
No external data was generated/is required outside of this manuscript
Conflict of Interests
The authors hereby state no conflict of interests.
Acknowledgments
We would like to thank the Lebanon Valley College Biology Department for financially supporting this research, as well as the Beta Beta Beta Biological Honors Society National Research Grant for the 2019-2020 grant received for this project.
Footnotes
Addition of primer position with respect to sourced sequence, Tables 1 and 2. Reformated figure placement and changed from two columns to one.
References



