
Abstract
The emergence of the Omicron variant (1) of SARS-CoV-2 in November 2021 in South Africa has raised concerns that, based on the large number of mutations in the spike protein and elsewhere on the virus (https://covdb.stanford.edu/page/mutation-viewer/#sec_b-1-351), this variant will have considerable escape from vaccine elicited immunity. Furthermore, several mutations in the receptor binding domain and S2 are predicted to impact transmissibility and affinity for ACE-2.
Here we investigated whether Omicron escapes antibody neutralization elicited by the Pfizer BNT162b2 mRNA vaccine and whether the virus still requires binding to the ACE2 receptor to infect cells. We used an early passage of isolated and sequence confirmed live Omicron virus isolated in South Africa. We used a human lung cell line clone (H1299-ACE2) engineered to express the ACE2 receptor (2) to both isolate the virus and test neutralization. We also tested growth in the parental H1299 which do not overexpress ACE2 and are not appreciably infectable with SARS-CoV-2 (Fig S1). The H1299-ACE2 cells were similar to Vero-E6 in titer dependent focus formation, but were considerably more sensitive (Fig S2).
We observed that Omicron infected the ACE2-expressing cells in a concentration dependent manner but did not infect the parental H1299 cells, indicating that ACE2 is required for Omicron entry (Fig. 1A). We then tested the ability of plasma from BNT162b2 vaccinated study participants to neutralize Omicron versus ancestral D614G virus in a live virus neutralization assay. We tested 14 plasma samples from 12 participants (Table S1), with 6 having no previous record of SARS-CoV-2 infection nor detectable nucleocapsid antibodies indicative of previous infection. For two of these participants, we used samples from two timepoints. The remaining 6 participants had a record of previous infection in the first SARS-CoV-2 infection wave in South Africa where infection was with ancestral D614G virus (Table S1). Geometric mean titer (GMT) FRNT50 (inverse of the plasma dilution required for 50% reduction in infection foci number) was 1321 for D614G. These samples therefore had very strong neutralization of D614G virus, consistent with sampling soon after vaccination. GMT FRNT50 for the same samples was 32 for Omicron, a 41-fold decline (Fig 1B). However, the escape was incomplete, with 5 of the participants, all previously infected, showing relatively high neutralization titers with Omicron.
Beta variant escape from BNT162b2 in a live virus neutralization assay has been reported to be substantial (3) and our own data confirmed these results (4), with about 3-fold reduction in FRNT50. The results we present here with Omicron show much more extensive escape. However, escape was incomplete in participants with higher FRNT50 due to previous infection. Previous infection, followed by vaccination or booster is likely to increase the neutralization level and likely confer protection from severe disease in Omicron infection.
Materials and Methods
Ethical statement
Blood samples were obtained from hospitalized adults with PCR-confirmed SARS-CoV-2 infection and/or vaccinated individuals who were enrolled in a prospective cohort study approved by the Biomedical Research Ethics Committee at the University of KwaZulu–Natal (reference BREC/00001275/2020). Use of residual swab sample was approved by the University of the Witwatersrand Human Research Ethics Committee (HREC) (ref. M210752).
Whole-genome sequencing, genome assembly and phylogenetic analysis
cDNA synthesis was performed on the extracted RNA using random primers followed by gene-specific multiplex PCR using the ARTIC V.3 protocol (https://www.protocols.io/view/covid-19-artic-v3-illumina-library-construction-an-bibtkann). In brief, extracted RNA was converted to cDNA using the Superscript IV First Strand synthesis system (Life Technologies) and random hexamer primers. SARS-CoV-2 whole-genome amplification was performed by multiplex PCR using primers designed using Primal Scheme (http://primal.zibraproject.org/) to generate 400-bp amplicons with an overlap of 70 bp that covers the 30 kb SARS-CoV-2 genome. PCR products were cleaned up using AmpureXP purification beads (Beckman Coulter) and quantified using the Qubit dsDNA High Sensitivity assay on the Qubit 4.0 instrument (Life Technologies). We then used the Illumina Nextera Flex DNA Library Prep kit according to the manufacturer’s protocol to prepare indexed paired-end libraries of genomic DNA. Sequencing libraries were normalized to 4 nM, pooled and denatured with 0.2 N sodium acetate. Then, a 12-pM sample library was spiked with 1% PhiX (a PhiX Control v.3 adaptor-ligated library was used as a control). We sequenced libraries on a 500-cycle v.2 MiSeq Reagent Kit on the Illumina MiSeq instrument (Illumina). We assembled paired-end fastq reads using Genome Detective 1.126 (https://www.genomedetective.com) and the Coronavirus Typing Tool. We polished the initial assembly obtained from Genome Detective by aligning mapped reads to the reference sequences and filtering out low-quality mutations using the bcftools 1.7-2 mpileup method. Mutations were confirmed visually with BAM files using Geneious software (Biomatters). P2 stock was sequenced and confirmed Omicron with the following substitutions: E:T9I,M:D3G,M:Q19E,M:A63T,N:P13L,N:R203K,N:G204R,ORF1a:K856R,ORF1a:L2084I,ORF1a:A2710T, ORF1a:T3255I,ORF1a:P3395H,ORF1a:I3758V,ORF1b:P314L,ORF1b:I1566V,ORF9b:P10S,S:A67V,S:T95I, S:Y145D,S:L212I,S:G339D,S:R346K,S:S371L,S:S373P,S:S375F,S:S477N,S:T478K,S:E484A,S:Q493R,S:G4 96S,S:Q498R,S:N501Y,S:Y505H,S:T547K,S:D614G,S:H655Y,S:N679K,S:P681H,S:Q954H,S:N969K,S:L981 F. Deletions were: N:E31-,N:R32-,N:S33-,ORF1a:S2083-,ORF1a:L3674-,ORF1a:S3675-,ORF1a:G3676-,ORF9b:E27-,ORF9b:N28-,ORF9b:A29-,S:H69-,S:V70-,S:G142-,S:V143-,S:Y144-,S:N211-.
SARS-CoV-2 nucleocapsid enzyme-linked immunosorbent assay (ELISA)
2 μg/ml nucleocapsid protein (Biotech Africa; Catalogue number: BA25-P was used to coat 96-well, high-binding plates and incubated overnight at 4°C. The plates were incubated in a blocking buffer consisting of 5% skimmed milk powder, 0.05% Tween 20, 1x PBS. Plasma samples were diluted to a 1:100 dilution in a blocking buffer and added to the plates. IgG secondary antibody was diluted to 1:3000 in blocking buffer and added to the plates followed by TMB substrate (Thermo Fisher Scientific). Upon stopping the reaction with 1 M H2SO4, absorbance was measured at a 450 nm wavelength.
Cells
Vero E6 cells (ATCC CRL-1586, obtained from Cellonex in South Africa) were propagated in complete DMEM with 10% fetal bovine serum (Hylone) containing 1% each of HEPES, sodium pyruvate, L-glutamine and nonessential amino acids (Sigma-Aldrich). Vero E6 cells were passaged every 3–4 days. The H1299-E3 cell line for first-passage SARS-CoV-2 expansion, derived as described in (2), was propagated in complete RPMI with 10% fetal bovine serum containing 1% each of HEPES, sodium pyruvate, L-glutamine and nonessential amino acids. H1299 cells were passaged every second day. Cell lines have not been authenticated. The cell lines have been tested for mycoplasma contamination and are mycoplasma negative.
Virus expansion
All work with live virus was performed in Biosafety Level 3 containment using protocols for SARS-CoV-2 approved by the AHRI Biosafety Committee. ACE2-expressing H1299-E3 cells were seeded at 4.5 × 105 cells in a 6 well plate well and incubated for 18–20 h. After one DPBS wash, the sub-confluent cell monolayer was inoculated with 500 μL universal transport medium diluted 1:1 with growth medium filtered through a 0.45-μm filter. Cells were incubated for 1 h. Wells were then filled with 3 mL complete growth medium. After 4 days of infection, cells were trypsinized, centrifuged at 300 rcf for 3 min and resuspended in 4 mL growth medium. Then 1 mL was added to Vero E6 cells that had been seeded at 2 × 105 cells per mL 18–20 h earlier in a T25 flask (approximately 1:8 donor-to-target cell dilution ratio) for cell-to-cell infection. The coculture of ACE2-expressing H1299-E3 and Vero E6 cells was incubated for 1 h and the flask was then filled with 7 mL of complete growth medium and incubated for 4 days. The viral supernatant (P2 stock) was used for experiments.
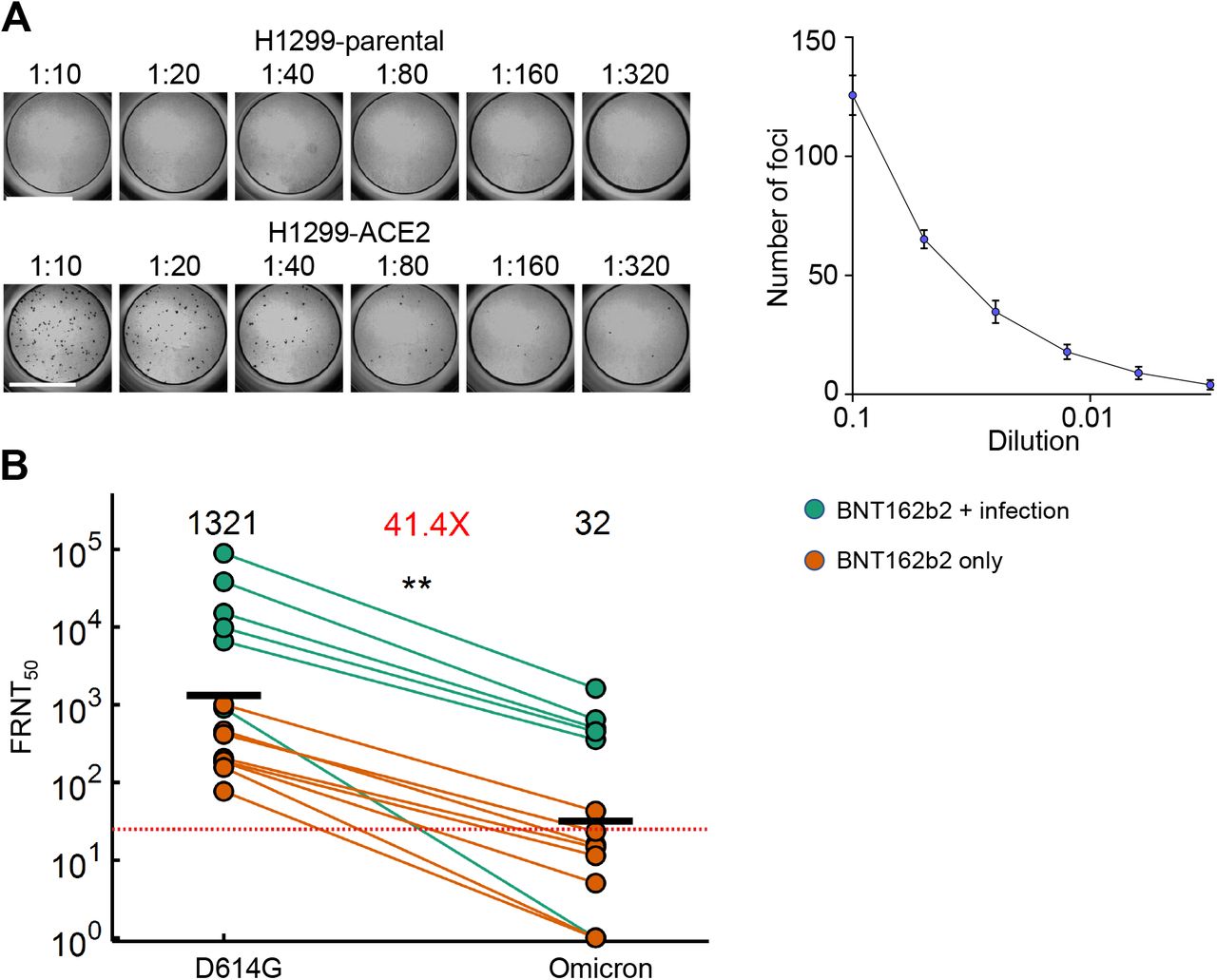
(A) Titration of live SARS-CoV-2 Omicron on H1299 parental cells and H1299-ACE2 cells. Plot shows result of titration on H1299-ACE2 cells. (B) Neutralization of the Omicron virus compared to D614G ancestral virus participants vaccinated with BNT162b2 and infected by ancestral SARS-CoV-2 (green) or vaccinated only. 14 samples from 12 participants were tested. Red horizontal line denotes most concentrated plasma tested. Numbers in black above each virus strain are geometric mean titers (GMT) of the reciprocal plasma dilution (FRNT50) causing 50% reduction in the number of infection foci. Number in red denote fold-change in GMT between virus strain on the left and the virus strain on the right of each panel. p=0.0018 as determined by the Wilcoxon rank sum test.
Live virus neutralization assay
H1299-E3 cells were plated in a 96-well plate (Corning) at 30,000 cells per well 1 day pre-infection. Plasma was separated from EDTA-anticoagulated blood by centrifugation at 500 rcf for 10 min and stored at −80 °C. Aliquots of plasma samples were heat-inactivated at 56 °C for 30 min and clarified by centrifugation at 10,000 rcf for 5 min. Virus stocks were used at approximately 50-100 focus-forming units per microwell and added to diluted plasma. Antibody–virus mixtures were incubated for 1 h at 37 °C, 5% CO2. Cells were infected with 100 μL of the virus–antibody mixtures for 1 h, then 100 μL of a 1X RPMI 1640 (Sigma-Aldrich, R6504), 1.5% carboxymethylcellulose (Sigma-Aldrich, C4888) overlay was added without removing the inoculum. Cells were fixed 18 h post-infection using 4% PFA (Sigma-Aldrich) for 20 min. Foci were stained with a rabbit anti-spike monoclonal antibody (BS-R2B12, GenScript A02058) at 0.5 μg/mL in a permeabilization buffer containing 0.1% saponin (Sigma-Aldrich), 0.1% BSA (Sigma-Aldrich) and 0.05% Tween-20 (Sigma-Aldrich) in PBS. Plates were incubated with primary antibody overnight at 4 °C, then washed with wash buffer containing 0.05% Tween-20 in PBS. Secondary goat anti-rabbit horseradish peroxidase (Abcam ab205718) antibody was added at 1 μg/mL and incubated for 2 h at room temperature with shaking. TrueBlue peroxidase substrate (SeraCare 5510-0030) was then added at 50 μL per well and incubated for 20 min at room temperature. Plates were imaged in an ELISPOT instrument with built-in image analysis (C.T.L).
Statistics and fitting
All statistics and fitting were performed using MATLAB v.2019b. Neutralization data were fit to Tx=1/1+(D/ID50).
Here Tx is the number of foci normalized to the number of foci in the absence of plasma on the same plate at dilution D and ID50 is the plasma dilution giving 50% neutralization. FRNT50 = 1/ID50. Values of FRNT50 <1 are set to 1 (undiluted), the lowest measurable value.
Data Availability
All data produced in the present work are contained in the manuscript

(A) The H1299 epithelial cell line with YFP labelled H2AZ was spinfected with the pHAGE2-EF1a-Int-ACE2 lentivector. Cells were single cell cloned by limiting dilution in a 384 well plate. Clones were expanded into duplicate 96-well plates, where one plate was used to select infectable clones based on mCherry signal from infection with SARS-CoV-2 mCherry expressing spike pseudotyped lentivirus. Clones were chosen based on infectability and expanded from the non-infected replicate 96-well plate. (B) Flow cytometry plots of SARS-CoV-2 mCherry expressing spike pseudotyped lentivirus infection in H1299-ACE2 cells. (C) Images of H1299-ACE2 cells stained with anti-ACE2 antibody and DAPI. Note membrane localization of ACE2. Scale bar is 50µm.

{kind=link}
{kind=link}
{kind=link}
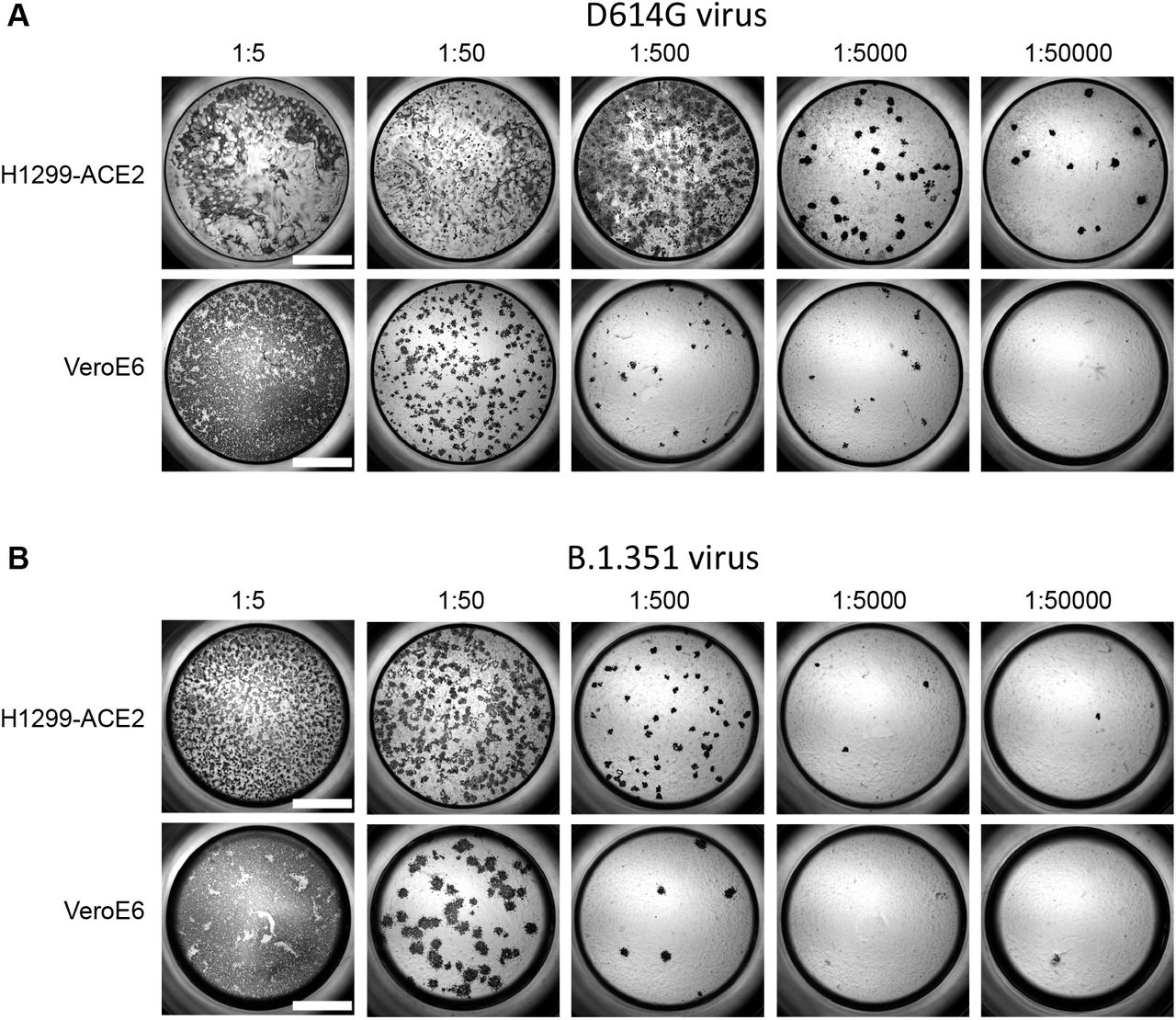
Both H1299-ACE2 and VeroE6 cells were infected with the same viral stock in the same experiment with D614G virus (A) or β virus (B) at different dilutions and a focus forming assay was performed. Scale bar is 2mm.
Summary Table of Participants:
Acknowledgements
This study was supported by the Bill and Melinda Gates award INV-018944 (AS), National Institutes of Health award R01 AI138546 (AS), and South African Medical Research Council awards (AS, TdO, PLM). PLM is also supported by the South African Research Chairs Initiative of the Department of Science and Innovation and the NRF (Grant No 9834). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.



