
ABSTRACT
Background Coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a respiratory illness that has led to millions of deaths worldwide and has been declared a pandemic. Most antiviral treatments are targeted toward patients with severe or moderate-to-severe illness or those at high risk of developing severe COVID-19. Limited options exist for patients with mild-to-moderate COVID-19, irrespective of vaccination history or the risk of developing severe illness. Ensitrelvir is a novel oral SARS-CoV-2 3C-like protease inhibitor that has shown promising results in phase 2 studies in treating mild-to-moderate COVID-19. Here we describe the protocol for a phase 3 study designed to evaluate the efficacy and safety of ensitrelvir in patients with mild-to-moderate COVID-19, regardless of risk status or history of vaccination.
Methods This is a multicenter, randomized, double-blind, placebo-controlled, phase 3 study. A total of 1590 patients with mild-to-moderate COVID-19 will be enrolled and randomized in 1:1:1 ratio into 3 treatment arms - ensitrelvir 125 mg (375 mg as loading dose on Day 1), ensitrelvir 250 mg (750 mg as loading dose on Day 1), or placebo. Patients will be administered the study interventions orally once daily for 5 days. Treatment outcomes will include efficacy and safety assessments. The primary endpoint will be the time to resolution of COVID-19 symptoms (including 12 symptoms listed in a partially modified index prescribed by the Food and Drug Administration). The three key secondary endpoints will be the change from baseline on Day 4 in the amount of SARS-CoV-2 viral RNA, the time to the first negative SARS-CoV-2 viral titer, and the proportion of participants without resolution of COVID-19 symptoms 3 weeks after administration. All safety assessments and adverse events will be reported.
Discussion Time to resolution of COVID-19 symptoms is a suitable endpoint to assess antiviral treatment in these patients. In phase 2a and 2b studies, ensitrelvir has been demonstrated to have antiviral efficacy against SARS-CoV-2 and a trend toward reducing time to resolution of symptoms in patients with mild-to-moderate COVID-19. Through this study, we seek to validate and further establish the clinical efficacy and safety of ensitrelvir in patients with mild-to-moderate COVID-19.
Trial registration Japan Registry of Clinical Trials (https://jrct.niph.go.jp): jRCT2031210350.
INTRODUCTION
Coronavirus disease 2019 (COVID-19) is an infectious respiratory illness caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1 and was declared a global pandemic by the World Health Organization (WHO) in March 2020.2 Globally, as of May 2022, over 500 million confirmed cases of COVID-19 have been reported, with more than 6 million associated deaths.3 Common symptoms of COVID-19 include fever, cough, fatigue, shortness of breath, headache, body aches, sore throat, runny or stuffy nose, diarrhea, nausea, and vomiting; some patients may also experience loss of smell and/or taste.4 High-risk patients such as the elderly and those with comorbidities and underlying health conditions such as cardiovascular disease, respiratory disease, renal disease, diabetes mellitus, obesity, or immunodeficiency are at a greater risk of developing severe COVID-19 symptoms.5,6 Also, some COVID-19 symptoms may persist even after cessation of SARS-CoV-2 shedding and recovery from the acute phase, and this causes a wide range of health problems as the post-acute COVID-19 syndrome.7,8
The SARS-CoV-2 Omicron (B.1.1.529) is a variant of concern (VOC) designated by the WHO.9 This variant is characterized by spike protein mutations that lead to increased transmissibility and to escape from SARS-CoV-2-neutralizing antibodies.10 A study from the United Kingdom indicated that common symptoms in patients infected with the Omicron variant of SARS-CoV-2 were mostly mild (runny nose, headache, sore throat, sneezing, persistent cough, and hoarse voice). The risk of hospitalization and hospitalization rates were lower during the prevalence of the Omicron variant compared with that of the Delta variant,11,12 in alignment with studies reporting that infection with the Omicron variant is less severe than that with the Delta variant.13-15 Although milder, several reports have indicated that infection with the Omicron variant is associated with a higher excess mortality compared with that with the Delta variant16 and also influenza.12 Although vaccination is progressing across the world, new variants that evade SARS-CoV-2-neutralizing antibodies, such as the Omicron variant, continue to be a threat of COVID-19.
As of April 2022, several therapeutic antiviral drugs against SARS-CoV-2 infection have been approved worldwide.17-20 In particular, remdesivir17 and molnupiravir,19 both ribonucleic acid (RNA) polymerase inhibitors, as well as nirmatrelvir, a SARS-CoV-2 3C-like (3CL) protease inhibitor, in combination with ritonavir as a pharmacokinetic booster,18 are antivirals recommended for treating patients at a high risk of developing severe illness. These have been approved for use in Japan as well.21 However, these drugs were mostly approved prior to the emergence of the Omicron variant and also assessed in clinical trials in unvaccinated individuals. Therefore, little evidence exists for their efficacy against the Omicron variant and in vaccinated patients. In Japan, remdesivir has been approved for moderate-to-severe COVID-19, regardless of patient risk for progression to severe COVID-19, and for mild COVID-19 patients at a high risk of severe complications.22 However, treatment options for patients with mild-to-moderate COVID-19 regardless of vaccination status and risk of developing severe complications are limited. Post-acute COVID-19 syndrome is found in anyone who have been infected with SARS-CoV-2, even people with mild or no symptoms8, and a new treatment or prevention of post-acute COVID-19 syndrome is needed.
Ensitrelvir fumaric acid (S-217622, hereafter ensitrelvir) is a novel oral SARS-CoV-2 3CL protease inhibitor that originated through collaborative research efforts between Shionogi & Co., Ltd., and Hokkaido University.23 Ensitrelvir has demonstrated antiviral efficacy against SARS-CoV-2, including the Omicron variant, and other VOCs in both in vitro and in vivo preclinical studies.24-26 The tolerability of 5-day administration of ensitrelvir has been confirmed in healthy adult patients in a phase 1 study (Japan Registry of Clinical Trials identifier: jRCT2031210202; Shimizu et al, unpublished). Promising results have been observed in the ongoing phase 2/3 study comprising several components - phase 2a, phase 2b, phase 2b/3, and phase 3 (SCORPIO-SR), designed to assess the antiviral efficacy of ensitrelvir. The phase 2a27 and 2b (H. Mukae et al, unpublished data, May 2022) studies showed that once-daily ensitrelvir administered for 5 days demonstrated a significant reduction in viral titer and viral RNA when compared to placebo in mild-to-moderate COVID-19. In the phase 2b study, while there was no significant difference between ensitrelvir and placebo in the time-weighted average change in the total score corresponding to the 12 COVID-19 symptoms, planned and post hoc analyses demonstrated that ensitrelvir produced favorable improvements in a subtotal of four respiratory symptoms (planned) or in the composite of respiratory symptoms and feverishness (post hoc) (H. Mukae et al, unpublished data, May 2022). Both studies demonstrated that ensitrelvir was safe and well tolerated. The phase 2b study was conducted during the dominance of the Omicron variant, and a majority of the patients had been vaccinated. (H. Mukae et al, unpublished data, May 2022).
In this study, we describe the protocol for a phase 3 study, with emphasis on the study rationale and key endpoints designed to assess the efficacy and safety of ensitrelvir in patients with mild-to-moderate COVID-19, irrespective of their risk status of developing severe illness.
MATERIALS AND METHODS
Study design
This is a multicenter, randomized, double-blind, placebo-controlled, phase 3 study to test the efficacy and safety of ensitrelvir in patients with mild-to-moderate COVID-19. The study will be conducted across several sites in Japan, Korea, Singapore, and Vietnam beginning in February 2022, and has been registered at the Japan Registry of Clinical Trials: jRCT2031210350. Patients will be randomized into three arms - ensitrelvir 125 mg, ensitrelvir 250 mg, and the placebo arm. The intervention period will span 5 days (Days 1–5), on which patients will be treated with the ensitrelvir or placebo, followed by a follow-up period of 23 days (Days 6–28; Figure 1). Patients who provide consent to participate in the exploratory period (Days 29–337) will be further evaluated over the follow-up period.

Study design
†Optional visit. However, administration of study intervention and entry of participant diary should continue.
††Assessments will be performed only for patients providing consent/assent to participate in the exploratory period. Op, optional; V, visit,
Study ethics
This study will be conducted in accordance with the protocol and consensus ethical principles derived from the Declaration of Helsinki, the Council for International Organizations of Medical Sciences (CIOMS) International Ethical Guidelines, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guidelines, and Good Clinical Practice guidelines. The protocol, protocol amendments, and other relevant documents will be submitted to institutional review boards of participating sites for approval. All patients will be required to provide written informed consent/assent to participate in the study before undergoing any evaluations or procedures. For minor patients, written informed consent will be obtained from a parent/legal guardian. All patients will be informed about the use of their personal, study-related data in accordance with data protection laws. The information and results of this study will be disclosed at the clinical study registration site and other sites. The results of this study may be published or presented at scientific meetings.
Randomization and blinding
Eligible patients will be randomly assigned to the 3 treatment arms in a 1:1:1 ratio using an interactive response technology, and a study intervention allocation table will be created. Randomization will be stratified by the time from the onset of COVID-19 to randomization (<72 hours vs ≥72 hours) and by SARS-CoV-2 vaccination history (vaccinated [first dose completed] vs unvaccinated). Placebo drugs will be indistinguishable in appearance, labeling, and packaging from treatment drugs. All relevant personnel, including the sponsor, patients, and the investigator and sub-investigator, will remain blinded to the treatments. Unblinding at the request of the investigator may be permitted only in the event of an emergency or adverse event (AE), when knowledge of the intervention may be needed for an appropriate course of therapy.
Eligibility criteria
Inclusion criteria
Patients must be capable of providing signed informed consent/assent and must be ≥12 years and <70 years of age at the time of providing informed consent/assent. All patients must (1) have been diagnosed as SARS-CoV-2 positive within 120 hours before randomization by either a nucleic acid detection test using a nasopharyngeal swab, a nasal swab, or saliva (qualitative/quantitative reverse transcription-polymerase chain reaction [RT-PCR] test or isothermal nucleic acid amplification method [e.g., the loop-mediated isothermal amplification method or the transcription-mediated amplification method]), a quantitative antigen test using a nasopharyngeal or nasal swab or saliva, or a qualitative antigen test using a nasopharyngeal or nasal swab; (2) have a time from COVID-19 onset to randomization of ≤120 hours (when at least one of the 14 symptoms occurs: low energy or tiredness, muscle or body aches, headache, chills or shivering, feeling hot or feverish, stuffy or runny nose, sore throat, cough, shortness of breath, nausea, vomiting, diarrhea, loss of smell, and loss of taste); and (3) have at least one moderate symptom (COVID-19 symptom score 2, Supplemental Table 1) or severe symptom (COVID-19 symptom score 3, Supplemental Table 1) among the COVID-19 symptoms at enrollment (excluding loss of smell or taste and any symptoms present prior to COVID-19 onset) or at least one moderate symptom or severe preexisting symptom (present prior to COVID-19 onset) which was considered to have worsened at baseline (before administration of study intervention). Both male and female patients will be eligible. Male patients must not donate sperm during the study intervention period and for ≥10 days after the administration of the last study intervention. They must also practice abstinence or use contraception during the study period and for ≥10 days after the administration of the last study intervention. Female patients must not be pregnant or breastfeeding. Women of child-bearing potential must use contraception/barrier during the study intervention period and for ≥10 days after the administration of the last study intervention, must have a negative result on a pregnancy test (urine or serum) 24 hours prior to the first dose of the study intervention, and must not donate eggs during the study. Patients’ body weight must be ≥40 kg (if patients are minor at the time of providing informed consent/assent).
Exclusion criteria
Patients will be excluded if they (1) have ≤93% (room air) saturation of percutaneous oxygen (SpO2) during wakefulness; (2) require oxygen administration or respirators; (3) are strongly suspected or expected to have worsening of symptoms associated with the SARS-CoV-2 infection within 48 hours after randomization in the opinion of the investigator or sub-investigator; (4) have suspected active and systemic infections (excluding SARS-CoV-2 infection) requiring treatment at the time of randomization; (5) have currently or chronic history of moderate or severe liver disease or known hepatic or biliary abnormalities (with the exception of Gilbert’s syndrome or asymptomatic gallstones), or have currently or chronic history of moderate or severe kidney disease (Grade 2 or higher based on Common Terminology Criteria for Adverse Events28); (6) have used approved or unapproved drugs (e.g., interferon, convalescent plasma, monoclonal antibodies, immunoglobulins, antirheumatic drugs, corticosteroids [oral, injection, inhaled], ivermectin, or favipiravir) for the treatment of SARS-CoV-2 infection within 7 days prior to randomization; (7) have used a strong cytochrome P450 3A (CYP3A) inhibitor, a strong CYP3A inducer, or products containing St. John’s wort within 14 days prior to randomization; (8) have previously received ensitrelvir; (9) have donated ≥400 ml of blood in the 12 weeks or ≥200 ml blood in the 4 weeks prior to providing informed consent/assent; (10) have been exposed to ≥4 new chemical entities in the 12 months prior to dosing; (11) are enrolled or have participated in any other clinical study involving an interventional drug or any other medical research within 28 days prior to providing informed consent; (12) have difficulty in entering details into the patient diary properly due to cognitive decline, have history of drug abuse, or are considered ineligible for the study by the investigator or sub-investigator due to sensitivity to any of the study interventions or their components thereof, have known allergic reactions to any drug, or have history of other allergies (except for seasonal allergies), or for any other reason.
Patients satisfying inclusion criteria 1–3 and not satisfying exclusion criteria 1–4 listed above are considered to have mild-to-moderate COVID-19.
Interventions
Drug dose and administration
Based on the results of the phase 2a27 and phase 2b (H. Mukae et al, unpublished data, May 2022) studies, patients will be orally administered a once-daily dosage of ensitrelvir 125 mg, ensitrelvir 250 mg, or a matching placebo. Two types of placebo doses will be administered – placebo-B, which is identical in appearance and packaging to ensitrelvir 125 mg, and placebo-D, which is identical in appearance and packaging to ensitrelvir 250 mg. For patients assigned to the placebo group, three tablets each of placebo-B and placebo-D will be administered on Day 1, followed by one tablet each of placebo-B and placebo-D administered on Days 2–5. For patients randomized to the ensitrelvir 125 mg group, a loading dose of 375 mg of ensitrelvir (three tablets of 125 mg each) along with three tablets of placebo-D will be administered orally on Day 1. Subsequently, a maintenance dose of ensitrelvir 125 mg (one tablet of 125 mg) along with one tablet of placebo-D will be administered orally, once daily on Days 2–5. For patients randomized to the ensitrelvir 250 mg group, a loading dose of 750 mg of ensitrelvir (three tablets of 250 mg each) along with three tablet of placebo-B will be administered on Day 1. Subsequently, a maintenance dose of ensitrelvir 250 mg (one tablet of 250 mg) along with one tablet of placebo-B will be administered orally, once daily on Days 2–5.
Criteria for discontinuation
The study intervention will be discontinued for patients who experience worsening of the SARS-CoV-2 infection, experience serious or intolerable AEs, are found to be ineligible for the study by the investigator or sub-investigator, request to discontinue the study interventions, are lost to follow-up, become pregnant, or for any other reason per the judgement of the investigator or sub-investigator.
Adherence monitoring
For each patient, the investigator or sub-investigator will assess and record the study adherence at each study visit and any deviation from the study protocol. A record of the number of study intervention tablets dispensed to and taken by each patient will be maintained and reconciled with adherence records.
Lifestyle considerations and prohibited concomitant therapy
Patients should refrain from excessive eating or drinking from the time of providing informed consent/assent to the end of the follow-up period (or completion of tests at discontinuation), and they should refrain from using tobacco and nicotine-containing products (including cigarettes, electronic cigarettes, pipes, cigars, chewing nicotine, nicotine patches, and nicotine gum) during the study intervention period. Because ensitrelvir has an inhibitory effect on CYP3A, patients must refrain from consuming any food (e.g., grapefruit, Seville oranges) and beverages containing CYP3A and also avoid consuming products containing St. John’s wort during the study intervention period. Details of prohibited concomitant drugs are provided in the Supplemental Appendix 1.
Outcome measures and endpoints
The primary endpoint for this study will be the time to resolution of COVID-19 symptoms, defined as the time from the start of the study intervention until the resolution of all symptoms of SARS-CoV-2 infection (with the exception of loss of smell or taste). Patients will assess their symptoms using a partially modified index prescribed by the Food and Drug Administration (FDA)29 (Supplemental Table 1). Loss of smell and/or taste will be assessed using a 3-point scale ranging from 0 to 2, where 0 indicates the same as usual, 1 indicates less than usual, and 2 indicates no sense of smell or taste. The remaining 12 symptoms will be assessed using a 4-point scale ranging from 0 to 3, where 0 indicates no symptoms, 1 indicates mild symptoms, 2 indicates moderate symptoms, and 3 indicates severe symptoms. Resolution of symptoms will be assessed as follows: (i) for pre-existing symptoms that were present before the onset of COVID-19 and considered by the patient to have worsened at baseline, severe symptoms at baseline must have improved to moderate or better, moderate symptoms at baseline must have improved to mild or better, and mild symptoms at baseline must have remained mild or better (no symptoms); (ii) for pre-existing symptoms that were present before the onset of COVID-19 and considered by the patient not to have worsened at baseline, severe symptoms at baseline must have remained severe or improved, moderate symptoms at baseline must have remained moderate or improved, mild symptoms at baseline must have remained mild or better (no symptoms). Symptoms other than the above (those not occurring before the onset of COVID-19 or those that occur at or after baseline) must have completely resolved. Patients will be considered to have achieved the primary endpoint if all 12 COVID-19 symptoms remain resolved for ≥24 hours.
The three key secondary endpoints for this study will be the change from baseline on Day 4 in the amount of SARS-CoV-2 viral RNA (key secondary endpoint 1), the time to first negative SARS-CoV-2 viral titer, defined as the time from the first administration of the study intervention until the first confirmation of SARS-CoV-2 viral titer below a pre-determined detection limit (key secondary endpoint 2), and the proportion of participants without resolution of COVID-19 symptoms 3 weeks after administration, defined as the proportion of participants with one or more of the 12 COVID-19 symptoms which has not been resolved at the final evaluation point 3 weeks after administration of the first dose (key secondary endpoint 3). Other secondary endpoints will include the time to resolution of COVID-19 symptoms (duration of resolution: 96 hours [4 days] or longer), change from baseline in SARS-CoV-2 viral titer at each time point, time to the first negative RT-PCR result, change from baseline in the amount of SARS-CoV-2 viral RNA at each time point, proportion of patients with positive SARS-CoV-2 viral titer or RT-PCR result at each time point, and the proportion of patients with a score of ≥1, ≥2, ≥3, ≥4, ≥5, ≥6, and 7 on the 8-point ordinal scale at each time point. (Supplemental Table 2). As part of the virological examinations, the SARS-CoV-2 viral titer, the amount of viral RNA, and the RT-PCR test results (whether positive or negative) will be determined using nasopharyngeal swabs. Endpoints including SpO2 levels, body temperature and quality of life assessed by the EQ-5D-5L questionnaire will be recorded in a diary maintained by patients. Other outcomes will include the proportion of patients with at least one post-acute COVID-19 syndrome at each time point, the proportion of patients for each post-acute COVID-19 syndrome at each time point, and the time to first onset of loss of smell or taste defined as the time from the start of the study intervention to the first occurrence of loss of smell or taste. Safety assessments will include laboratory tests related to hematology, blood chemistry, coagulation, serology, urinalysis, and others; physical examination; and measurement of vital signs (Supplemental Table 3).
Rationale for the primary endpoint and key secondary endpoints
An appropriate endpoint for evaluating the clinical efficacy of therapeutics for SARS-CoV-2 infection has not yet been established. The phase 2a27 and 2b (H. Mukae et al, unpublished data, May 2022) studies demonstrated decreased viral load with ensitrelvir treatment. In the phase 2a and 2b, time to improvement of COVID-19 symptoms were applied for the clinical assessment which has been used for assessment of influenza treatment. FDA recommends the assessment in the time to clinical recovery for COVID-19 treatment29, and in a post hoc analysis of the phase 2a study, compared with placebo, the ensitrelvir 125 mg and 250 mg groups demonstrated a trend toward shorter median (95% confidence interval [CI]) time to resolution of COVID-19 symptoms (placebo, 335.5 [146.9, -] hours; ensitrelvir 125 mg, 195.2 [120.5, -] hours, P = .67 [vs placebo]; ensitrelvir 250 mg, 166.7 [81.9, 350.2] hours, P = .11 [vs placebo]; Figure 2A). The results were further confirmed in post hoc analysis of the subsequent phase 2b study. Compared with placebo, a trend toward a shorter median (95% CI) time to resolution of COVID-19 symptoms was observed in the ensitrelvir 125 mg group, whereas a statistically significant difference was observed in the ensitrelvir 250 mg group (placebo, 243.4 [159.5, 338.4] hours; ensitrelvir 125 mg, 169.4 [133.5, 204.8] hours, P = .09 [vs placebo]; ensitrelvir 250 mg, 154.7 [135.6, 196.9] hours, P = .04 [vs placebo]; Figure 2B). COVID-19 symptoms may be prolonged, regardless of risk factors or severity of illness. A more stringent endpoint such as the time to resolution of symptoms may be a meaningful clinical target for patients with SARS-CoV-2 infection. Therefore, the time to resolution of symptoms is proposed as the primary endpoint to assess the clinical benefit of antiviral treatment in patients with SARS-CoV-2 infection.

Proportion of patients showing resolution of symptoms in the phase 2a [A] and 2b [B] studies
mg, milligrams.
Post-acute COVID-19 syndrome causes a wide range of health problems, and it is reported that symptoms at 3-4 weeks is associated with an increased risk of post-acute COVID-19 syndrome months later.30, 31 In the post-hoc analysis of phase 2b, the proportion of participants without resolution of 12 COVID-19 symptoms 3 weeks after administration in the ensitrelvir 125 mg and 250 mg groups were lower than that in placebo group (ensitrelvir 125 mg, P = .0461; ensitrelvir 250 mg, P = .2635; Figure 3A). This trend was also observed in 14 COVID-19 symptoms (ensitrelvir 125 mg, P = .0300; ensitrelvir 250 mg, P = .2025; Figure 3B), which together suggest that ensitrelvir treatment may reduce the risk of developing post-acute COVID-19 syndrome. Hence, the proportion of participants without resolution of COVID-19 symptoms 3 weeks after administration is newly proposed as an endpoint for assessment of post-acute COVID-19 syndrome and is included in key secondary endpoints.

Proportion of participants without resolution of COVID-19 symptoms 3 weeks after administration in the phase 2b study; 12 protracted symptoms [A], 14 protracted symptoms [B] mg, milligrams.
Patient timelines
The schedule of activities for each patient will include efficacy and safety assessments (Table 1). Efficacy assessments will include virological examinations, maintenance of patient diaries, and assessing disease severity. Patients will self-assess all 14 symptoms twice daily (morning and evening) from before the first dose of study intervention on Day 1 to Day 9 and once daily (evening) from Day 10 to Day 21. All endpoints assessed using the SARS-CoV-2 nasopharyngeal swab will be evaluated as per the schedule of activities (Table 1). All samples for viral titer from Days 1–6 will be assessed, and only select samples will be assessed from Days 9–21 based on the RT-PCR test result. SpO2 levels and body temperature will be measured and recorded in the diary twice daily (morning and evening) from before the first dose of study intervention on Day 1 until Day 9 and once daily (evening) from Day 10 to Day 21. If acetaminophen is taken as an antipyretic or analgesic, the COVID-19 symptom score will not be evaluated, and body temperature will not be measured until 4 hours after taking it. Patients will also complete the EQ-5D-5L questionnaire twice daily (morning and evening) from before the first dose of study intervention on Day 1 until Day 9 and once daily (evening) from Day 10 until Day 21 and record the assessments in the diary. Disease severity will be scored by the investigator or sub-investigator using the 8-point ordinal scale as per the schedule of activities (Table 1). Safety assessments will be performed as per the schedule of activities (Table 1).
Schedule of activities
Statistical analysis
Sample size estimation
In the phase 2b study, the median time to resolution of COVID-19 symptoms was 10.1 days in the placebo group, 7.1 days in the ensitrelvir 125 mg group, and 6.4 days in the ensitrelvir 250 mg group. Most of the patients in the phase 2b study were enrolled in Japan. Given that the current phase 3 study will be expanded to other Asian countries, the median time to resolution for each ensitrelvir group was conservatively assumed to be 8 days, while that for the placebo group was assumed to be 10 days. As such, the hazard ratio of the ensitrelvir groups to placebo group in time to resolution of COVID-19 symptoms was assumed to be 0.8. As multiplicity adjustment using the Bonferroni method will be performed, the sample size required to detect differences in median time to resolution with 80% power using the log-rank test at a one-sided significance level of .0125 was estimated to be 1428 patients (476 per group). Assuming a dropout rate due to a negative RT-PCR result of 10% before the enrollment, the sample size of patients to be enrolled was estimated to be 1590 patients (530 per group).
Analysis populations
Primary and secondary endpoints except for endpoints related to SARS-CoV-2 viral titer and efficacy assessments will be assessed in the intention-to-treat (ITT) population, comprising all patients randomly assigned to the study intervention and had SARS-CoV-2 infection confirmed by RT-PCR result based on nasopharyngeal swab sample on Visit 1 (pre-intervention). Secondary endpoints related to SARS-CoV-2 viral titer will be assessed in the modified intention-to-treat (mITT) population, comprising all patients randomly assigned to the study intervention with SARS-CoV-2 viral titer detected at baseline. All safety assessments will be conducted in the safety analysis population (SAP) comprising patients who were randomly assigned to a study intervention and who received at least one dose of the study intervention.
Statistical tests
For discrete variables, summary statistics such as the number and proportion of patients in each group will be calculated. For continuous variables, summary statistics such as the number of patients, arithmetic mean (mean), standard deviation (SD), minimum, median, and maximum values will be calculated for each group. The 95% confidence intervals will be calculated using the Clopper-Pearson method and the bootstrap percentile method for the difference in median time. All statistical tests will be performed at a two-sided significance level of .05, unless otherwise specified.
As the primary analysis for the primary endpoint, a pairwise comparison of the time to resolution of COVID-19 symptoms will be performed between each ensitrelvir treatment group and the placebo group using a log-rank test stratified by time from COVID-19 onset to randomization (<72 hours, ≥72 hours), and SARS-CoV-2 vaccination history at a one-sided significance level of .025. Type I error rate control/multiplicity-adjusted comparisons for the primary endpoint will be conducted; the Bonferroni method with a one-sided significance level of .0125 for pairwise comparisons between each ensitrelvir treatment group and the placebo group will be applied.
The primary analyses for each of the three key secondary endpoints are as follows. For the key secondary end point 1, a pairwise comparison of the change from baseline on Day 4 in the amount of SARS-CoV-2 viral RNA will be performed between each ensitrelvir treatment group and the placebo group using an analysis of covariance (ANCOVA) with change from baseline in the amount of SARS-CoV-2 viral RNA as response, and time from the onset of COVID-19 to randomization (< 72 hours, ≥ 72 hours), SARS-CoV-2 vaccination history, and the amount of SARS-CoV-2 viral RNA at baseline as covariates at a one-sided significance level of .025. For the key secondary endpoint 2, a pairwise comparison of the time to the first negative SARS-CoV-2 viral titer will be performed between each ensitrelvir treatment group and the placebo group using a log-rank test stratified by time from COVID-19 onset to randomization (<72 hours, ≥72 hours), and SARS-CoV-2 vaccination history at a one-sided significance level of .025. For the key secondary endpoint 3, a pairwise comparison of the proportion of participants without resolution of COVID-19 symptoms 3 weeks after administration will be performed between each ensitrelvir treatment group and the placebo group using a Mantel-Haenszel test stratified by time from COVID-19 onset to randomization (< 72 hours, ≥ 72 hours) and SARS-CoV-2 vaccination history at a one-sided significance level of .025. Type 1 error rate control/multiplicity-adjusted pairwise comparisons between each ensitrelvir treatment group and the placebo group for each of the three key secondary endpoints will be performed using the Bonferroni method with a one-sided significance level of .0125 as with the primary endpoint. In terms of type 1 error control induced by statistical tests for the three key secondary endpoints, a fixed sequence procedure will be applied with hierarchical order of the key secondary endpoint 1, the key secondary endpoint 2 and the key secondary endpoint 3 in each pairwise comparison between ensitrelvir 125 mg group and the placebo group. In this procedure, if a precedent statistical test is not statistically significant at .0125, the subsequent statistical tests will not be performed. The same will be true for the three key secondary endpoints in each pairwise comparison between ensitrelvir 250 mg group and the placebo group.
The other analyses for the primary and key secondary endpoints are as follows. A pairwise comparison of the time to resolution of COVID-19 symptoms and the time to the first negative SARS-CoV-2 viral titer will be performed between each ensitrelvir treatment group and the placebo group using the Peto-Prentice’s generalized Wilcoxon test stratified by time from COVID-19 onset to randomization (<72 hours, ≥72 hours) and SARS-CoV-2 vaccination history at a two-sided significance level of .05. Kaplan-Meier curves will be plotted for each treatment group. The median time to resolution of COVID-19 symptoms and first negative SARS-CoV-2 viral titer along with the respective 95% CIs will be calculated; the difference in median time between treatment groups along with 95% CIs will be calculated. The hazard ratio for the time to resolution of COVID-19 symptoms and the time to first negative SARS-CoV-2 viral titer of each ensitrelvir treatment group to the placebo group will be estimated using a Cox proportional hazard model stratified by time from COVID-19 onset to randomization (<72 hours, ≥72 hours) and SARS-CoV-2 vaccination history. Additionally, restricted mean survival time with a 21-day investigation period will be estimated for the time to resolution of COVID-19 symptoms and the time to first negative SARS-CoV-2 viral titer for each treatment group, and pairwise comparisons between each ensitrelvir treatment group and the placebo will be performed. Multiplicity adjustments will not be performed for the above analyses.
Analysis of the time to resolution of COVID-19 symptoms (duration of resolution, 96 hours [4 days] or longer) will be performed by changing the definition of the duration of resolution of COVID-19 symptoms from 24 hours to 96 hours. The same analysis as the primary endpoint (time to resolution of COVID-19 symptoms) (except for multiplicity adjustment) will be performed in the ITT population. For analyzing the change from baseline in SARS-CoV-2 viral titer and the amount of viral RNA at each time point, least squares means and difference between the ensitrelvir groups and the placebo group based on the ANCOVA model will be calculated. The time from onset to randomization of COVID-19 (<72 hours or ≥72 hours), SARS-CoV-2 vaccination history (Yes or No) and the amount of SARS-CoV-2 viral RNA at baseline (SARS-CoV-2 viral titer at baseline for analyzing the change from baseline in SARS-CoV-2 viral titer) will be used as the covariate in the ANCOVA model. Time to the first negative RT-PCR result will be analyzed in a manner similar to the key secondary endpoint. To evaluate the proportion of patients with positive SARS-CoV-2 viral titer or RT-PCR results at each time point and the proportion of patients with a score of ≥1, ≥2, ≥3, ≥4, ≥5, ≥6, and 7 on the 8-point ordinal scale at each time point, a pairwise Mantel-Haenszel test will be conducted to compare each ensitrelvir treatment group with the placebo group at a two-sided significance level of .05. The same stratification factors used for the primary endpoint apply. Additionally, for the proportion of patients with a score of ≥1, ≥2, ≥3, ≥4, ≥5, ≥6, and 7 on the 8-point ordinal scale at each time point, the risk ratio and risk difference of each ensitrelvir treatment group to the placebo group will be estimated using the Mantel-Haenszel method.
For the proportion of patients with at least one post-acute COVID-19 syndrome at each time point and the proportion of patients for each post-acute COVID-19 syndrome at each time point, a pairwise Mantel-Haenszel test will be conducted to compare each ensitrelvir treatment group with the placebo group at a two-sided significance level of .05, stratified by time from COVID-19 onset to randomization (<72 hours, ≥72 hours) and SARS-CoV-2 vaccination history. Additionally, for the proportion of patients for each post-acute COVID-19 syndrome at each time point, the risk ratio and risk difference of each ensitrelvir treatment group to the placebo group will also be estimated using the Mantel-Haenszel method.
For the time to first onset of loss of smell or taste, a pairwise comparison will be performed between each ensitrelvir treatment group and the placebo group using a log-rank test, Peto-Prentice’s generalized Wilcoxon test stratified by time from COVID-19 onset to randomization (<72 hours, ≥72 hours) and SARS-CoV-2 vaccination history at a one-sided significance level of .025.
Imputation of missing data
Missing data will not be imputed; all statistical analyses will be based on observed cases, unless otherwise specified. The missing assessment times of a patient’s diary after the initial administration of the study intervention will be imputed: morning times will be imputed as 11:59:59, and evening times will be imputed as 23:59:59 (only evening times apply for Days 10–21).
Data management and monitoring
No interim analyses will be performed for this phase 3 study. An Independent Data Monitoring Committee will not be established. A Data and Safety Monitoring Board will be established for the purpose of third-party evaluation of safety throughout the study period. All patient data relating to the study will be recorded on electronic case report forms (eCRFs) unless electronically transferred to the sponsor or designee (e.g., laboratory data). After the follow-up period is complete, all data in the eCRFs for each patient will be locked. Source data verification will be performed to confirm that the data entered into the eCRFs by authorized site personnel are accurate, complete, and verifiable from source documents, that the safety and rights of patients are being protected, and that the study is being conducted in accordance with the currently approved protocol and any other study agreements and applicable regulatory requirements.
All AEs and SAEs will be monitored from the time of providing informed consent/assent until the end of the follow-up period, as per the schedule of activities (Table 1). The investigator or sub-investigator or any qualified designee will be responsible for detecting, documenting, reporting AEs and SAEs, and will following up with patients for AEs and SAEs considered related to the study intervention or study procedures, or those that caused the participant to discontinue the intervention.
DISCUSSION AND CONCLUSIONS
We described the protocol for a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial to assess the efficacy and safety of ensitrelvir, a novel oral SARS-CoV-2 3CL protease inhibitor, in expediting the resolution of symptoms in patients with mild-to-moderate COVID-19. We aimed to test the hypothesis that COVID-19 symptoms resolve sooner when patients are treated with ensitrelvir compared with placebo. Through this study, we seek to validate its potential antiviral efficacy observed in previous studies and to further establish its clinical efficacy and safety.
Approximately 81%–85% of individuals with COVID-19 experience mild-to-moderate symptoms.32,33 However, up to 14% of patients with mild-to-moderate illness can develop severe complications within a week.32 Specific groups of patients such as those above 75 years of age and those having comorbidities such as obesity, chronic kidney disease, diabetes mellitus, hypertension, and heart failure, are predisposed to progressing from mild-to-moderate illness to critical forms.32 Therefore, several treatment strategies are directed toward protecting these vulnerable subgroups.17-19 Treating patients with mild disease early in the disease course is not only critical for preventing progression of the infection to severe forms but also for containing the spread of the virus.32 Treatments for individuals who are at a low risk of developing severe illness are limited, and new medicines are awaited to treat such patients.
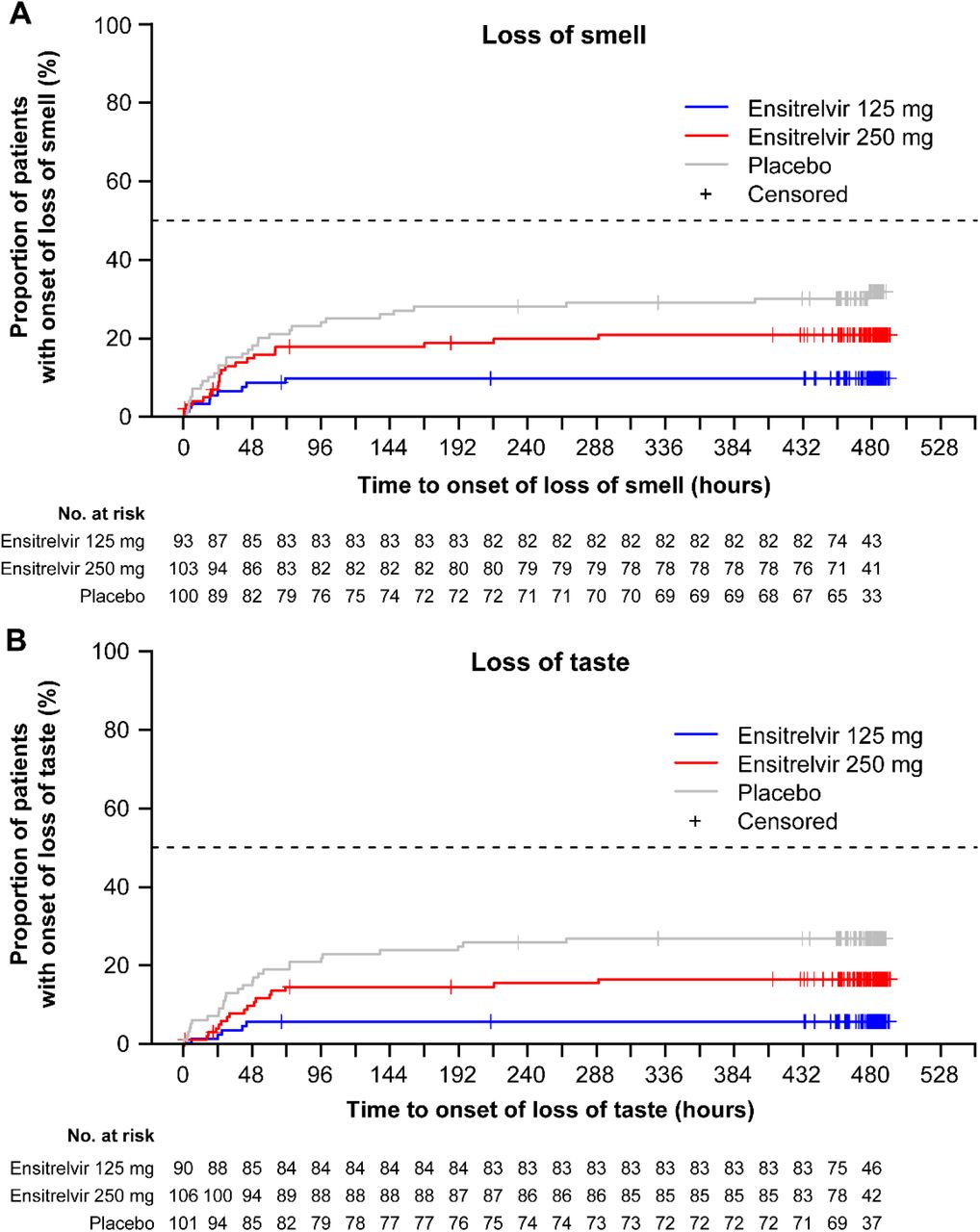
Ensitrelvir has previously been demonstrated to have antiviral efficacy against SARS-CoV-2. In both the phase 2a27 and 2b (H. Mukae et al, unpublished data, May 2022) studies evaluating its antiviral effect, once-daily ensitrelvir was shown to significantly reduce SARS-CoV-2 viral titer and viral RNA compared with placebo. In post hoc analyses of the phase 2a study data, a trend toward a difference in the time to resolution of symptoms was observed between the ensitrelvir groups and the placebo group. This finding was further confirmed in post hoc analyses of the phase 2b study data; the time to resolution of symptoms was shorter in the ensitrelvir groups than in the placebo group. Additional analysis of the phase 2b study on loss of smell and taste revealed that compared with placebo, ensitrelvir reduced the number of patients with onset of both loss of smell (ensitrelvir 125 mg, P < .001; ensitrelvir 250 mg, P = .10; Figure 4A) and loss of taste (ensitrelvir 125 mg, P < .001; ensitrelvir 250 mg, P = .07; Figure 4B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of patients showing onset of loss of smell [A] and/or taste [B] in the phase 2b study
mg, milligrams
Irrespective of risk factors and the severity of the infection, symptoms of COVID-19 may persist for a prolonged duration. Therefore, a stringent endpoint such as the time to resolution of symptoms may be suitable for evaluating the clinical efficacy of ensitrelvir in mild-to-moderate infections. Moreover, this endpoint is also appropriate in light of the disease modification due to vaccination and the emergence of new variants of the virus. Since there is no established evidence on how symptoms in the mild-to-moderate COVID-19 progress over time, the endpoint of time to resolution of COVID-19 symptoms will provide clarity on how symptoms resolve with or without an antiviral treatment.
One of the strengths of this study is that it includes patients with COVID-19, irrespective of whether they are at a high risk of developing severe illness. Additionally, this study will enroll both vaccinated and unvaccinated individuals, unlike other studies assessing oral antivirals that mainly recruited unvaccinated patients. Since vaccination has a large impact on how the infection manifests and modifies in individuals, including vaccinated patients in the study will provide a comprehensive understanding of the antiviral treatment.
It is hoped that this phase 3 study will be able to confirm the clinical efficacy and safety of ensitrelvir in the treatment of mild-to-moderate COVID-19, and the results will help provide evidence about symptom progression or improvement.
AUTHOR CONTRIBUTIONS
Conceptualization: All authors.
Data curation: Hiroshi Yotsuyanagi, Norio Ohmagari, Yohei Doi, Genki Ichihashi, Takao Sanaki, and Hiroshi Mukae
Formal analysis: Takumi Imamura and Takao Sanaki
Visualization: Takumi Imamura
Methodology: Takao Sanaki
Supervision: Takumi Imamura, Takuhiro Sonoyama, Yuko Tsuge, Takeki Uehara, and Hiroshi Mukae
Project administration: Genki Ichihashi, Yuko Tsuge, and Takeki Uehara
Writing – review and editing: All authors.
FUNDING
This study was funded by Shionogi & Co., Ltd. Employees of Shionogi & Co., Ltd. participated in and approved the design and conduct of the study, wrote the protocol, and were involved in the collection, management, analysis, and interpretation of data. Institutional authors reviewed and approved the protocol and collected and interpreted the data.
CONFLICTS OF INTEREST
HY reports consulting fees from Shionogi, lecture fees from Shionogi and ViiV Healthcare, and travel support from Shionogi outside the submitted work. He serves as an advisory board member of Shionogi and President of the Japanese Society of Infectious Diseases. NO declares no conflict of interest. YD reports grants from Shionogi and Entasis; consulting fees from Shionogi, Meiji Seika Pharma, Gilead Sciences, GSK, MSD, Chugai, and bioMerieux; and lecture fees from MSD, AstraZeneca, Shionogi, and Teijin Healthcare outside the submitted work and serves as an advisory board member of FujiFilm. TI, T Sonoyama, GI, T Sanaki, YT, and TU are full-time employees of Shionogi & Co., Ltd. and may own stocks or stock options. HM has received funding relevant to the submitted work from Shionogi and grants from Taisho Pharma; lecture fees from Pfizer, MSD, Shionogi, and Taisho Pharma; and advisory fees from Pfizer, MSD, and Shionogi outside the submitted work.
SUPPLEMENTAL MATERIAL
SUPPLEMENTAL APPENDIX 1
Prohibited concomitant drugs
Drugs to be avoided from the time of providing informed consent/assent until the end of the follow-up period (Day 28) include approved drugs and unapproved drugs (e.g., interferons, convalescent plasma, monoclonal antibodies, immunoglobulins, antirheumatic drugs, oral, injectable, or inhaled corticosteroids, ivermectin, and favipiravir) for the treatment of SARS-CoV-2 infection; CYP3A substrates; antiviral, antibacterial, or antifungal drugs (excluding those for external use); antipyretic analgesics other than acetaminophen; antitussives and expectorants; combination cold remedy; and investigational drugs as part of other clinical trials.
Drugs to be avoided from the time of providing informed consent/assent until 10 days after the last study intervention administration include strong CYP3A inhibitors or inducers; P-glycoprotein (P-gp) inhibitors; breast cancer resistance protein (BCRP) inhibitors; and substrates for P-gp, BCRP, organic anion transporter polypeptide (OATP) 1B1, OATP 1B3, organic cation transporter (OCT) 1, organic anion transporter (OAT) 3, and multidrug and toxin extrusion (MATE) 1 transporters.
SUPPLEMENTAL TABLES
COVID-19 symptom score
The 8-point ordinal scale for assessing symptom severity
List of safety laboratory assessments
ACKNOWLEDGMENTS
We would like to thank Shintaro Tanaka, Masahiro Kinoshita, Satoshi Kojima and Manami Yoshida for their contribution toward the development and review of this manuscript. Medical writing and editorial support were provided by Varsha Sreenivasan, PhD, of Cactus Life Sciences (part of Cactus Communications Pvt. Ltd.) and funded by Shionogi & Co., Ltd.
Footnotes
Name and contact of trial sponsor: Shionogi & Co., Ltd.
ABBREVIATIONS
- AE
- adverse event
- CI
- confidence interval
- COVID-19
- coronavirus disease 2019
- CYP3A
- cytochrome P450 3A
- eCRF
- electronic case report form
- RNA
- ribonucleic acid
- RT-PCR
- reverse transcription-polymerase chain reaction
- SAE
- serious adverse event
- SARS-CoV-2
- severe acute respiratory syndrome coronavirus
- 2 SpO2
- saturation of percutaneous oxygen
- VOC
- variant of concern
REFERENCES




Subject Area
Reviews and Context
0
Comment
0
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
0
Blogs/Media
Author Videos