
Abstract
Molnupiravir, an antiviral medication that has been widely used against SARS-CoV-2, acts by inducing mutations in the virus genome during replication. Most random mutations are likely to be deleterious to the virus, and many will be lethal. Molnupiravir-induced elevated mutation rates have been shown to decrease viral load in animal models. However, it is possible that some patients treated with molnupiravir might not fully clear SARS-CoV-2 infections, with the potential for onward transmission of molnupiravir-mutated viruses. We set out to systematically investigate global sequencing databases for a signature of molnupiravir mutagenesis. We find that a specific class of long phylogenetic branches appear almost exclusively in sequences from 2022, after the introduction of molnupiravir treatment, and in countries and age-groups with widespread usage of the drug. We calculate a mutational spectrum from the AGILE placebo-controlled clinical trial of molnupiravir and show that its signature, with elevated G-to-A and C-to-T rates, largely corresponds to the mutational spectrum seen in these long branches. Our data suggest a signature of molnupiravir mutagenesis can be seen in global sequencing databases, in some cases with onwards transmission.
Introduction
Molnupiravir is an antiviral drug, licensed in some countries for the treatment of COVID-19. In the body, molnupiravir is ultimately converted into a nucleotide-analog, molnupiravir triphosphate (MTP)1. MTP is capable of being incorporated into RNA during strand synthesis, particularly by viral RNA-dependent RNA polymerases, where it can result in errors of sequence fidelity during viral genome replication. These errors in RNA replication result in many viral progeny that are non-viable, and so reduce the virus’s effective rate of growth – molnupiravir was shown to reduce viral replication in 24 hours by 880-fold in vitro, and to reduce viral load in animal models (Rosenke et al., 2021). Molnupiravir initially showed some limited efficacy as a treatment for COVID-19 (Jayk Bernal et al., 2022; Extance, 2022), but subsequent larger clinical trials found that molnupiravir did not reduce hospitalisation or death rates in high risk groups (Butler, 2022). As one of the first orally bioavailable antivirals on the market, molnupiravir has been widely adopted by many countries, most recently China (Reuters, 2022). However, recent trial results and the approval of more efficacious antivirals have since led to several countries recommending against molnupiravir usage on the basis of limited effectiveness (NICE Guidance ; NC19CET, 2022).
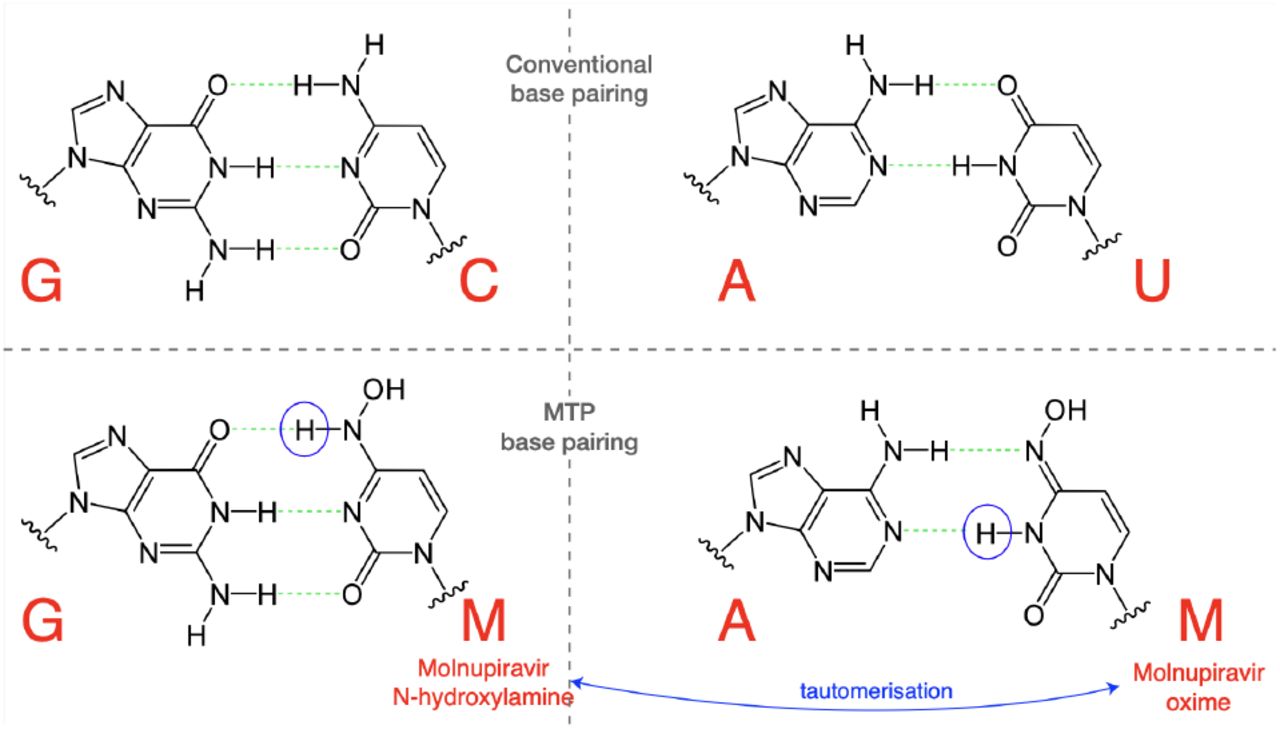
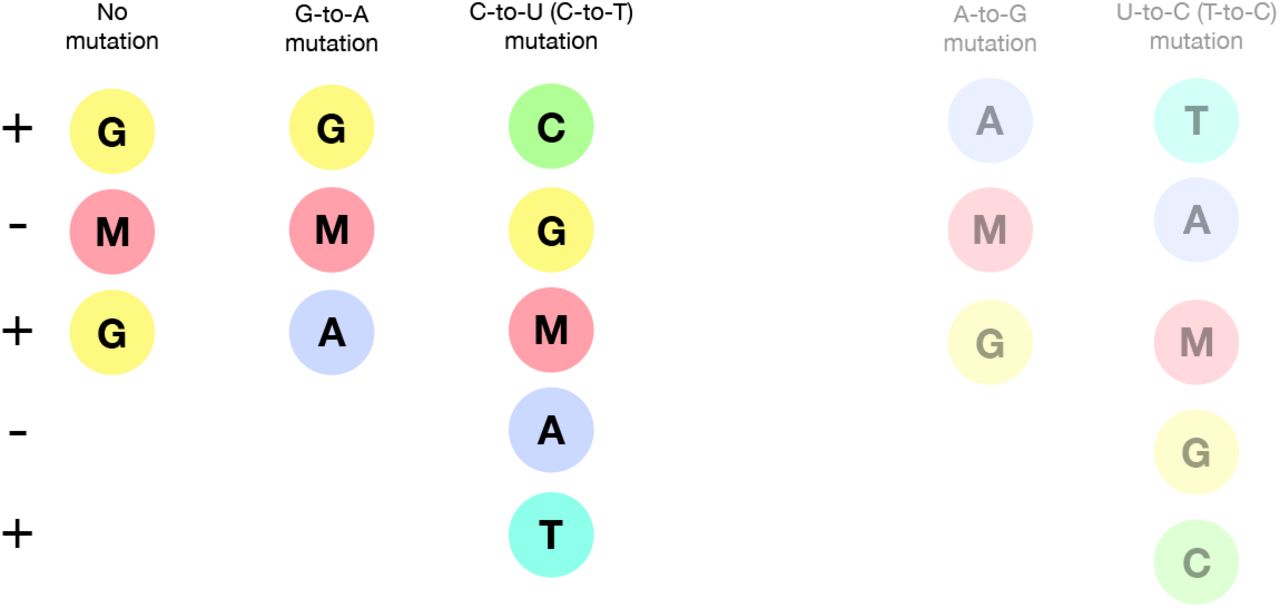
MTP appears to be incorporated into nascent RNA primarily by acting as an analogue of cytosine (C), pairing opposite guanine (G) bases (Fig. 1). However, once incorporated, the molnupiravir (M)-base can transition into an alternative tautomeric form which resembles uracil (U) instead. This means that in the next round of replication, to give the positive-sense SARS-CoV-2 genome, the M base can pair with adenine (A), resulting in a G-to-A mutation, as shown in Fig. 2. Incorporation of MTP can also occur during the second step synthesis of the positive-sense genome. In this case, an initial positive-sense C correctly pairs with a G in the first round of replication, but this G then pairs with an M base during positive-sense synthesis. In the next round of replication this M can then pair with A, which will result in a U in the final positive sense genome, with the overall process producing a C-to-U mutation (Fig. S1).

The N-hydroxylamine form resembles cytosine, while the oxime form more closely resembles uracil. They therefore pair with guanine and adenine respectively. (Figure adapted in part from Malone and Campbell (2021).)

In the most common scenario, shown on the left-hand side, M is incorporated opposite G nucleotides. It can then pair to A in subsequent replication, creating a G-to-A mutation. If the original G resulted from negative strand synthesis from a coding-strand C, then the G-to-A change ultimately creates a C-to-U coding change (Fig. S1). In the second, less common, scenario shown at the right hand side M is initially incorporated by pairing with A, which can result in an A-to-G mutation or, if the original A came from a coding U, a U-to-C mutation.
The free nucleotide MTP is less prone to tautomerisation than when incorporated into RNA, and so this directionality of mutations is the most likely (Gordon et al., 2021). However it is also possible for some MTP to bind, in place of U, to A bases and undergo the above processes in reverse, causing A-to-G and U-to-C mutations (Scenario 2 in Fig. 2, also Fig. S1).
It has been proposed that many major SARS-CoV-2 variants emerged from long-term chronic infections. This model explains several peculiarities of variants such as a general lack of genetic intermediates, rooting with much older sequences, long phylogenetic branch lengths, and the level of convergent evolution with known chronic infections (Rambaut et al., 2020; Viana et al., 2022; Hill et al., 2022; Harari et al., 2022).
During analysis of long phylogenetic branches in the SARS-CoV-2 tree, recent branches exhibiting potential hallmarks of molnupiravir-driven mutagenesis have been noted, including clusters of sequences indicating onward transmission. We therefore aimed to systematically identify sequences that might have been influenced by molnupiravir and characterise their mutational profile to examine the extent to which these signatures appear in global sequencing databases.
Results
During examination of SARS-CoV-2 phylogenetic branches with large numbers of mutations, a subset that contained skewed ratios of mutation classes and very few transversion substitutions were identified (Hisner, 2022). These patterns appeared very different to typical SARS-CoV-2 mutational spectra (Ruis et al., 2022; Bloom et al., 2022; Masone et al., 2022; Tonkin-Hill et al., 2021).
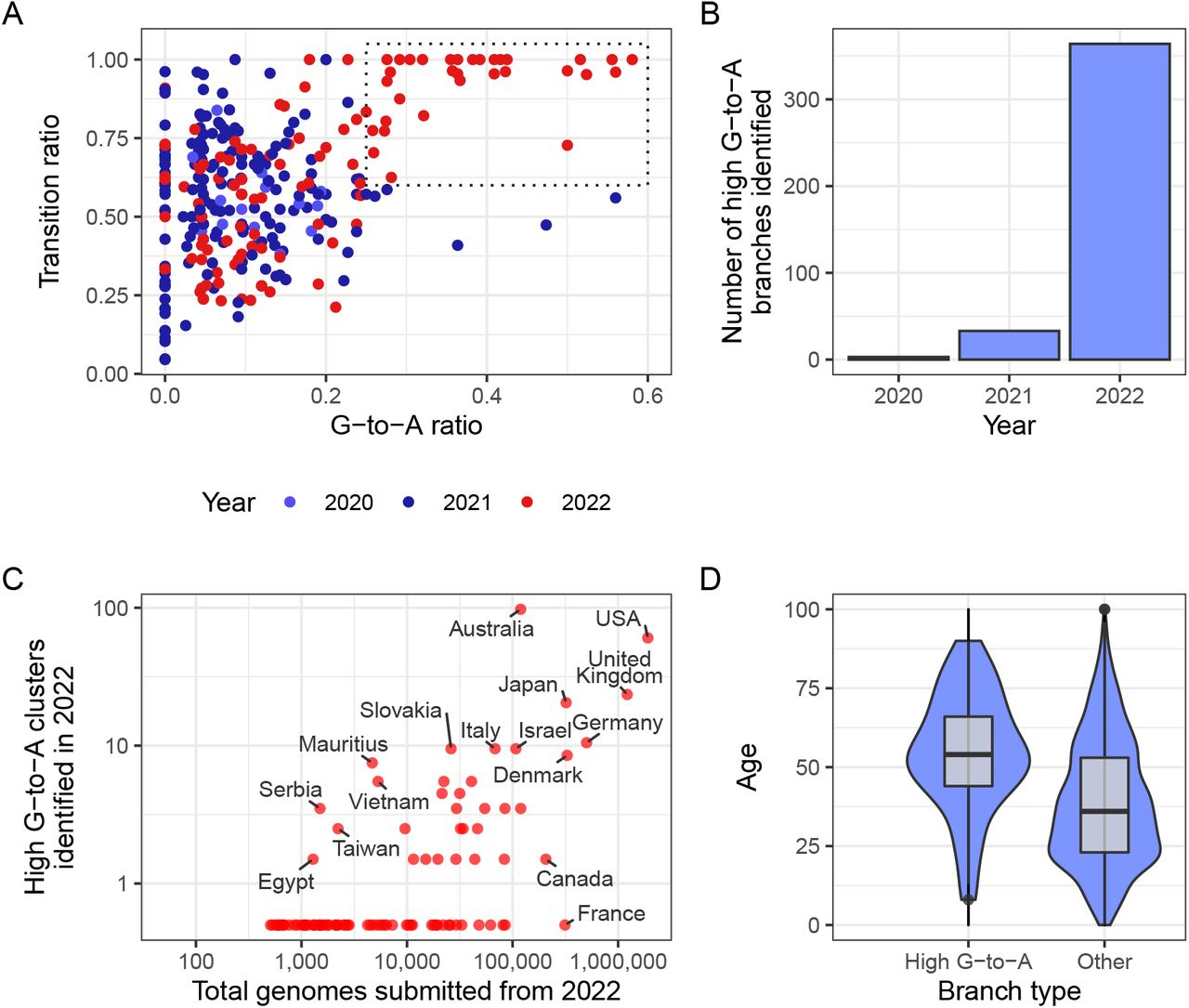
To investigate this pattern more systematically we analysed a mutation-annotated tree, derived from McBroome et al. (2021), containing >13 million SARS-CoV-2 sequences from GISAID (Elbe and BucklandMerrett, 2017) and the INSDC databases (Cochrane et al., 2011). For each branch of the tree we counted the number of each substitution class (A-to-T, A-to-G, etc. – we use T instead of U for the remainder of this manuscript as in sequencing databases). Filtering this tree to branches involving at least 20 substitutions, and plotting the proportion of substitution types revealed a region of this space with higher G-to-A and almost exclusively transition substitutions2, that only contained branches sampled in 2022 (Fig. 3A), suggesting some change (either biological or technical) had resulted in a new mutational signature.

(A) Each point in this scatterplot represents a branch from the mutation-annotated tree with >20 substitutions. Points are positioned according the proportion of the branch’s mutations that are G-to-A (x) or any transition mutation (y), and coloured by the year in which they occurred. A boxed region with higher G-to-A and almost exclusively transition mutations occurs only in 2022. (B) A count of the number of branches which satisfy a specific criterion (G-to-A ratio >= 25%, C-to-T ratio >= 20% transition ratio > 95%, total mutations > 10). (C) A comparison of number of total genomes with number of identified high G-to-A clusters (using the same criterion as B). Note logarithmic axes (semi-log, with the truncated line representing zero). For example, Australia has 97 clusters from a total of 119,194 genomes, whereas France has 0 clusters from 313,680 genomes. (D) A comparison of the age distributions for clusters with >10 mutations from the USA, partitioned by whether or not they satisfy the high G-to-A criterion. High G-to-A clusters correspond to older individuals.
Noticing that this signature also involved a high proportion of C-to-T mutations, we created a criterion for branches of interest, which we refer to as “ high G-toA” branches: we selected branches involving at least 10 substitutions, of which at least 25% were G-to-A, at least 20% were C-to-T and at most 5% were transversions. Again, these branches were almost all sampled in 2022 (Fig. 3B). The branches were predominantly sampled from a small number of countries, which could not be explained by differences in sequencing efforts (Fig. 3C, Table 1). Many countries which exhibited a high proportion of high G-to-A branches use molnupiravir: >380,000 prescriptions had occurred in Australia by the end of 2022 (Department of Aged Care Webinar, 2022), >30,000 in the UK (NHS, 2023; Butler, 2022), and >240,000 in the US within the early months of 2022 (Gold et al., 2022). Countries with high levels of total sequencing but a low number of G-to-A branches (Canada, France) have not authorised the use of molnupiravir (Goverment of Canada, 2022; Spencer et al., 2021). Age metadata from the US showed a significant bias towards patients with older ages for these high G-to-A branches, compared to control branches with similar numbers of mutations but without selection on substitution-type (Fig. 3D). Where age data was available in Australia it also identified long branches primarily in an aged population. This is consistent with the prioritised use of molnupiravir to treat older individuals, who are at greater risk from severe infection, in these countries. In Australia, molnupiravir was pre-placed in agedcare facilities, and it was recommended that it be considered for all patients aged 70 or older, with or without symptoms (Australian Department of Health and Aged Care, 2022).
Number of high G-to-A (G-to-A ratio >= 25%, C-to-T ratio >= 20% transition ratio > 95%, total mutations > 10) identified by country in 2022, set against total number of genomes. Only countries with >10,000 genomes are included.
We found that high G-to-A branches had a different distribution of branch lengths from other types of branches, with an enrichment for longer branch lengths (Fig. 4). We next sought to see whether these high G-to-A branches were associated with a different mutation rate to other branches of similar length. We assigned a date to each node of the tree using Chronumental (Sanderson, 2021) and observed that the branch length measured in time was shorter for branches with a high G-toA signature than for branches of matched length without this signature (Fig. S2), suggesting an increased mutation rate.

Plot is limited to branch lengths greater than 10, in order to be confident in the branch type, and to branch lengths less than 20 due to the exclusion of long branch samples from the UShER MAT. Branches are filtered to those from 2022.
Most of the long branches sampled have just a single descendant tip sequence in sequencing databases, but in some cases branches have given rise to clusters with a significant number of descendant sequences. For example, a cluster in Australia in August 2022 involves 20 tip sequences, with distinct age metadata indicating that they truly derive from multiple individuals (Fig. 5). This cluster involves 25 substitutions in the main branch of which all are transitions, 44% are C-to-T and 36% are G-to-A. Closely related outgroups date from July 2022, suggesting that these mutations emerged in a period of 1-2 months. There are many other examples of high G-to-A branches with multiple descendant sequences, including from the United Kingdom (Fig. 6A,B).

This cluster involves a saltation of 25 mutations occuring within a period of perhaps under a month, all of which are transition substitutions, with an elevated G-to-A rate. Sequences are annotated with age metadata suggestive of an outbreak in an aged-care facility.

(A) A cluster of four sequences from the UK from Feb-March 2022 with 13 shared muations with the high G-to-A signature. (B) A cluster of four sequences from the UK from Feb 2022 with 31 shared muations with the high G-to-A signature. (C) A singleton sequence from Australia with a high G-to-A signature and a total of 133 mutations. Just 2 of the 133 mutations observed are transversions and transitions include numerous G-to-A events.
During the construction of the daily-updated mutationannotated tree (McBroome et al., 2021), samples highly divergent from the existing tree are excluded. This is a necessary step given the technical errors in some SARS-CoV-2 sequencing data, but it also means that highly divergent molnupiravir-induced sequences might be excluded. To examine this effect, we created a comprehensive mutation-annotated-tree for Australia, allowing identification of even the most divergent sequences in this subset. This analysis allowed the identification of mutational events involving up to 130 substitutions (Fig. 6C, Fig. S3), with the same signature of elevated G-to-A mutation rates and almost exclusively transition substitutions. The cases we identified with these very high numbers of mutations involved single sequences, and could represent sequences resulting from chronically infected individuals who have been treated with multiple courses of molnupiravir.
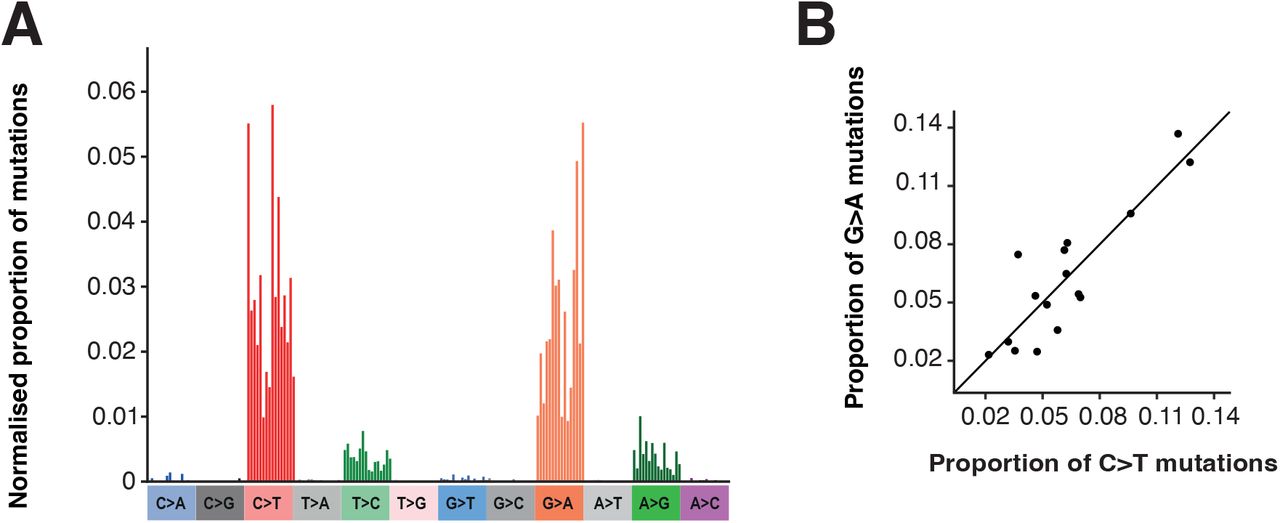
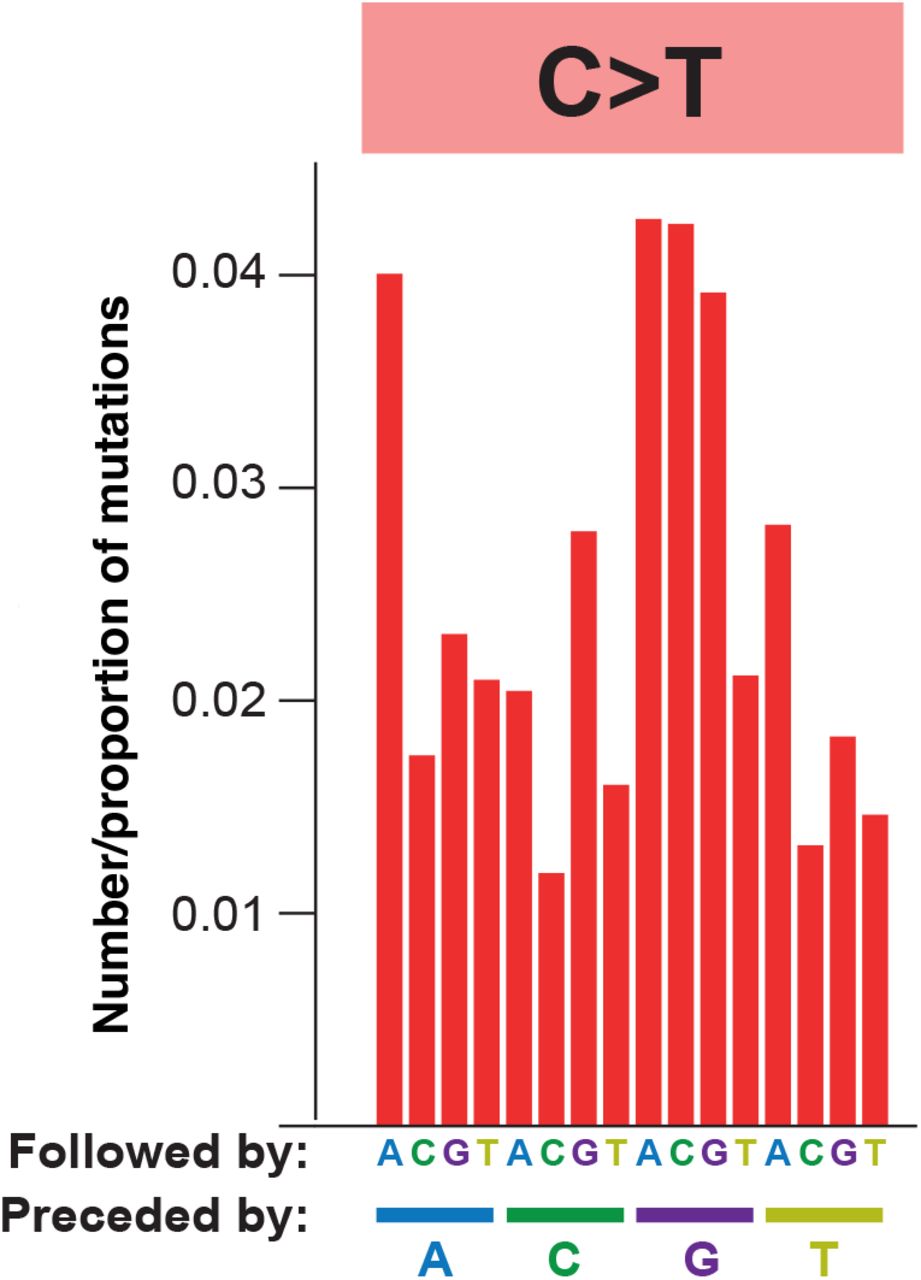
We next assessed the mutational spectrum (the patterns of contextual nucleotide substitutions) on these branches. The spectrum we identified (Fig. 7A) is dominated by G-to-A and C-to-T transition mutations with smaller contributions from A-to-G and T-to-C transitions. This pattern is consistent with the known mechanisms of action for molnupiravir (Fig. 2). These transitions exhibit preference for particular surrounding nucleotide contexts, for example G-to-A mutations occur most commonly in TGT and TGC contexts. This may represent a preference for molnupiravir binding adjacent to particular surrounding nucleotides, a preference of the viral RdRp to incorporate molnupiravir adjacent to specific nucleotides, or a preference of the viral proofreading exonuclease to remove molnupiravir in specific contextual surroundings.

(A) Single base substitution (SBS) spectrum of long phylogenetic branches. (B) Comparison of contextual mutation preferences in C-to-T and G-to-A mutations on long phylogenetic branches. The proportion of C-to-T or G-to-A mutations in the equivalent (reverse) contexts are plotted. There is a significant correlation, showing similar contextual preferences for C-to-T and G-to-A mutations. Proportions are normalised for the number of times the context occurs in the genome.
The dominance of both C-to-T and G-to-A mutations in the spectrum is likely due to molnupiravir-induced G-toA mutations during synthesis of different strands during virus replication. Incorporation of molnupiravir during negative strand synthesis will result in G-to-A mutations in the virus consensus sequence while incorporation during positive strand synthesis will be read as C-to-T mutations (Fig. S1). Consistent with this, we observe a strong positive correlation between the mutational biases in equivalent contextual patterns within C-to-T and G-to-A mutations (Pearson’s r = 0.88, 95% CI 0.68-0.96, p < 0.001, Fig. 7B) (e.g. a C-to-T mutation in the ACG context on one strand is the equivalent of a G-to-A mutation in the CGT context on the other strand).
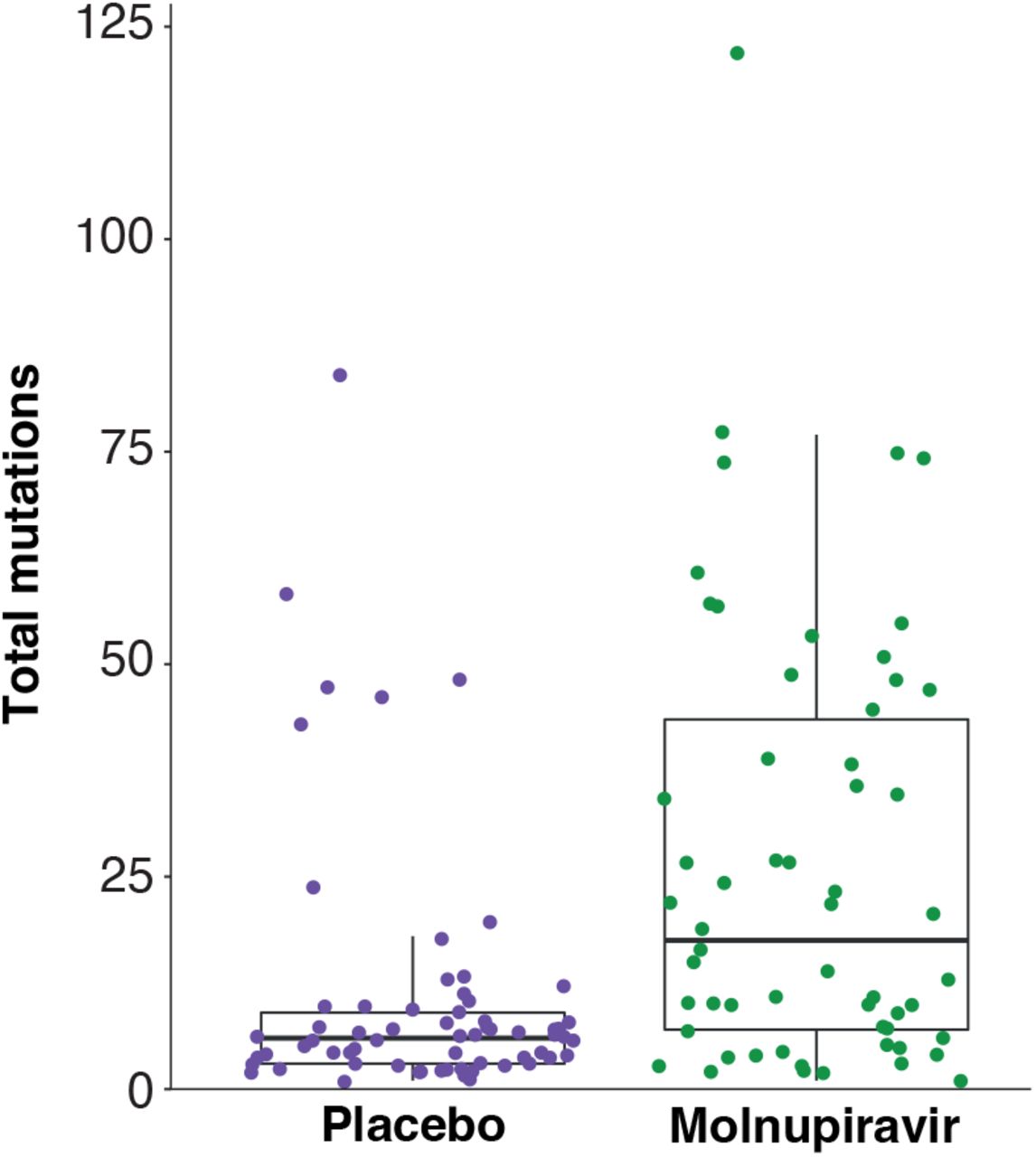
To compare the observed signatures to mutations in known molnupiravir-exposed individuals, we reexamined a genomic dataset from the AGILE Phase IIa clinical trial (Donovan-Banfield et al. (2022), NCT04746183). For the first time we analysed the spectrum of likely molnupiravir-induced mutations by comparing day 1 samples (taken just before treatment initiation) to day 5 samples from the same patient. This dataset had the advantage that it also included individuals treated with placebo, providing a control for mutational spectra in the absence of molnupiravir. We found that molnupiravir-treated patients exhibited significantly higher mutational burdens than patients treated with placebo (ANOVA p < 0.001, Fig. S6). The spectrum of mutations was highly different between placebo and molnupiravir (Fig. 8A, cosine similarity between spectra = 0.68). Assuming that the mutational processes within the placebo patients also occurred in the molnupiravir treatment, we subtracted the placebo spectrum to obtain the mutations specifically induced by molnupiravir (Fig. 8A). Again, we found a significant enrichment of transition mutations (Fig. 8B-C).

(A) SBS spectra of mutations in patients treated with placebo or molnupiravir in the AGILE trial. The right hand panel shows the mutations induced by molnupiravir, calculated by subtracting the placebo spectrum (left panel) from the spectrum of all mutations in molnupiravir treated patients (centre panel). Spectra are shown as the number of mutations per available context per patient. (B) Comparison of the number of each mutation type (summed across all contexts) between placebo and molnupiravir treatments. Error bars show confidence intervals from mutation bootstrapping (see methods). (C) The ratio of each mutation type between treatments was calculated by dividing the mutational burden in molnupiravir by that in placebo. The red dashed line shows an equal burden.
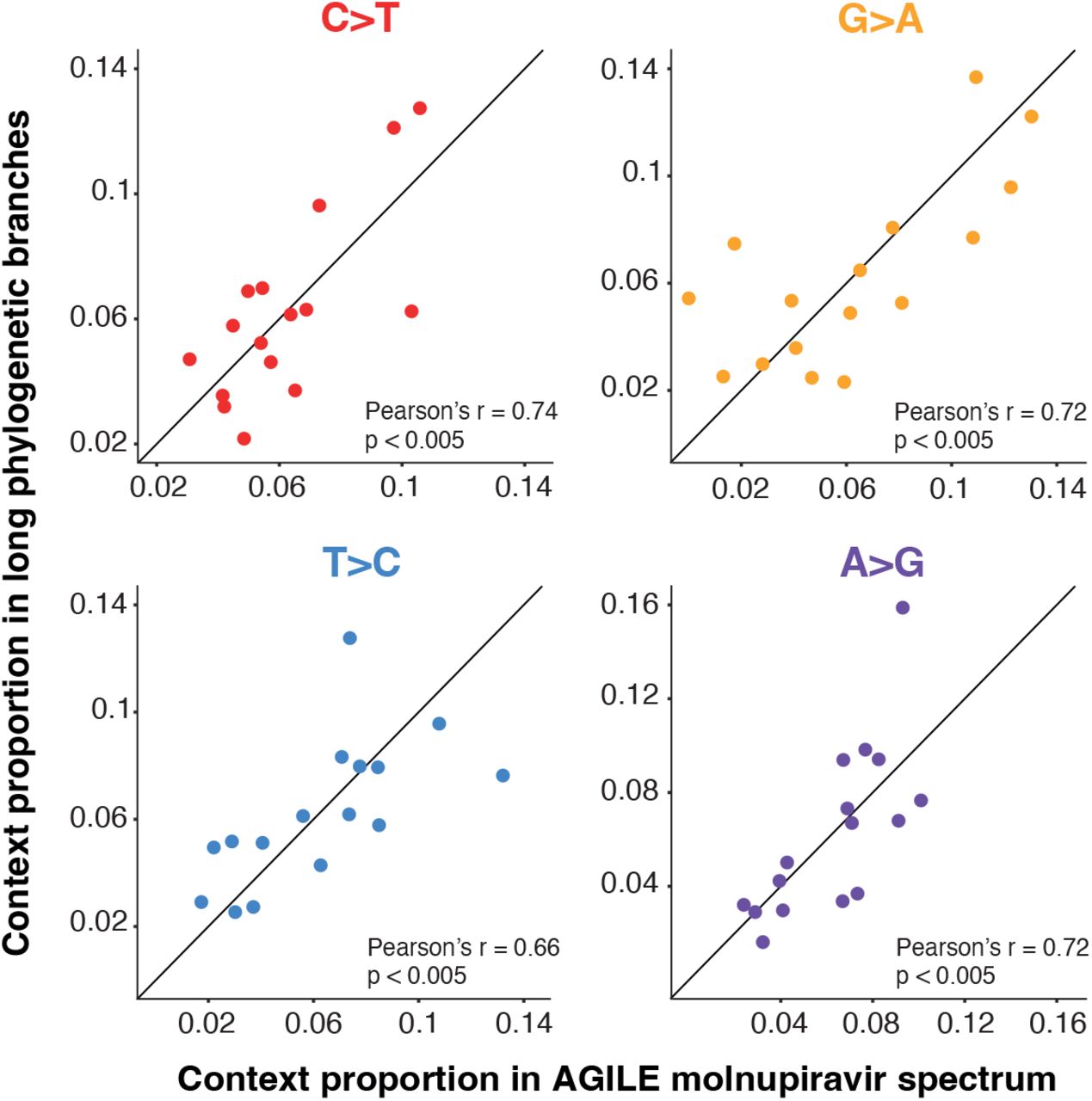
We used the calculated spectrum to examine whether the long phylogenetic branches identified above are likely to be molnupiravir-driven. The overall patterns of mutations in the long phylogenetic branches are qualitatively similar to the AGILE trial patients treated with molnupiravir, with a preponderance of transition mutations, most commonly C-to-T and G-to-A (Fig. 7, Fig. 8).
The contextual patterns within each transition mutation are highly similar between the known molnupiravir spectrum and the long phylogenetic branches (Fig. S7). This suggests a shared driver of transition mutations and therefore supports the long phylogenetic branches being driven by treatment with molnupiravir.
While the transition patterns are highly similar, the AGILE trial molnupiravir spectrum contains a high rate of G-to-T mutations that is not present in the long phylogenetic branches (Fig. 7, Fig. 8). This high rate is also present in the placebo-treated patients, although the rate appears higher in molnupiravir treatment (Fig. 8A). The rate of G-to-T mutations in both placebo and molnupiravir groups appears higher than that calculated from within patient mutations from untreated individuals (Tonkin-Hill et al. (2021), Fig. S8), for reasons that are not clear.
Discussion
We have shown a variety of lines of evidence that together suggest that a signature of molnupiravir treatment is visible in global sequencing databases. We identified a set of long phylogenetic branches that exhibit a high number of transition mutations. The number of these branches increased dramatically in 2022 and they are enriched for countries and age groups known to be exposed to molnupiravir. Mutation rates on these branches, were elevated, consistent with a recent study from Fountain-Jones et al. (2022) in immunocompromised patients. The branches exhibited a mutational spectrum that is highly similar to that in patients known to be treated with molnupiravir. The sequencing data suggest that in at least some cases, viruses with a large number of molnupiravir-induced substitutions have been transmitted to other individuals, at least in a limited manner.
Molnupiravir’s mode of action is often described using the term “ error catastrophe” – the concept that there is an upper limit on the mutation rate of a virus beyond which it is unable to maintain self-identity (Eigen, 1971). This model has been criticised on its own terms (Summers and Litwin, 2006), but is particularly problematic in the case of molnupiravir treatment. The model assumes a steady-state condition, with the mutation rate fixed at a particular level. The threshold for error catastrophe is the mutation rate at which, according to the model, the starting sequence will ultimately be lost after an infinite time at this steady-state. However molnupiravir treatment does not involve a steady state, but a temporarily elevated mutation rate over a short treatment period. Therefore there isn’t any particular threshold point, or well-defined “ catastrophe” condition. We suggest that the use of this term in the context of mutagenic antiviral drugs is unhelpful. It is enough to think of these drugs as acting through mutagenesis to reduce the number of viable progeny that each virion is able to produce – particularly given that much of the reduction in fitness will be due to “ single-hit” lethal events (Summers and Litwin, 2006).
New variants of SARS-CoV-2 are generated through acquisition of mutations that enhance properties including immune evasion and intrinsic transmissibility (Telenti et al., 2022; Carabelli et al., 2023). The impact of molnupiravir treatment on the trajectory of variant generation and transmission is difficult to predict. On the one hand, molnupiravir increases the amount of sequence diversity in the surviving viral population in the host and this might be expected to provide more material for selection to act on during intra-host evolution towards these properties that increase fitness. However, a high proportion of induced mutations are likely to be deleterious or neutral, and it is necessary to consider the counterfactual to molnupiravir treatment. As molnupiravir results in a modest reduction in viral load in treated patients (Khoo et al., 2022), it is possible that in the absence of treatment the total viral load would be higher and chronic infections might persist for longer. Variants generated through chronic infections might be fitter than those that have accumulated mutations during molnupiravir treatment, albeit taking a much longer period of time to accumulate the same number of mutations and therefore usually being derived from older, rather than contemporary lineages. At the time of writing we have not identified a molnupiravir-implicated cluster that had spread to more than 21 individuals.
There are some limitations of our work. Detecting a particular branch as involving a molnupiravir-like signature is a probabilistic rather than absolute judgement: where molnupiravir creates just a handful of mutations (which trial data suggests is often the case), branch lengths will be too small to assign the cause of the mutations with confidence. We therefore limited our analyses here to long branches. This approach may also fail to detect branches which feature a substantial number of molnupiravir-induced mutations alongside a considerable number of mutations from other causes (which might occur in chronic infections). We discovered drastically different rates of molnupiravirassociated sequences by country, and suggest that this reflects in part whether, and how, molnupiravir is used in different geographical regions – however there will also be contributions from the rate at which genomes are sequenced in settings where molnupiravir is used. For example, if molnupiravir is used primarily in aged-care facilities and viruses in these facilities are significantly more likely to be sequenced than those in the general community this will elevate the ascertainment rate of such sequences. Furthermore, it is likely that some included sequences were specifically analysed due to representing continued test positivity after molnupiravir treatment as part of specific studies. Such effects are likely to differ based on sequencing priorities in different locations.
We would recommend public health authorities in countries showing these patterns perform investigations to determine if these sequences or clusters can indeed be directly linked back to use of molnupiravir. These data will be useful for ongoing assessments of the risks and benefits of this treatment, and may guide the future development of mutagenic agents as antivirals, particularly for viruses with high mutational tolerances such as coronaviruses.
Methods
Processing of mutation-annotated tree
To identify clusters in global sequence databases with a molnupiravir associated signature we analysed a regularly updated mutation-annotated tree built using UShER (Turakhia et al., 2021) with almost all global data – a version of the McBroome et al. (2021) tree. We parsed the tree using a custom script adapted from Taxonium Tools (Sanderson, 2022). The script added metadata from sequencing databases to each node, then passed these metadata to parent nodes using simple heuristics: (1) a parent node was annotated with a year if all of its descendants were annotated with that year, (2) a parent node was annotated with a particular country if all of its descendants were annotated with that country, (3) a parent node was annotated with the mean age of its (age-annotated) descendants.
Mutation rate analysis
We used Chronumental (Sanderson, 2021) to assign dates to each node in the mutation-annotated tree. We ran Chronumental for 300 steps, then extracted length of time in days from the output Newick tree, and compared against the number of mutations on the branch, splitting by whether the branch satisfied our criteria to be a high G-to-A branch.
Generation of cluster trees
For the cluster of 20 individuals, we observed small imperfections in UShER’s representation of the mutationannoated tree within the cluster resulting from missing coverage at some positions. We therefore recalculated the tree that we display here. We took the 20 sequences in the cluster, and the three closest outgroup sequences, we aligned using Nextclade (Aksamentov et al., 2021), calculated a tree using iqtree (Minh et al., 2020) and reconstructed the mutation-annotated tree using TreeTime (Sagulenko et al., 2018). We visualised the tree using FigTree (Rambaut, 2018). We also visualised Fig. S3 with NextStrain (Hadfield et al., 2018).
Adding back excluded divergent sequences to the mutation-annotated tree
We used GISAIDR (Wirth and Duchene, 2022) to download all Australian sequences in 2022 from the GISAID database. We then filtered those based on sequence ID to those absent from the existing mutation-anotated tree (MAT). We aligned these sequences to the Hu-1 reference using flowalign. We pruned the existing MAT to retain only Australian sequences, and then added all sequences missing from the tree using UShER (Turakhia et al., 2021), without filtering on parsimonious placements or path length, to achieve a complete MAT for Australia.
Calculation of mutational spectrum of long phylogenetic branches
To calculate the SBS mutational spectrum of the long phylogenetic branches, we extracted the path of mutations leading to each branch from the 2022-12-18 UShER phylogenetic tree. Internal and tip branches in these paths containing at least ten mutations were identified and their mutations extracted. The context of each mutation was identified using the Wuhan-Hu-1 genome (accession NC_045512.2), incorporating mutations acquired earlier in the path. Mutation counts were rescaled by genomic content by dividing the number of mutations by the count of the starting triplet in the Wuhan-Hu-1 genome. MutTui (https://github.com/chrisruis/MutTui) was used to rescale and plot mutational spectra.
Calculation of molnupiravir and placebo mutational spectra using AGILE trial data
We calculated molnupiravir and placebo SBS spectra using previously published variant data (DonovanBanfield et al., 2022). We used deep sequencing data from samples collected on day one (pre-treatment) and day five (post-treatment) from 65 patients treated with placebo and 58 patients treated with molnupiravir. For each patient, we used the consensus sequence of the day one sample as the reference sequence and identified mutations as variants in the day five sample away from the patient reference sequence in at least 5% of reads at genome sites with at least 100-fold coverage. The surrounding nucleotide context of each mutation was identified from the patient reference sequence.
We converted mutation counts to mutational burdens by dividing each mutation count by the number of the starting triplet in the Wuhan-Hu-1 genome (accession NC_045512.2) and by the number of patients in the treatment group (65 for placebo and 58 for molnupiravir). This therefore rescales by the number of opportunities for each mutation to occur across the genome and the number of patients in each group.
To ensure that any spectrum differences between placebo and molnupiravir treatments are not due to previously observed differences in spectrum between SARS-CoV-2 variants (Ruis et al., 2022; Bloom et al., 2022), we compared the distribution of variants between the treatments (Fig. S4). The distributions were highly similar.
To calculate the total mutational burden of each mutational class, we summed the mutational burden of the 16 contextual mutations within the class. Confidence intervals were calculated by bootstrapping mutations. Here, the original set of mutations within the treatment was resampled with replacement before rescaling by triplet availability and number of patients. 1000 bootstraps were run and 95% confidence intervals calculated.
To enable comparison with an additional SARS-CoV-2 dataset containing mutations acquired during within host infections, we obtained variants from a previous deep sequencing study Tonkin-Hill et al. (2021). We identified the surrounding nucleotide context of each mutation using the Wuhan-Hu-1 genome and rescaled by context availability.
Comparison of mutational spectra
The cosine similarity between placebo and molnupiravir spectra was calculated using MutTui (https://github.com/chrisruis/MutTui).
We compared contextual patterns within each transition mutation between the long phylogenetic branch and AGILE molnupiravir spectra through regression of the proportion of mutations within the mutational class that are in each context. To assess significance of the correlation, we randomised the proportions within the mutational class within each spectrum and recalculated the correlation. 1000 randomisations were carried out and the p-value calculated as the proportion of randomisations with a correlation at least as large as in the real data.
Data Availability
No new primary data was generated for this study. We used data from international sequencing databases (GISAID and INSDC), and from the AGILE clinical trial, where genomic data were obtained from BioProject PRJNA854613 at the SRA. Our GitHub repository is available at https://github.com/theosanderson/molnupiravir.
DATA AVAILABILITY
No new primary data was generated for this study. We used data from international sequencing databases (Elbe and Buckland-Merrett, 2017; Cochrane et al., 2011), and from the AGILE clinical trial (Donovan-Banfield et al., 2022), where genomic data were obtained from BioProject PRJNA854613 at the SRA. Our GitHub repository is available at https://github.com/theosanderson/molnupiravir.
The findings of this study are based on metadata associated with 14,449,737 sequences available on GISAID up to December 2022, and accessible at 10.55876/gis8.230110wz and 10.55876/gis8.230110db (see also, Supplemental Tables). The findings of this study are also based on 6,594,478 sequences from INSDC – authors, metadata, and sequences are available here. Data present in both databases are deduplicated before the construction of the mutation-annotated tree.
AUTHOR CONTRIBUTIONS
RH identified initial branches, and their likely connection to molnupiravir. TS performed analyses of mutation-annotated tree and global metadata. CR performed all mutational spectra analyses. ID-B created bioinformatic pipelines for the AGILE trial data. All authors participated in mansuscript writing.
FUNDING
TS was supported by the Wellcome Trust (210918/Z/18/Z) and the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001043), the UK Medical Research Council (FC001043), and the Wellcome Trust (FC001043). This research was funded in whole, or in part, by the Wellcome Trust [210918/Z/18/Z, FC001043]. For the purpose of Open Access, the authors have applied a CC-BY public copyright licence to any Author Accepted Manuscript resulting from this preprint.
ID-B is supported by PhD funding from the National Institute for Health and Care Research (NIHR) Health Protection Research Unit (HPRU) in Emerging and Zoonotic Infections at University of Liverpool in partnership with Public Health England (PHE) (now UKHSA), in collaboration with Liverpool School of Tropical Medicine and the University of Oxford (award 200907). The views expressed are those of the authors and not necessarily those of the Department of Health and Social Care or NIHR. Neither the funders or trial sponsor were involved in the study design, data collection, analysis, interpretation, nor the preparation of the manuscript.
TP was funded by the G2P-UK National Virology Consortium funded by the MRC (MR/W005611/1).
CR was supported by a Fondation Botnar Research Award (Programme grant 6063) and UK Cystic Fibrosis Trust (Innovation Hub Award 001).
Supplementary Information

This figure depicts some of the mutational pathways related to MTP incorporation into MTP. The first column shows what may be a common event, but is not detectable by sequencing. MTP can be incorporated into RNA (pairing with G) and then pair with G again in the next round of synthesis, which will result in no mutation in the final sequence. However if the MTP takes on an alternative tautomeric form after incorporation it can bind to A, creating a G-to-A mutation. The third column shows that if the positive-sense base is C, then this will bind to a G in the formation of the negative-sense genome. In subsequent replication this negative sense genome can undergo the same G-to-A mutation seen in the second column, which ultimately results in a positive sense C-to-T mutation. Although the biases of tautomeric forms for the free and incorporated MTP nucleotides appear to favour these directionalities of mutations, the reverse is also possible, resulting in A-to-G and T-to-C mutations.

We used Chronumental to assign a date to all nodes in the global UShER mutation annotated tree, then isolated branch lengths >10 and <=20, and plotted the mean length of time and its 95% confidence interval by number of mutations, as well as a fitted linear model, for Australia, Japan, the UK and the USA. This analysis should be seen only as semi-quantitative due to the fact that the algorithm will attempt to reconcile its chronology with the overall SARS-CoV-2 mutation rate and therefore be conservative, and that sequencing errors and recombination can also drive a large apparent rate of mutations in a short time.

A complete MAT from Australian sequences was made, and genomes descended from high G-to-A events were identfied. Usher.bio was then used to create a tree with these sequences highlighted on a downsampled global tree, with visualisation performed using Nextstrain.

The proportion of patients infected with each variant is shown. The proportions are similar suggesting that differences between placebo and molnupiravir spectra will not be influenced by previously observed spectrum differences between variants (Ruis et al., Bloom et al.). VOC = variant of concern.

The RNA mutational spectrum contains 12 mutation types, for example C-to-T. The spectrum also captures the nucleotides surrounding each mutation. There are four potential upstream nucleotides and four potential downstream nucleotides. This figure shows the location of each of the 16 contexts within an example mutation type. For example, the leftmost bar represents C-to-T mutations in the ACA context while the second leftmost bar represents C-to-T mutations in the ACC context.

The total number of mutations across all substitution classes is plotted in the day 5 sample of each patient in the AGILE trial.
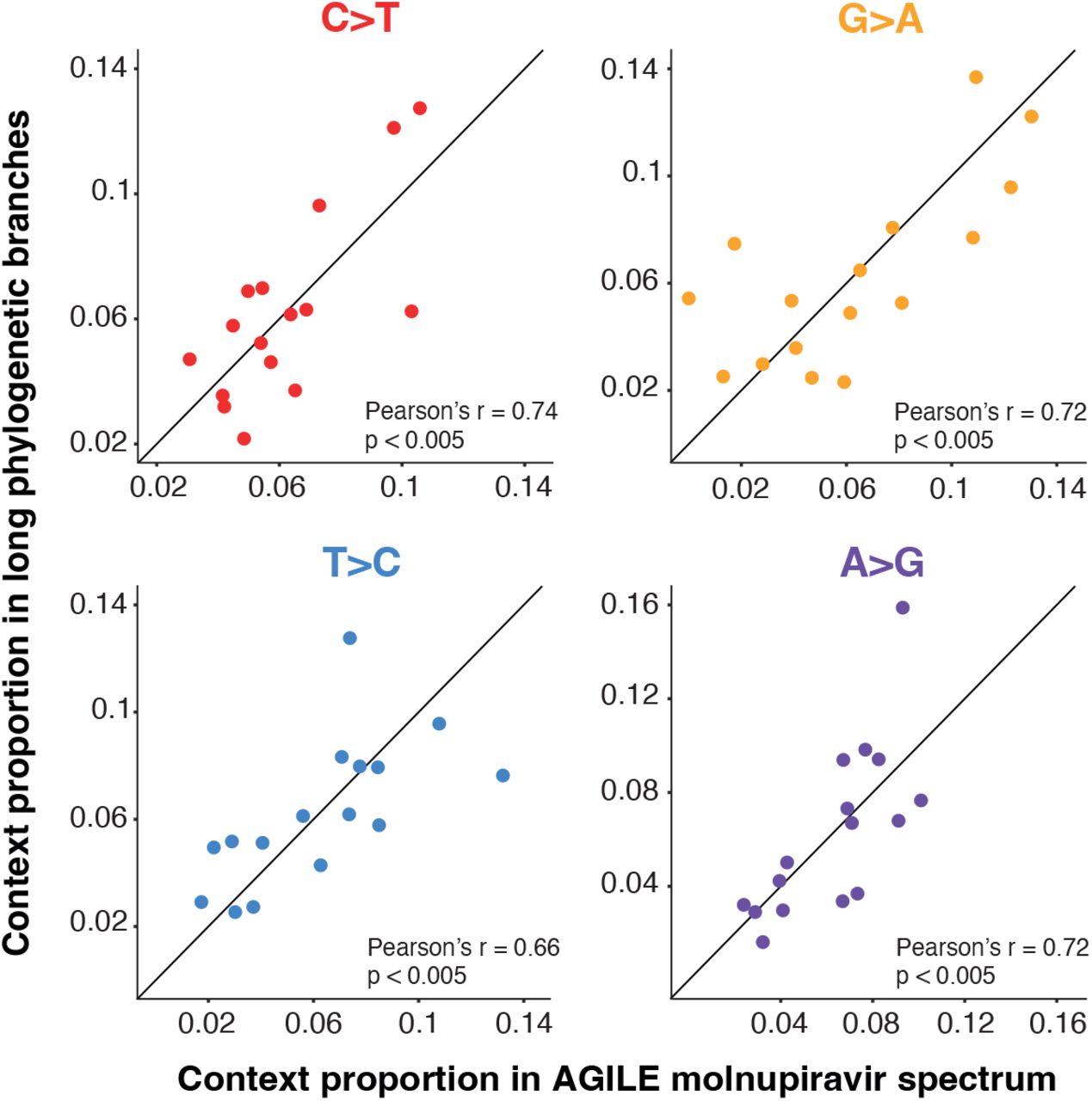
The proportion of mutations within the respective transition (for example the proportion of C>T mutations that are in the ACA context) is shown. P-values represent the proportion of context randomisations with a Pearson’s r correlation at least as great as with the real data (see methods). Proportions are normalised for the number of times the context occurs in the genome.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
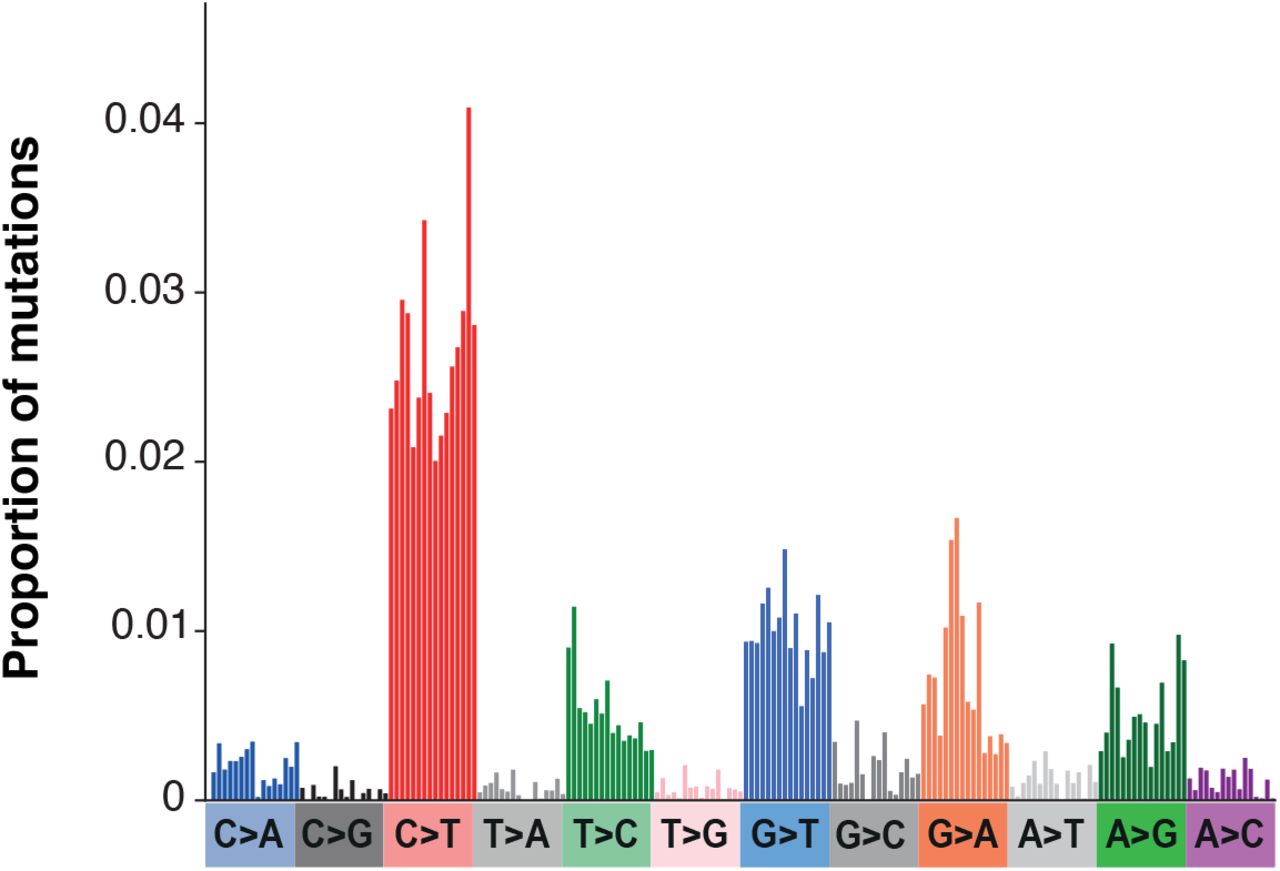
Mutations are from Tonkin-Hill et al. (2021).
ACKNOWLEDGEMENTS
We gratefully acknowledge all data contributors, i.e., the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. We are also very grateful to everyone who has contributed to the generation of the genomes that have been deposited in the INSDC databases, on which this research is also based. We thank Angie Hinrichs and colleagues for access to an UShER mutationannotated tree built with all available genomic data. We thank Jesse Bloom, Michael Lin, Richard Neher, and Kelley Harris for useful discussions. This preprint uses a LaTeX template from Stephen Royle and Ricardo Henriques.
Footnotes
Bibliography



