
Abstract
The apparent lack of antigenic evolution by the Delta variant (B.1.617.2) of SARS-CoV-2 during the COVID-19 pandemic is puzzling. The combination of increasing immune pressure due to the rollout of vaccines and a relatively high number of infections following the relaxation of non-pharmaceutical interventions should have created perfect conditions for immune escape variants to evolve from the Delta lineage. Instead, the Omicron variant (B.1.1.529), which is hypothesised to have evolved in an immunocompromised individual, is the first major variant to exhibit significant immune escape following vaccination programmes and is set to become globally dominant in 2022. Here, we use a simple mathematical model to explore possible reasons why the Delta lineage did not exhibit antigenic evolution and to understand how and when immunocompromised individuals affect the emergence of immune escape variants. We show that when the pathogen does not have to cross a fitness valley for immune escape to occur, immunocompromised individuals have no qualitative effect on antigenic evolution (although they may accelerate immune escape if within-host evolutionary dynamics are faster in immunocompromised individuals). But if a fitness valley exists between immune escape variants at the between-host level, then persistent infections of immunocompromised individuals allow mutations to accumulate, therefore facilitating rather than simply speeding up antigenic evolution. Our results suggest that better global health equality, including improving access to vaccines and treatments for individuals who are immunocompromised (especially in lower- and middle-income countries), may be crucial to preventing the emergence of future immune escape variants of SARS-CoV-2.
Lay Summary We study the role that immunocompromised individuals may play in the evolution of novel variants of the coronavirus responsible for the COVID-19 pandemic. We show that immunocompromised hosts can be crucial for the evolution of immune escape variants. Targeted treatment and surveillance may therefore prevent the emergence of new variants.
Introduction
Understanding how and when variants of SARS-CoV-2, the causative agent of COVID-19, are likely to evolve is key to managing the future of the pandemic. Multiple variants of concern have evolved since the start of the pandemic, with higher transmissibility evolving on at least two occasions, by the Alpha (B.1.1.7) variant (relative to the wildtype) [1], and by the Delta (B.1.617.2) variant (relative to Alpha) [2,3], with the latter becoming the globally dominant strain in 2021 [4]. Other variants such as Beta (B.1.351) and Omicron (B.1.1.529) have instead shown evidence of immune escape, indicating antigenic evolution [5–7]. With increasing numbers of people acquiring immunity to SARS- CoV-2, either through infection or vaccination, we should expect a shift towards antigenic evolution rather than higher transmissibility or greater virulence as the primary driver of new variants of concern [8]. The extent to which SARS-CoV-2 may evolve antigenically in future, thereby allowing it to evade host immunity partially or fully, is currently unknown. However, the emergence and rapid spread of Omicron towards the end of 2021 has demonstrated that antigenic evolution is both possible and under strong selection. The unusual nature of Omicron (possessing a large number of mutations in the spike protein but only distantly related to the dominant variant at the time, Delta [9]) has led to speculation that it underwent long-term within-host evolution in an immunocompromised individual who was unable to clear the infection [10]. We explore this hypothesis using a simple mathematical model to understand the potential importance of immunocompromised individuals for the antigenic evolution of SARS-CoV-2.
A fundamental tenet of evolutionary epidemiology is that the rate of antigenic evolution depends on a balance between immune pressure and mutation supply [11–13]. The greater the proportion of the population that is immune, the greater the strength of selection for immune escape but mutation supply is constrained as few hosts can be infected. Conversely, if many hosts are susceptible to infection, then mutation supply may be plentiful but selection for immune escape is relatively weak. Hence, the rate of antigenic evolution should be maximised at an intermediate level of immune pressure, whereby moderate pathogen prevalence leads to a plentiful supply of mutations for selection to act upon, and the strength of selection for immune escape is reasonably strong.
Rapid deployment of vaccinations against SARS-CoV-2 combined with the relaxation of non- pharmaceutical interventions in many countries led to both strong immune pressure and high numbers of infections in the latter half of 2021. For example, by the end of November 2021 the UK had fully vaccinated 68% of the population while still experiencing over 620 confirmed cases per million (approximately 70% of the previous peak in January 2021) [14]. At the time, the Delta variant was dominant globally and accounted for over 99% of infections in the UK [14]. Yet, despite apparently favourable evolutionary conditions for immune escape there were no indications of the Delta variant exhibiting antigenic evolution in the UK or elsewhere. Instead, the Omicron variant, first detected in South Africa and reported to the World Health Organization on November 24, 2021 [9], was able to substantially escape host immunity and evolved from a different clade. Omicron contains 30 mutations to the spike protein (used for binding to host cell receptors) and has been shown to evade over 85% of neutralizing antibodies [7]. Relative to Delta, Omicron exhibits substantially lower vaccine effectiveness [15] and is estimated to be over five times as likely to lead to reinfection [6]. Omicron became the dominant variant in the UK within a month and is on course to replace Delta in many countries in early 2022 [14].
The Omicron variant confirms that substantial immune escape is not only possible for SARS-CoV-2 but also that selection for immune escape towards the end of 2021 was very strong. According to the conceptual model of antigenic evolution as a balance between immune pressure and mutation supply [11], this suggests that the lack of adaptation to evade host immunity by the Delta lineage was simply due to insufficient mutation supply. However, this is difficult to reconcile with the high number of cases at the time, implying mutation supply was plentiful. Furthermore, if mutation supply was the key constraint, how did an immune escape variant appear from an obscure clade that was responsible for few infections?
A promising hypothesis is that the Omicron variant arose due to long-term within-host evolution in an immunocompromised individual, who was most likely infected between March and August 2021 [9]. While an immunocompetent individual would be expected to clear infection after a relatively short period, an immunocompromised person may fail to fully clear the infection, allowing the virus to coevolve with the immune system [16]. Indeed, longitudinal sequencing from an immunocompromised patient who was infected for over 150 days with SARS-CoV-2 revealed rapid accumulation of mutations [17]. These mutations appeared to be adaptive at the within-host level due to their concentration in the spike protein, with several common to other variants of concern. Similar results have been observed in other patients with long-term infections of SARS-CoV-2 [18,19], including those who have been treated with convalescent plasma, indicating antigenic evolution within the host [20].
It is currently unclear how important immunocompromised individuals are for the antigenic evolution of SARS-CoV-2. Do infections of immunocompromised individuals simply accelerate antigenic evolution or do they play a key role in facilitating immune escape? In the first case, infection of immunocompromised individuals speeds up antigenic evolution due to faster within- host dynamics, leading to the emergence of new immune escape variants on shorter timescales than would be possible in an immunocompetent population. Such a scenario would suggest that although immunocompromised individuals might speed up the process, they are not essential for antigenic evolution to occur. In the second case, long-term infections of immunocompromised individuals allow the virus to accumulate mutations that are advantageous at the within-host level but may be disadvantageous at the between-host level. If there is epistasis between mutations at the between host level (i.e. fitness depends on the context of which other mutations are present), then sustained adaptation within immunocompromised individuals may allow the virus to traverse valleys in the fitness landscape, which would otherwise be very difficult to cross, to reach another peak (Fig. 1b). The second scenario would therefore suggest that long-term infections in immunocompromised individuals play a crucial role in the antigenic evolution of SARS-CoV-2.

(a) Cross-immunity,σij, for variants i and j, with lighter colours corresponding to greater cross-immunity. (b) Illustration of the normalised transmission rate for each variant, showing a fitness valley. (c) Model schematic.
Here, we analyse a simple phenomenological model to explore the potential importance of immunocompromised hosts for the antigenic evolution of SARS-CoV-2. We show that in the absence of epistasis, antigenic evolution readily occurs regardless of the frequency of immunocompromised individuals in the population. If epistasis is present, however, such that the virus must traverse a fitness valley at the between-host level to escape host immunity, then immunocompromised hosts are crucial for antigenic evolution to occur. These patterns are robust irrespective of whether within- host evolutionary dynamics are faster in immunocompromised individuals and for a wide range of parameters affecting cross-immunity, the strength of epistasis, the proportion of the population that is immunocompromised and their duration of infection relative to immunocompetent hosts.
Model description
We adapt the model of antigenic evolution presented by Gog and Grenfell [21] to incorporate immunocompromised individuals and epistasis. The model assumes that there are n =30 variants equally spaced in a line, with adjacent variants differing by a single mutation. Hosts are classed as either entirely susceptible to a variant, or entirely immune to it. Cross-immunity between variants is therefore ‘polarising’, which means that when an individual is infected by variant i, a proportion σ ij of those currently susceptible to variant j become fully immune to it for life (no waning immunity) and a proportion 1 − σ ij remain fully susceptible to variant j. This assumption greatly reduces the complexity of the model as it means we do not need to track all infection histories, which would require at least (2+n)2n ≈ 137 billion classes with n =30. The strength of cross-immunity between variants i and j is given by
 where η > 0 controls the breadth of cross-immunity (large values of η give broad cross-immunity between distant variants, whereas small values of η limit cross-immunity to closely related variants; Fig. 1a). We assume that the population is large, well-mixed, and of constant size (n =107), with a proportion p of individuals who are immunocompromised (only able to produce a weak immune response; subscript C) and a proportion 1 − p who are immunocompetent (able to produce a normal or “healthy” immune response; subscript H). For simplicity, we ignore host demographics (births and deaths) and mortality from infection, as we are only interested in the antigenic evolution of the virus over a relatively short timescale.
where η > 0 controls the breadth of cross-immunity (large values of η give broad cross-immunity between distant variants, whereas small values of η limit cross-immunity to closely related variants; Fig. 1a). We assume that the population is large, well-mixed, and of constant size (n =107), with a proportion p of individuals who are immunocompromised (only able to produce a weak immune response; subscript C) and a proportion 1 − p who are immunocompetent (able to produce a normal or “healthy” immune response; subscript H). For simplicity, we ignore host demographics (births and deaths) and mortality from infection, as we are only interested in the antigenic evolution of the virus over a relatively short timescale.
Let 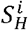 (respectively
(respectively  ) be the proportion of the population that is immunocompetent (resp. immunocompromised) and susceptible to variant i ∈{1,…,n}, and
) be the proportion of the population that is immunocompetent (resp. immunocompromised) and susceptible to variant i ∈{1,…,n}, and 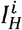 (resp.
(resp. 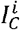 ) be the proportion of the population that is immunocompetent (resp. immunocompromised) and infected with variant i. To incorporate a fitness valley at the between-host level, we assume that the transmission rate of variant i is given by
) be the proportion of the population that is immunocompetent (resp. immunocompromised) and infected with variant i. To incorporate a fitness valley at the between-host level, we assume that the transmission rate of variant i is given by
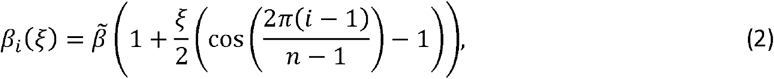 where
where 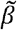 is the maximum transmission rate and ξ controls the strength of epistasis (Fig. 1b). Preliminary analysis revealed that other functional forms with qualitatively similar properties produce results consistent with those presented below. When ξ = 0, there is no epistasis as
is the maximum transmission rate and ξ controls the strength of epistasis (Fig. 1b). Preliminary analysis revealed that other functional forms with qualitatively similar properties produce results consistent with those presented below. When ξ = 0, there is no epistasis as 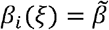 for all variants. When 0 < ξ < 1, epistasis reduces the transmission rate for variants intermediate between 1 and n, reaching a minimum of βi (ξ) =1 − ξ with
for all variants. When 0 < ξ < 1, epistasis reduces the transmission rate for variants intermediate between 1 and n, reaching a minimum of βi (ξ) =1 − ξ with 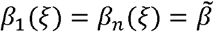 for all ξ (Fig. 1b).
for all ξ (Fig. 1b).
To allow for random mutations, we simulate our model using the stochastic τ-leaping method [22] for the underlying ordinary differential equations (ODEs)

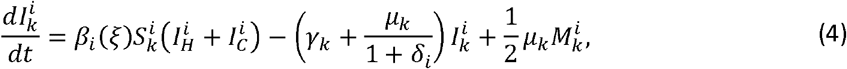 where
where  is the set of variants adjacent to i in the one-dimensional antigenic space
is the set of variants adjacent to i in the one-dimensional antigenic space 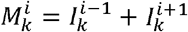 for i ∈{2...,n − 1}, with boundary conditions
for i ∈{2...,n − 1}, with boundary conditions 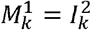 and
and 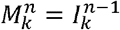 , and δi = 1 if i ∈{1,n} and is 0 otherwise to control the rate of antigenic evolution at the boundaries. A schematic for this system can be found in Fig. 1c.
, and δi = 1 if i ∈{1,n} and is 0 otherwise to control the rate of antigenic evolution at the boundaries. A schematic for this system can be found in Fig. 1c.
We run 10 simulations for each parameter combination up to tmax = 1460 time steps (days), as preliminary analysis revealed that either antigenic evolution reaches the boundary of antigenic space within this timeframe, or the infection is driven extinct. Note however, that this duration is arbitrary and varies inversely with µC and µH. We say that a variant is ‘observed’ if it exceeds a threshold of 0.01. In each simulation, we summarise the dynamics by measuring the total number of variants observed and the maximum distance in antigenic space between observed variants.
Results
We focus our analysis on the strength of epistasis on transmissibility, the strength of cross- immunity, the proportion of the population that is immunocompromised, the relative rate of adaptation in immunocompromised hosts. In the absence of epistasis (or when epistasis is sufficiently weak), the virus diffuses gradually through antigenic space (Figs. 2a and 2c). As the host population accumulates immunity to the current dominant variant, selection favours the next variant in line that can substantially escape immunity, leading to successive epidemic waves at regular intervals. This occurs regardless of whether within-host evolution is assumed to be faster in immunocompromised individuals (Fig. 3a).

(a) No epistasis in an entirely immunocompetent population. (b) Strong epistasis in an entirely immunocompetent population. (c) No epistasis and a small immunocompromised subpopulation. (d) Strong epistasis and a small immunocompromised subpopulation. All other parameter values given in Table A3. Dynamics are shown for a single simulation.

Antigenic evolution when the rate of within-host evolution is faster in immunocompromised individuals. In both plots, there is a small immunocompromised proportion and the rate of within-host evolution in the immunocompromised population is 5 times higher than in the immunocompetent population. (a) No epistasis and (b) strong epistasis. All other parameters as in Figure 2. Dynamics are shown for a single simulation.
When epistasis is sufficiently strong, however, the proportion of the population that is immunocompromised plays a crucial role in antigenic evolution (Figs. 2b and 2d). If very few individuals are immunocompromised, the epidemic quickly burns out with little antigenic evolution, as the virus is unable to cross the fitness valley caused by epistasis at the between-host level (Fig. 2b). But if a sufficient proportion of the population is immunocompromised, then the virus can cross this fitness valley due to within-host evolution in this subpopulation (Fig. 2d). Immunocompromised hosts experience longer infections, on average, which allows the virus to gradually accumulate mutations and cross the fitness valley. When the virus has acquired enough mutations in the immunocompromised such that between-host fitness is restored, it is able to spread in the rest of the host population. Again, this process is sped up if the within-host evolutionary dynamics are assumed to be faster in immunocompromised individuals, but the qualitative dynamics are unchanged (Fig. 3b).
Our results are robust to variation in key model parameters, although our sensitivity analysis reveals two notable interactions (Figure 4). When varying the strength of epistasis and the extent of cross- immunity between variants, we find that, intuitively, immunocompromised individuals are especially important for traversing the fitness valley if epistasis is stronger or if cross-immunity is broader (Figure 4a). This is because stronger epistasis makes the fitness valley deeper and broader cross- immunity reduces the pool of susceptible hosts across a wider range of variants. However, if epistasis is sufficiently strong (around ξ = 0.9 in Figure 4a) a large jump in antigenic space to a distant variant occurs regardless of the strength of cross-immunity. Our sensitivity analysis also reveals that as the proportion of the population that is immunocompromised decreases, a jump in antigenic space becomes less likely and requires a longer relative infectious period in immunocompromised hosts (Figure 4b). This suggests that better treatment of immunocompromised hosts (to reduce the average duration of infection) and better prevention and treatment of pre-existing conditions (to reduce the proportion of the population that is immunocompromised) may greatly reduce the likelihood of new variants emerging at distant fitness peaks.

Top row: maximum distance between observed variants (darker shading indicates larger jumps in antigenic space); bottom row: total number of variants observed. (a) Varying the strength of cross-immunity (η) and epistasis (ξ) when 5% of the population is immunocompromised (p=0.05). (b) Varying the percentage of the population that is immunocompromised and the relative recovery periods (with η = 5 and ξ = 0.8). All other parameters as in Table A3. All datapoints are averaged over 10 simulations.
Discussion
The presence of immunocompromised individuals has been suggested as an important driver behind not only the emergence of the Omicron variant, but also other variants of concern, including Alpha and Delta [17]. Using a simple model of antigenic evolution, we have shown that prolonged infections of immunocompromised individuals allow pathogens to accumulate sufficient mutations to overcome epistasis at the between-host level, facilitating the emergence of novel immune escape variants. Our model was motivated by the surprising lack of antigenic evolution arising from the Delta lineage in 2021. Given relatively high levels of infection (and hence mutation supply) combined with rapidly increasing immune pressure in mid- to late-2021, conditions for the Delta lineage to exhibit antigenic evolution seemed to be favourable. The local mutational space appears to have been thoroughly explored by the Delta variant (Fig. 5), which suggests that mutation supply was not the fundamental constraint for the lack of antigenic evolution. Indeed, our model suggests that novel immune escape variants readily evolve when epistasis is relatively weak. When epistasis is stronger, reducing transmissibility for variants between fitness peaks, we find that immunocompromised individuals play a key role in antigenic evolution, effectively allowing the pathogen to traverse the fitness valley to reach a new peak. Note that while faster within-host adaptation in immunocompromised individuals speeds up the rate of antigenic evolution, unlike epistasis it does not qualitatively affect the outcome. Crucially, we have also shown that improving treatment for those who are immunocompromised can greatly reduce the likelihood of new variants emerging.

Three variants of concern (Alpha, Delta and Omicron) are highlighted to illustrate that there has been high mutation supply for the Delta variant. Data downloaded from Nextstrain (nextstrain.org) [23,24] and provided by the Global Initiative for Sharing All Influenza Data (GISAID, gisaid.org) [25–27]. Data plotted using the ggtree software package in R [28–30].
While our model does not capture the full complexity of antigenic evolution, it has important implications for our understanding of future immune escape variants of SARS-Cov-2. Crucially, our model suggests that the lack of antigenic evolution by Delta followed by the emergence of Omicron is consistent with epistasis constraining immune escape, but epistasis may be overcome when immunocompromised individuals are infected for sufficiently long periods. Hence, rather than simply accelerating antigenic evolution, prolonged infections of immunocompromised individuals may be critical for the evolution of immune escape variants. Based on the lack of antigenic evolution by Delta, we tentatively speculate that it may be difficult for SARS-CoV-2 to evolve antigenically through incremental mutations, and future variants may require multiple (epistatic) mutations to substantially escape host immunity. If true, a possible implication is that vaccines against SARS-CoV- 2 may not need to be regularly updated in a similar manner to seasonal influenza vaccines, but if new variants do emerge, they may substantially escape prior immunity and be harder to predict.
However, it is also possible that Delta was unusual in being limited in its scope for antigenic evolution and that other variants may not experience similar constraints in the fitness landscape.
Our results agree with previous models which suggest that immunocompromised individuals are more likely to facilitate or accelerate within-host pathogen evolution, for example due to a longer average duration of infection or higher viral load [31,32]. However, while we find immunocompromised hosts to play a crucial role in pathogen evolution at the population level, other studies have concluded the opposite as these individuals only make up a small proportion of infections [31,32]. The reason for this discrepancy is likely due to contrasting assumptions regarding within-host fitness, immunity, and traits under selection. For example, van Egeren et al. [32] assume that a fitness valley exists at the within-host level with two or three mutations required to cross, whereas our model assumes that the fitness valley only exists at the between-host level (transmission) but may require many more mutations to traverse. If a fitness valley exists at the within-host level, then intuitively the importance of immunocompromised individuals for pathogen evolution will be lower. Their model also focused on a static measure of relative fitness and did not consider antigenic evolution explicitly, whereas in our model the fitness of a particular variant depends on the level of immunity in the population, and so will vary over the course of the epidemic. Nevertheless, both models concur that longer duration infections, especially those of immunocompromised individuals, can play a disproportionate role in the evolution of novel variants, and are of particular concern for SARS-CoV-2 evolution.
We assumed that the rate of antigenic evolution during an infection was constant (but may vary by host type), which was motivated by the within-host model discussed in the appendix. For immunocompetent hosts, who typically clear infection within two weeks [33], this means that there is relatively little time for new variants to emerge for onwards transmission, which slows down adaptation and can prevent epistatic mutations accumulating. But for immunocompromised hosts, who may experience much longer infections (upwards of 150 days [17]), the coevolutionary dynamics between the virus and the host immune system could allow many (potentially epistatic) mutations to accumulate. Interestingly, this hypothesis is consistent with previous theoretical [34] and experimental [35–37] studies showing that coevolution can both accelerate adaptation and allow a pathogen to cross fitness valleys caused by epistasis.
In this study we have focused on evolution in immunocompromised individuals as a source of immune escape variants such as Omicron, but two alternative explanations must also be considered [10]. The first is that Omicron evolved early in the pandemic in a remote population (e.g., in southern Africa) before eventually taking off globally in late 2021. However, the combination of a large number of mutations in the spike protein and strong selection for immune escape in the wider population renders this explanation unsatisfactory, as it does not explain why these mutations did not appear in places where mutation supply and immune pressure were both high, such as the UK in late 2021. The second is that Omicron evolved in an animal host following transmission from humans, before crossing the species barrier into humans once again. This is a plausible but requires crossing the species barrier twice and selection to favour mutations that are beneficial in both the animal species and humans. We believe it is more plausible that Omicron evolved in an immunocompromised individual, especially since this is consistent with longitudinal sequencing of within-host evolution [17,18].
We stress that while our results suggest that infected immunocompromised individuals may play a significant role in the antigenic evolution of SARS-CoV-2, we urge caution in how this message is interpreted and communicated. While our model is informative, it does not capture the true complexity of antigenic space, the impact of vaccinations and non-pharmaceutical interventions, variation in disease outcomes, and the evolution of other disease characteristics such as transmissibility and virulence. This is by design so that our model requires as few assumptions as possible. We did not attempt to capture these effects, as our results are intended to be illustrative of the key roles that epistasis and immunocompromised individuals may play in the antigenic evolution of SARS-CoV-2 (and other pathogens). We urge particular caution with regards to the implications of our results for people who are immunocompromised. People may be immunocompromised for a variety of reasons, including uncontrolled HIV, undergoing treatment for cancer, or as a transplant recipient, and some conditions still wrongly attract stigma. Although Omicron was first detected in South Africa, which is estimated to have the highest HIV prevalence in the world (7.7 million people, with many infections uncontrolled [38]), this variant may have evolved in an individual without HIV and may have evolved elsewhere. Rather than stigmatising people who are immunocompromised, our results emphasise the need for global health equality. Improving access to vaccines and treatments, especially in lower- and middle-income countries, and facilitating wider surveillance for new variants is crucial for controlling the COVID-19 pandemic.
Appendix
Within-host model
The model in the main text focuses on population-level dynamics and implicitly models within-host dynamics by assuming that: (1) immunocompetent and immunocompromised hosts differ in terms of their average infectious period; and (2) antigenic evolution occurs at a constant rate. Here, we consider the dynamics of a simple within-host model to justify the implicit within-host dynamics in our population-level model.
Let Vi be the viral abundance of variant i ∈ {1,…, n} within a single infected host and let Ri be the strength of the corresponding immune response. The virus grows exponentially with rate r in the absence of an immune response and decreases through the immune response at rate 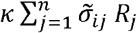 , where k is the per-capita rate of virus removal by the host immune system and
, where k is the per-capita rate of virus removal by the host immune system and 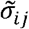 is the probability that an immune response for variant j causes cross-immunity to variant i such that
is the probability that an immune response for variant j causes cross-immunity to variant i such that
 where
where 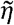 controls the breadth of cross-immunity between variants (similar to η in the main text). The virus also mutates to adjacent variants in the antigenic space with rate
controls the breadth of cross-immunity between variants (similar to η in the main text). The virus also mutates to adjacent variants in the antigenic space with rate 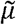 The immune response to variant i increases at per-capita rate kqVi and decays with rate d. The parameter q controls the strength of host immune system such that larger values indicate an immune system that can respond well to infection (immunocompetent) and smaller values indicate a weaker immune response (immunocompromised).
The immune response to variant i increases at per-capita rate kqVi and decays with rate d. The parameter q controls the strength of host immune system such that larger values indicate an immune system that can respond well to infection (immunocompetent) and smaller values indicate a weaker immune response (immunocompromised).
As with the between-host model, we use the stochastic τ-leaping method [22] to simulate the within-host dynamics, corresponding to the following set of ODEs
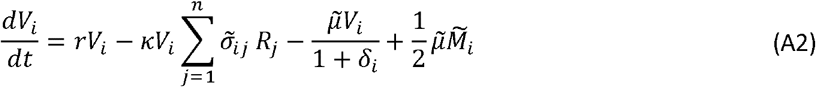

Where  is the set of variants adjacent to i in the one-dimensional antigenic space
is the set of variants adjacent to i in the one-dimensional antigenic space 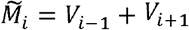 for i ∈ {2,…, n −1}, with boundary conditions
for i ∈ {2,…, n −1}, with boundary conditions 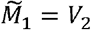 and
and 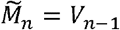 , and δ =1 if i ∈ {1,n} and is 0 otherwise to control the mutation rate at the boundaries.
, and δ =1 if i ∈ {1,n} and is 0 otherwise to control the mutation rate at the boundaries.
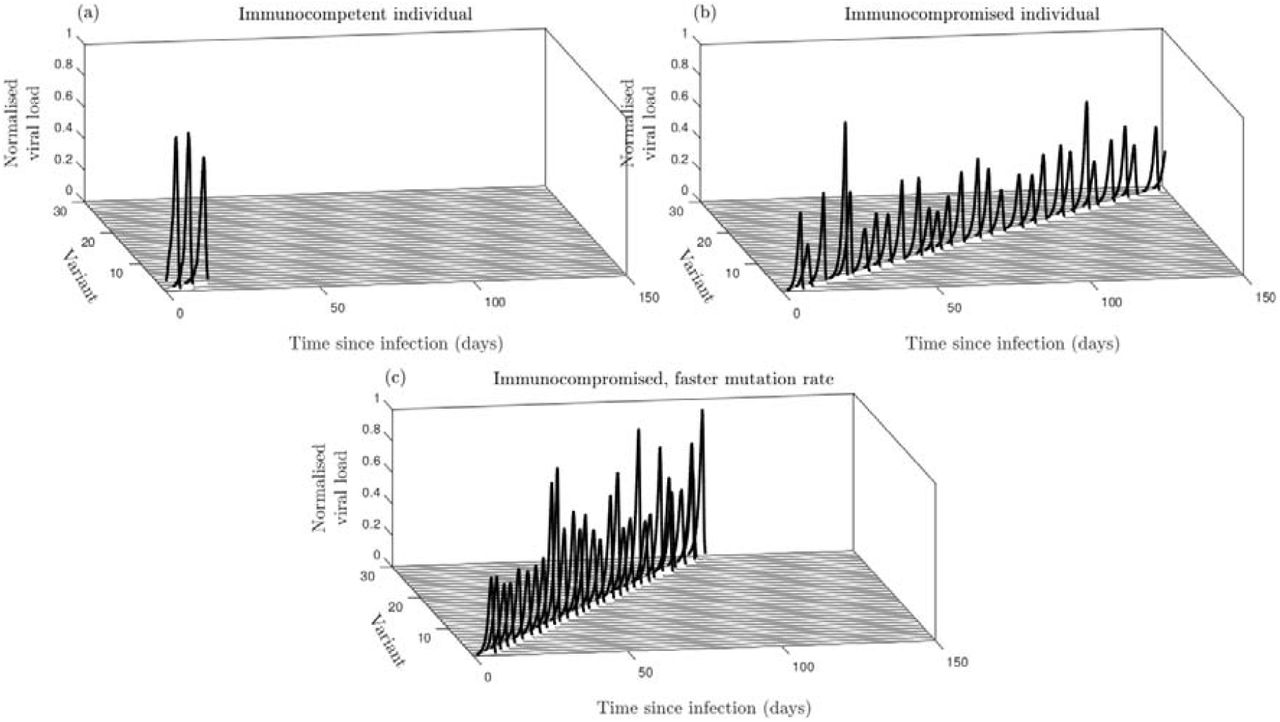
When the host is immunocompetent (large), the infection is rapidly cleared, with little within-host evolution (Fig. A1a). But when the host is immunocompromised (small), the infection persists over much longer timescales, with immune pressure leading to successive selective sweeps as the virus diffuses through the antigenic space at a constant rate (Fig. A1b). If the mutation rate is faster in immunocompromised hosts (larger), the coevolutionary dynamics of the virus and the immune response are accelerated but the rate of antigenic evolution remains constant (Fig. A1c). These results justify the simplifying assumptions in our population-level model regarding within-host dynamics, where we assume that there is a constant rate of antigenic evolution, which may differ between host types.
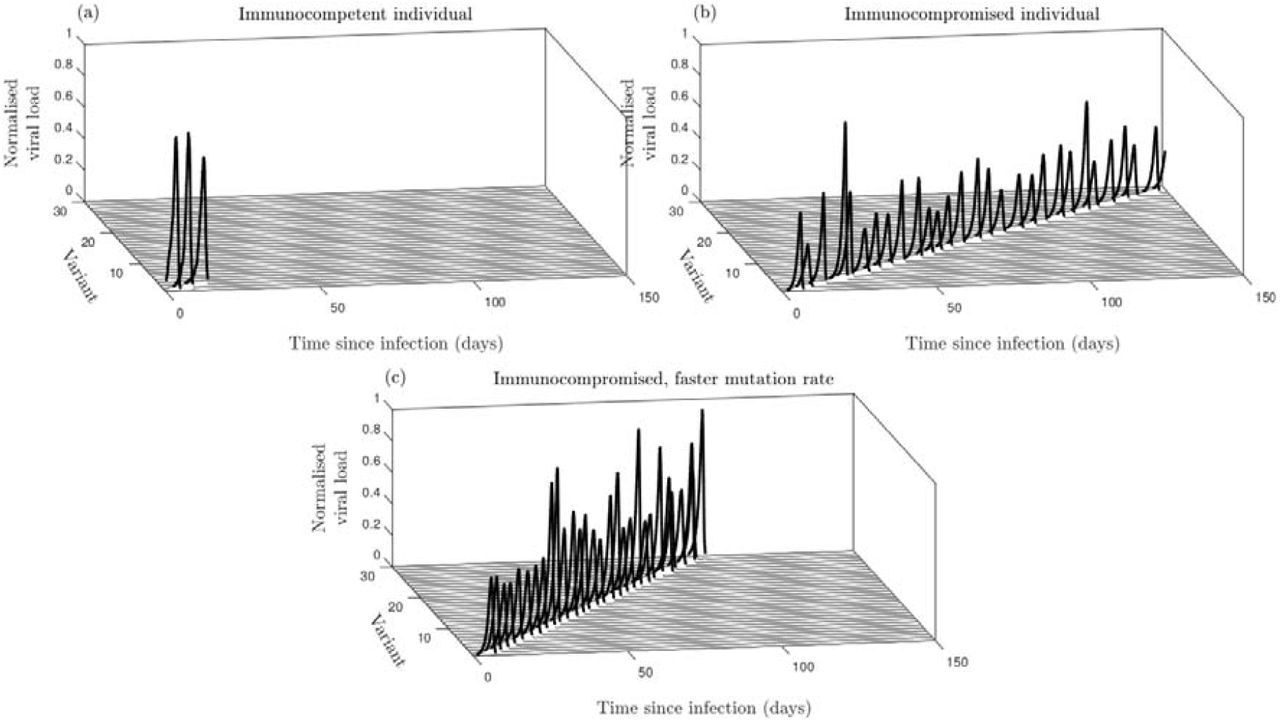
(a) Immunocompetent host, (b) Immunocompromised host, (c) immunocompromised host with faster mutation. Values of the viral load are normalised by the maximum value attained. All other parameters as in Table A4. Dynamics are shown for a single simulation.
Simulation algorithm
We simulate our within-host and population-level models using the τ-leaping method [22], which is an approximate stochastic simulation algorithm. We define the propensity functions 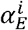 in Table A1, which give the rates of event type E for each variant index i. These propensity functions are then used to update the system synchronously at a time interval of one day using random numbers from the Poisson distribution
in Table A1, which give the rates of event type E for each variant index i. These propensity functions are then used to update the system synchronously at a time interval of one day using random numbers from the Poisson distribution 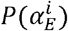 . Source code for the simulations is available in the Supplementary Material and at:
. Source code for the simulations is available in the Supplementary Material and at:
https://github.com/ecoevotheory/Smith_and_Ashby_2022.
The propensity functions for each of the event types in the population-level model.
The propensity functions for the various event types for the within-host model.
Default parameters for the between-host model.
Default parameters for the within-host model.
Data Availability
Source code for the simulations is available in the Supplementary Material and at: https://github.com/ecoevotheory/Smith_and_Ashby_2022
Author contributions
BA conceived the study, CAS carried out the analysis, and both co-authored the manuscript.
Data accessibility
Source code for the simulations is available in the Supplementary Material and at: https://github.com/ecoevotheory/Smith_and_Ashby_2022
Acknowledgements
We thank Angus Buckling for helpful discussions. CAS is funded by the Natural Environment Research Council (NE/V003909/1). BA is funded by the Natural Environment Research Council (NE/N014979/1 and NE/V003909/1).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}