
Abstract
SARS-CoV-2 Spike protein is critical for virus infection via engagement of ACE2, and amino acid variation in Spike is increasingly appreciated. Given both vaccines and therapeutics are designed around Wuhan-1 Spike, this raises the theoretical possibility of virus escape, particularly in immunocompromised individuals where prolonged viral replication occurs. Here we report fatal SARS-CoV-2 escape from neutralising antibodies in an immune suppressed individual treated with convalescent plasma, generating whole genome ultradeep sequences by both short and long read technologies over 23 time points spanning 101 days. Little evolutionary change was observed in the viral population over the first 65 days despite two courses of remdesivir. However, following convalescent plasma we observed dynamic virus population shifts, with the emergence of a dominant viral strain bearing D796H in S2 and ΔH69/ΔV70 in the S1 NTD of the Spike protein. As serum neutralisation waned, viruses with the escape genotype diminished in frequency, before returning during a final, unsuccessful course of convalescent plasma. In vitro, the Spike escape variant conferred decreased sensitivity to multiple units of convalescent plasma/sera from different recovered patients, whilst maintaining infectivity similar to wild type. These data reveal strong positive selection on SARS-CoV-2 during convalescent plasma therapy and identify the combination of Spike mutations D796H and ΔH69/ΔV70 as a broad antibody resistance mechanism against commonly occurring antibody responses to SARS-CoV-2.
Introduction
SARS-CoV-2 is an RNA betacoronavirus, with closely related viruses identified in pangolins and bats 1,2. RNA viruses have inherently higher rates of mutation than DNA viruses such as Herpesviridae 3. The capacity for successful adaptation is exemplified by the Spike D614G mutation, that arose in China and rapidly spread worldwide 4, now accounting for more than 90% of infections. The mutation appears to increase infectivity and transmissibility in animal models 5. Although the SARS-CoV-2 Spike protein is critical for virus infection via engagement of ACE2, substantial Spike amino acid variation is being observed in circulating viruses 6. Logically, mutations in the receptor binding domain (RBD) of Spike are of particular concern due to the RBD being targeted by neutralising antibodies and therapeutic monoclonal antibodies.
Deletions in the N-terminal domain (NTD) of Spike S1 are also being increasingly recognised, both within hosts 7 and across individuals 8. The evolutionary basis for the emergence of deletions is unclear at present, and could be related to escape from immunity or to enhanced fitness/transmission. The most notable deletion in terms of frequency is ΔH69/ΔV70. This double deletion has been detected in multiple unrelated lineages, including the recent ‘Cluster 5’ mink related strain in the North Jutland region of Denmark (https://files.ssi.dk/Mink-cluster-5-short-report_AFO2). There it was associated with the RBD mutation Y453F in almost 200 individuals. Another European cluster in GISAID includes ΔH69/ΔV70 along with the RBD mutation N439K.
Although ΔH69/ΔV70 has been detected multiple times, within-host emergence remains undocumented and the reasons for its selection are unknown. Here we document real time SARS-CoV-2 emergence of ΔH69/ΔV70 in response to convalescent plasma therapy in an immunocompromised human host treated with the B cell depletion agent rituximab, demonstrating broad antibody escape in combination with the S2 mutation D796H.
Results
Clinical case history of SARS-CoV-2 infection in setting of immune-compromised host
These details are available on request from the authors.
Negative SARS-CoV-2 specific antibodies and requirement for convalescent plasma (CP)
We measured blood parameters including serum SARS-CoV-2 specific antibodies over time (Supplementary table 1, Supplementary Figure 1, 2). Imaging was consistent with COVID-19 disease (Supplementary Figure 3). Total serum antibodies to SARS-CoV-2 were tested at days 44 and 50 by S protein immunoassay (Siemens). Results were negative. Three units (200mL each) of convalescent plasma (CP) from three independent donors were obtained from the NHS Blood and Transfusion Service and administered on compassionate use basis. These had been assayed for antibody titres using the validated Euroimmun assay (Supplementary figure 3). Patient serum was subsequently positive for SARS-CoV-2 specific antibodies by S protein immunoassay (Siemens) in the hospital diagnostic laboratory on days 68, 90 and 101.
Virus genomic comparative analysis of 23 sequential respiratory samples over 101 days
The majority of samples were respiratory samples from nose and throat or endotracheal aspirates during the period of intubation. Ct values ranged from 16-34 and all 23 respiratory samples were successfully sequenced by standard long read approach as per the ARTIC protocol implemented by COG-UK; of these 20 additionally underwent short-read deep sequencing using the Illumina platform. There was generally good agreement between the methods, though as expected nanopore demonstrated greater error at low variant frequencies (<10%) (Supplementary Figure 4). We detected no evidence of recombination, based on two independent methods.
Maximum likelihood analysis of patient-derived whole genome consensus sequences demonstrated clustering with other local sequences from the same region (Figure 1A). The infecting strain was assigned to lineage 20B bearing the D614G Spike variant. Environmental sampling showed evidence of virus on surfaces such as telephone and call bell but not in air on days 59, 92 and 101. Sequencing of these surface viruses showed clustering with those derived from the respiratory tract (Figure 1B). All samples were consistent with having arisen from a single viral population. In our phylogenetic analysis, we included sequential sequences from three other local patients identified with persistent viral RNA shedding over a period of 4 weeks or more (Supplementary Table 2). Viruses from these individuals showed very little divergence in comparison to the case patient (Figure 1B) and none showed amino acid changes in Spike over time. We additionally inferred a maximum likelihood phylogeny comparing sequences from these three local individuals and two long term immunosuppressed SARS-CoV-2 ‘shedders’ recently reported7,9, (Figure 1B). While the sequences from Avanzato et al showed a pattern of evolution more similar to two of the three other local patients, the case patient showed significant diversification with a mutation rate of 30 per year (Supplementary table 2).

A. Circularised maximum-likelihood phylogenetic tree rooted on the Wuhan-Hu-1 reference sequence, showing a subset of 250 local SARS-CoV-2 genomes from GISAID. This diagram highlights significant diversity of the case patient (green) compared to three other local patients with prolonged shedding (blue, red and purple sequences). All SARS-CoV-2 genomes were downloaded from the GISAID database and a random subset of 250 local sequences selected. B. Close-view maximum-likelihood phylogenetic tree indicating the diversity of the case patient and three other long-term shedders from the local area (red, blue and purple), compared to recently published sequences from Choi et al (orange) and Avanzato et al (gold). Control patients generally showed limited diversity temporally, though the Choi et al sequences were found to be even more divergent than the case patient. Environmental samples are indicated. 1000 ultrafast bootstraps were performed and support at nodes is indicated.
Further investigation of the sequence data suggested the existence of an underlying structure to the viral population in our patient, with samples collected at days 93 and 95 being rooted within, but significantly divergent from the original population (Figure 1B and 2A). The relationship of the divergent samples to those at earlier time points rules out the possibility of superinfection. The increased divergence of sequences does not necessarily indicate selection; a spatially compartmentalised subset of viruses, smaller in number than the main viral population, would be expected to evolve more quickly than the main population due to the increased effect of genetic drift 10,11.
Virus population structure changes following convalescent plasma and remdesivir
All samples tested positive by RT-PCR and there was no sustained change in Ct values throughout the 101 days following the first two courses of remdesivir (days 41 and 54), or the first two units of convalescent plasma (days 63 and 65). According to nanopore data, no polymorphisms occurred over the first 60 days at consensus level (Figure 2A). However, short read deep sequence Illumina data revealed minority polymorphisms in the viral population during this period (Figure 2B). For example, T39I in ORF7a reached a majority frequency of 77% on day 44, arising de novo and increased in frequency during the first period of the infection (Supplementary Figure 6).

A. Highlighter plot indicating nucleotide changes at consensus level in sequential respiratory samples compared to the consensus sequence at first diagnosis of COVID-19. Each row indicates the timepoint the sample was collected (number of days from first positive SARS-CoV-2 RT-PCR). Black dashed lines indicate the RNA-dependent RNA polymerase (RdRp) and Spike regions of the genome. Of particular interest, at consensus level, there were no nucleotide substitutions on days 1-57, despite the patient receiving two courses of remdesivir. The first major changes in the spike genome occurred on day 82, following convalescent plasma given on days 63 and 65. The double amino acid deletion in S1, ΔH69/V70 is indicated by black lines. All samples are nose and throat swabs unless indicated with ETA (endotracheal aspirate). B. Whole genome amino acid variant trajectories based on Illumina short read ultra deep sequencing at 1000x coverage. All variants which reached a frequency of at least 10% in at least two samples were plotted. Black dashed line represents Δ69/70. CP, convalescent plasma; RDV, Remdesivir.
In contrast to the early period of infection, between days 66 and 82 a dramatic shift in virus population structure was observed, with the near-fixation of D796H in S2 along with ΔH69/ΔV70 in the S1 N-terminal domain (NTD) at day 82. This was identified in a nose and throat swab sample with high viral load as indicated by Ct of 23 (Figure 3). The deletion was not detected at any point prior to the day 82 sample, even as minority variants by short read deep sequencing.
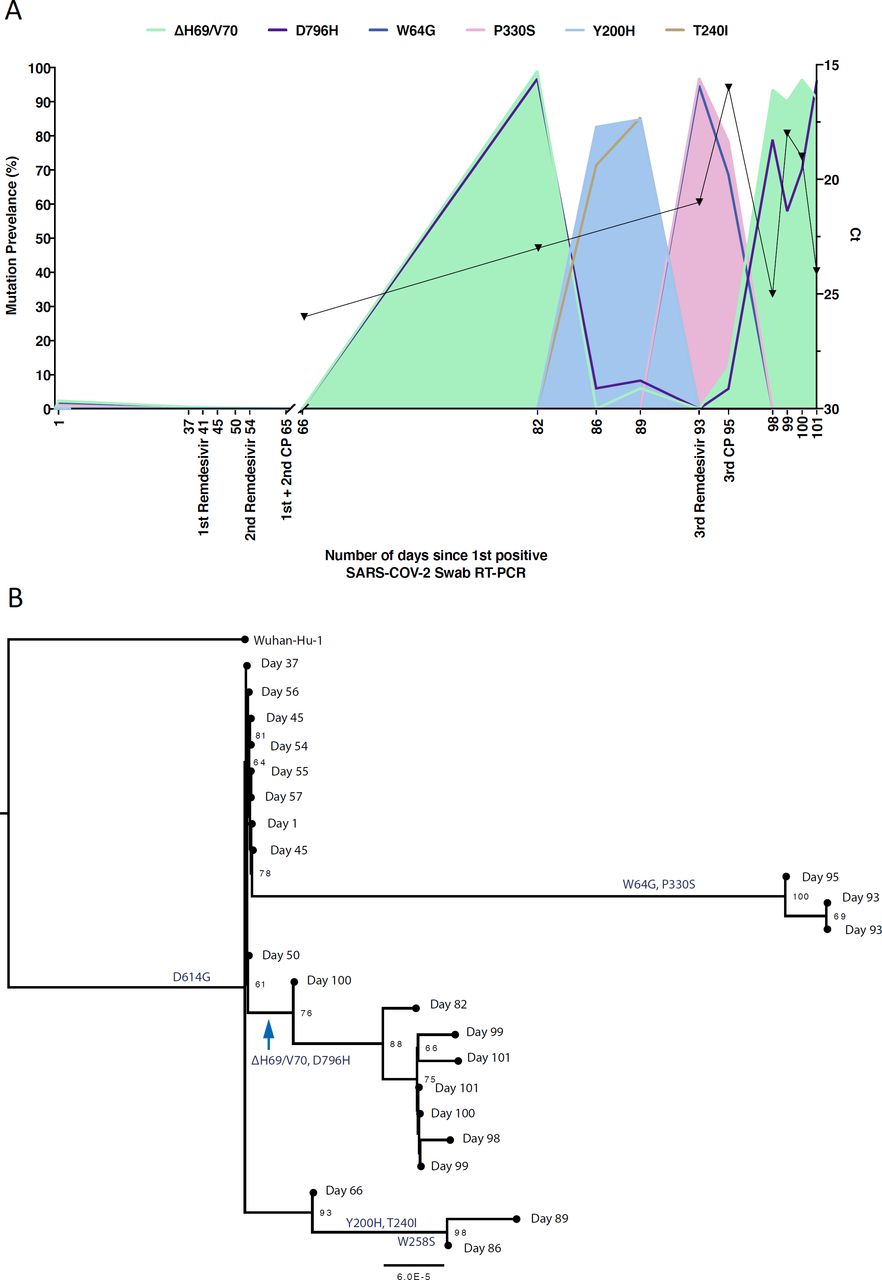
A. At baseline, all four S variants (Illumina sequencing) were absent (<1% and <20 reads). Approximately two weeks after receiving two units of convalescent plasma (CP), viral populations carrying ΔH69/V70 and D796H mutants rose to frequencies >90% but decreased significantly four days later. This population was replaced by a population bearing Y200H and T240I, detected in two samples over a period of six days. These viral populations were then replaced by virus carrying W64G and P330S mutations in Spike, which both reached near fixation at day 93. Following a 3rd course of remdesivir and an additional unit of convalescent plasma, the ΔH69/V70 and D796H virus population re-emerged to become the dominant viral strain reaching variant frequencies of >90%. Pairs of mutations arose and disappeared simultaneously indicating linkage on the same viral haplotype. B. Maximum likelihood phylogenetic tree of the case patient with day of sampling indicated. Spike mutations defining each of the clades are shown ancestrally on the branches on which they arose. Number at node denotes support by ultrafast bootstrapping consisting of 1000 replicates.
On Days 86 and 89, viruses collected were characterised by the Spike mutations Y200H and T240I, with the deletion/mutation pair observed on day 82 having fallen to very low frequency. Sequences collected on these days formed a distinct branch at the bottom of the phylogeny in Figure 3, but were clearly associated with the remainder of the samples, suggesting that they did not result from superinfection (Supplementary Figure 7), and further were not significantly divergent from the bulk of the viral population (Supplementary Figure 5).
Sequencing of a nose and throat swab sample at day 93 again showed D796H along with ΔH69/ΔV70 at <10% abundance, along with an increase in a virus population characterised by Spike mutations P330S at the edge of the RBD and W64G in S1 NTD. This new lineage reached near 100% abundance at day 93. Viruses with the P330S variant were detected in two independent samples from different sampling sites, ruling out the possibility of contamination. The divergence of these samples from the remainder of the population (Figure 3B), noted above, suggests the possibility of their resulting from the stochastic emergence, in the upper respiratory tract, of a previously unobserved subpopulation of viruses (Supplementary Figure 5).
Following the third course of remdesivir (day 93) and third CP (day 95), we observed a re-emergence of the D796H + ΔH69/ΔV70 viral population. The inferred linkage of D796H and ΔH69/ΔV70 was maintained as evidenced by the highly similar frequencies of the two variants, suggesting that the third unit of CP led to the re-emergence of this viral strain under renewed positive selection. Ct values remained low throughout this period with hyperinflammation, eventually leading to multi-organ failure and death at day 102. The repeated increase in frequency of the novel viral strain during CP therapy strongly supports the hypothesis that the deletion/mutation combination conferred antibody escape properties.
ΔH69/ΔV70 + D796H confers impaired neutralisation by multiple convalescent plasma units and sera from recovered COVID-19 patients
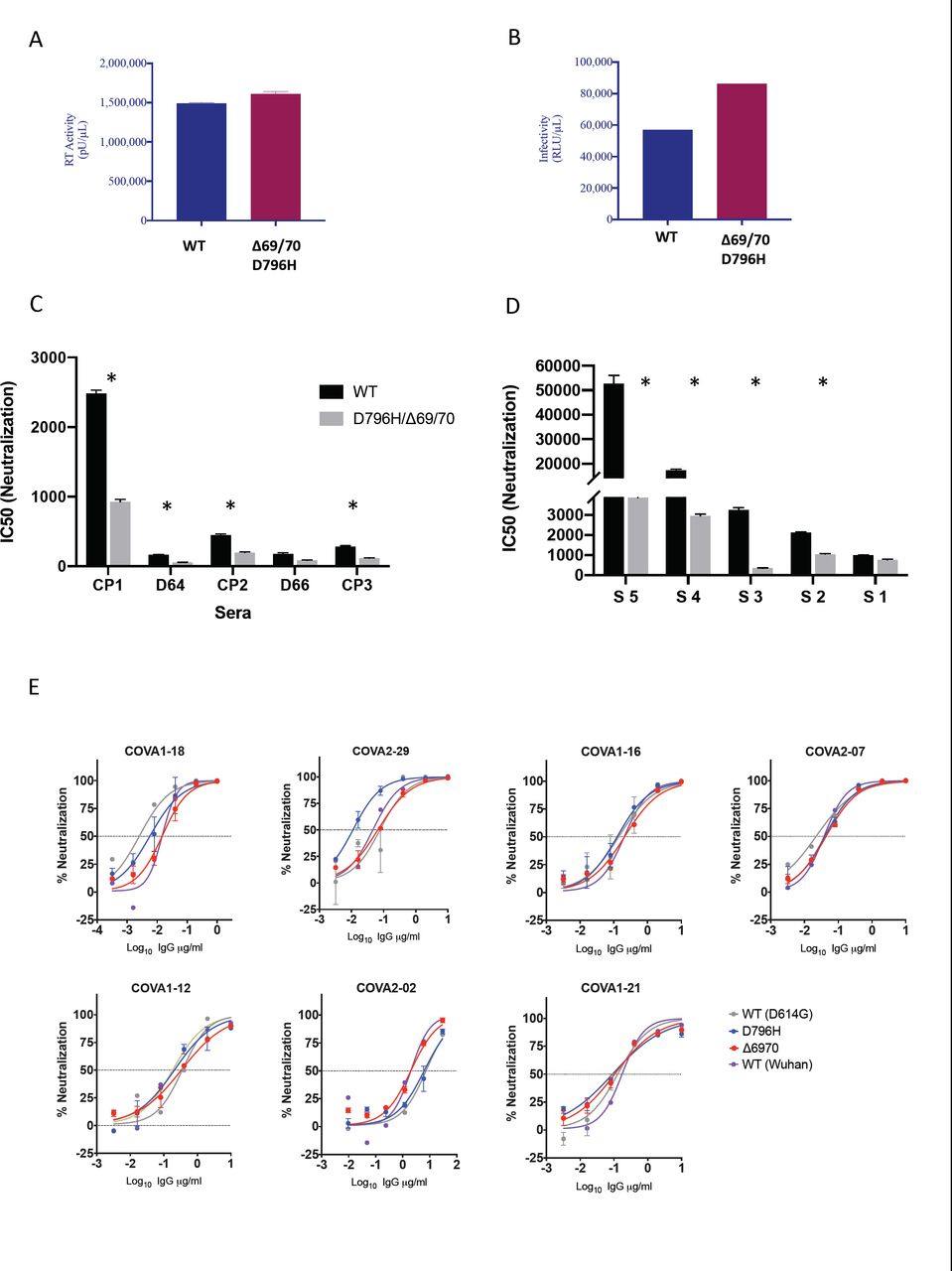
Using lentiviral pseudotyping we expressed wild type and ΔH69/ΔV70 + D796H mutant Spike protein in enveloped virions and compared neutralisation activity of CP against these viruses. This system has been shown to give similar results to replication competent virus12,13. We first tested infection capacity over a single round of infection and found that ΔH69/ΔV70 + D796H had similar infectivity to wild type (both in a D614G background, Figure 4A, B). The ΔH69/ΔV70 + D796H mutant was partially resistant to the first two CP units (Figure 4C, Table 1A). In addition, patient derived serum from days 64 and 66 (one day either side of CP2 infusion) similarly showed lower potency against the mutant (Figure 4C, Table 1A). The repeated observation of D796H + ΔH69/ΔV70 emergence and positive selection strengthens the hypothesis that these variants were the key drivers of antibody escape. Experimentally, the D796H + ΔH69/ΔV70 mutant also demonstrated reduced susceptibility to the CP3 administered on day 95, explaining its re-emergence (Figure 4C, Table 1A).
Shown are IC50 dilution titres - the CP or serum reciprocal dilution at which 50% of virus infectivity is lost against wild type (baseline) or D796H + Δ69/70 mutant Spike protein expressed on the surface of a lentiviral particle. Fold change is the difference between the two viruses, and values below 1 indicate that the D796H/Δ69/70 mutant is less sensitive to the CP or serum. Only the D66 serum sample failed to demonstrate significantly higher resistance of the D796H/Δ69/70 mutant.

A. Reverse transcriptase activity of virus supernatants containing lentivirus pseudotyped with SARS-CoV-2 Spike protein (WT versus mutant) B. Single round Infectivity of luciferase expressing lentivirus pseudotyped with SARS-CoV-2 Spike protein (WT versus mutant) on 293T cells co-transfected with ACE2 and TMPRSS2 plasmids. C. Patient serum (taken at indicated Day (D)) and convalescent plasma (CP units 1-3) neutralization potency against Spike mutant D796H + ΔH69/V70 measured using lentivirus pseudotyped with SARS-CoV-2 Spike protein (WT versus mutant). Indicated is serum dilution required to inhibit 50% of virus infection. D. Neutralization potency of sera from 5 unselected convalescent patients (with previous confirmed SARS-CoV-2 infection) against WT versus mutant virus as in panel C. E Neutralization potency of a panel of monoclonal antibodies against lentivirus pseudotyped with SARS-CoV-2 Spike protein (WT Wuhan versus single mutants D614G, D796H, ΔH69/V70). Data are representative of at least two independent experiments * p<0.05
Given reduced susceptibility of the mutants to at least two units of CP, and the expansion of sequences bearing ΔH69/ΔV70, we hypothesised that this represented a broad escape mechanism. We therefore screened antiviral neutralisation activity in sera from five recovered patients against the mutant and wild type viruses (Figure 4D). We observed that the mutant was indeed significantly less susceptible to four of five randomly selected sera, with the fifth showing reduced susceptibility that did not reach statistical significance (Table 1B). Fold change reductions in susceptibility of the mutant were as high as ten-fold compared to wild type (Table 1B).
In order to probe the impact of the D796H and ΔH69/ΔV70 mutations on potency of monoclonal antibodies (mAbs) targeting Spike, we screened neutralisation activity of a panel of seven neutralizing mAbs across a range of epitope clusters13 (Figure 4E). We observed no differences in neutralisation between single mutants and wild type, suggesting that the mechanism of escape was likely outside these epitopes.
In order to understand the mechanisms that might confer resistance to antibodies we examined a published Spike structure and annotated it our residues of interest (Figure 5). This analysis showed that ΔH69/ΔV70 is in a disordered, glycosylated loop at the very tip of the NTD, and therefore could alter binding of antibodies. ΔH69/V70 is close to the binding site of the polyclonal antibodies derived from COV57 plasma, indicating the sera tested here may contain similar antibodies 14,15. D796H is in an exposed loop in S2 (Figure 5), and appears to be in a region frequently targeted by antibodies16, despite mutations at position 796 being rare (Supplementary table 4).

Amino acid residues H69 and Y70 deleted in the N-terminal domain (red) and D796H in subunit 2 (orange) are highlighted on a SARS-CoV-2 spike trimer (PDB: 6ZGE Wrobek et al., 2020). Each of the three protomers making up the Spike homotrimer are coloured separately in shades of grey (centre). Close-ups of ΔH69/Y70 (above) and D796H (below) are shown in cartoon, stick representation. Both mutations are in exposed loops.
Discussion
Here we have documented a repeated evolutionary response by SARS-CoV-2 against antibody therapy during the course of a persistent and eventually fatal infection in an immunocompromised host. The observation of potential selection for specific variants coinciding with the presence of antibodies from convalescent plasma is supported by the experimental finding of reduced susceptibility of these viruses to plasma. Further, we were able to document real-time emergence of a variant ΔH69/ΔV70 in the NTD of Spike that has been increasing in frequency in Europe. In the case we report that it was not clear that the emergence of the antibody escape variant was the primary reason for treatment failure. However, given that both vaccines and therapeutics are aimed at Spike, our study raises the possibility of virus evasion, particularly in immune suppressed individuals where prolonged viral replication occurs.
Our observations represent a very rare insight, and only possible due to lack of antibody responses in the individual following administration of the B cell depleting agent rituximab for lymphoma, and an intensive sampling course undertaken due to concerns about persistent shedding and risk of nosocomial transmission. Persistent viral replication and the failure of antiviral therapy allowed us to define the viral response to convalescent plasma, similar to a recent report on asymptomatic long term shedding with four sequences over 105 days9, although the reported shifts in genetic composition of the viral population could not be explained phenotypically. Another common finding is the very low neutralisation activity in serum post transfusion of CP with waning as expected. Apart from the difference in the outcome of infection (severe, fatal disease versus asymptomatic disease and clearance), critically important differences in our study include: 1. The intensity of sampling and use of both long and short read sequencing to verify variant calls, thereby providing a unique scientific resource for longitudinal population genetic analysis. 2. The close alignment between the genetic composition of the viral population and CP administration, with an experimentally verified resistant strain emerging, falling to low frequency, and then rising again under CP selection. 3. Real time detection of emergence of a variant, ΔH69/ΔV70, that is increasing in frequency in Europe, and shown here to affect neutralization by multiple COVID-19 patient derived sera.
We have noted in our analysis the potential influence of compartmentalised viral replication upon the sequences recovered in upper respiratory tract samples. Both population genetic and small animal studies have shown a lack of reassortment between influenza viruses within a single host during an infection, suggesting that acute respiratory viral infection may be characterised by spatially distinct viral populations17,18. In the analysis of data it is important to distinguish genetic changes which occur in the primary viral population from apparent changes that arise from the stochastic observation of spatially distinct subpopulations in the host. While the samples we observe on days 93 and 95 of infection are genetically distinct from the others, the remaining samples are consistent with arising from a consistent viral population, supporting the finding of a reversion and subsequent regain of antibody resistance. We note that in a study of SARS-CoV-2, Choi et al reported the detection in post mortem tissue of viral RNA not only in lung tissue, but also in the spleen, liver and heart7. Mixing of virus from different compartments, for example via blood, or movement of secretions from lower to upper respiratory tract, could lead to fluctuations in viral populations at particular sampling sites. Experiments in animal models with sampling of different replication sites could allow a better understanding of SARS-CoV-2 population genetics and enable prediction of escape variants following antibody based therapies.
This is a single case report and therefore limited conclusions can be drawn about generalisability.
In addition to documenting the emergence of SARS-CoV-2 Spike ΔH69/ΔV70 + D796H in vivo, conferring broad reduction in susceptibility to serum/plasma polyclonal, but no effect on a set of predominantly RBD-targeting monoclonal antibodies, these data highlight that infection control measures need to be specifically tailored to the needs of immunocompromised patients. The data also highlight caution in interpretation of CDC guidelines that recommend 20 days as the upper limit of infection prevention precautions in immune compromised patients who are afebrile19. Due to the difficulty with culturing clinical isolates, use of surrogates for infectious virus such as sgRNA are warranted20. However, where detection of ongoing viral evolution is possible, this serves as a clear proxy for the existence of infectious virus. In our case we detected environmental contamination whilst in a single occupancy room and the patient was moved to a negative-pressure high air-change infectious disease isolation room.
The clinical efficacy of CP has been called into question recently21, and our data suggest caution in use of CP in patients with immune suppression of both T cell and B cell arms. In such cases, the antibodies administered have little support from cytotoxic T cells, thereby reducing chances of clearance and raising the potential for escape mutations. Whilst we await further data, CP administered for clinical need in immune suppression, should ideally be undertaken in single occupancy rooms with enhanced infection control precautions, including SARS-CoV-2 environmental sampling and real-time sequencing.
Ethics
The study was approved by the East of England – Cambridge Central Research Ethics Committee (17/EE/0025). The patient and family provided written informed consent. Additional controls with COVID-19 were enrolled to the NIHR BioResource Centre Cambridge under ethics review board (17/EE/0025).
Author contributions
Conceived study: RKG, SAK, DAC, AS, TG, EGK
Designed experiments: RKG, SAK, DAC, LEM, JAGB, EGK, AC, NT, AC, CS
Performed experiments: SAK, DAC, LEM, RD, CRS, AJ, IATMF, KS, TG, CJRI, BB, JS, MJvG
Interpreted data: RKG, SAK, DAC, PM, LEM, JAGB, PM, SG, KS, TG, JB, KGCS, IG, CJRI, JAGB, IUL, DR, JS, BB
Methods
Clinical Sample Collection and Next generation sequencing
Serial samples were collected from the patient periodically from the lower respiratory tract (sputum or endotracheal aspirate), upper respiratory tract (throat and nasal swab), and from stool. Nucleic acid extraction was done from 500µl of sample with a dilution of MS2 bacteriophage to act as an internal control, using the easyMAG platform (Biomerieux, Marcy LEtoile) according to the manufacturers’ instructions. All samples were tested for presence of SARS-CoV-2 with a validated one-step RT q-PCR assay developed in conjunction with the Public Health England Clinical Microbiology 22. Amplification reaction were all performed on a Rotorgene™ PCR instrument. Samples which generated a CT of ≤36 were considered to be positive.
Sera from recovered patients in the COVIDx study23 were used for testing of neutralisation activity by SARS-CoV-2 mutants.
For viral genomic sequencing, total RNA was extracted from samples as described. Samples were sequenced using MinION flow cells version 9.4.1 (Oxford Nanopore Technologies) following the ARTICnetwork V3 protocol (https://dx.doi.org/10.17504/protocols.io.bbmuik6w) and BAM files assembled using the ARTICnetwork assembly pipeline (https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html). A representative set of 10 sequences were selected and also sequenced using the Illumina MiSeq platform. Amplicons were diluted to 2 ng/µl and 25 µl (50 ng) were used as input for each library preparation reaction. The library preparation used KAPA Hyper Prep kit (Roche) according to manufacturer’s instructions. Briefly, amplicons were end-repaired and had A-overhang added; these were then ligated with 15mM of NEXTflex DNA Barcodes (Bio Scientific, Texas, USA). Post-ligation products were cleaned using AMPure beads and eluted in 25 µl. Then, 20 µl were used for library amplification by 5 cycles of PCR. For the negative controls, 1ng was used for ligation-based library preparation. All libraries were assayed using TapeStation (Agilent Technologies, California, USA) to assess fragment size and quantified by QPCR. All libraries were then pooled in equimolar accordingly. Libraries were loaded at 15nM and spiked in 5% PhiX (Illumina, California, USA) and sequenced on one MiSeq 500 cycle using a Miseq Nano v2 with 2x 250 paired-end sequencing. A minimum of ten reads were required for a variant call.
Bioinformatics Processes
For long-read sequencing, genomes were assembled with reference-based assembly and a curated bioinformatics pipeline with 20x minimum coverage across the whole-genome 24. For short-read sequencing, FASTQs were downloaded, poor-quality reads were identified and removed, and both Illumina and PHiX adapters were removed using TrimGalore v0.6.6 25. Trimmed paired-end reads were mapped to the National Center for Biotechnology Information SARS-CoV-2 reference sequence MN908947.3 using MiniMap2-2.17 with arguments −ax and sr 26. BAM files were then sorted and indexed with samtools v1.11 and PCR optical duplicates removed using Picard (http://broadinstitute.github.io/picard). Single nucleotide polymorphisms (SNPs) were called using Freebayes v1.3.2 27 with a ploidy setting of 1, minimum allele frequency of 0.20 and a minimum depth of five reads. Finally, a consensus sequences of nucleic acids with a minimum whole-genome coverage of at least 20× were generated with BCFtools using a 0% majority threshold.
Phylogenetic Analysis
All available full-genome SARS-CoV-2 sequences were downloaded from the GISAID database (http://gisaid.org/) 28 on 17th September. Duplicate and low-quality sequences (>5% N regions) were removed, leaving a dataset of 138,472 sequences with a length of >29,000bp. All sequences were sorted by name and only sequences sequenced with United Kingdom / England identifiers were retained. From this dataset, a subset of 250 sequences were randomly subsampled using seqtk (https://github.com/lh3/seqtk). These 250 sequences were aligned to the 23 patient sequences, as well as three other control patients (persistent long-term shedders from the same hospital) (Supplementary Table 2) and the SARS-CoV-2 reference strain MN908947.3, using MAFFT v7.473 with automatic flavour selection 29. Major SARS-CoV-2 clade memberships were assigned to all sequences using the Nextclade server v0.8 (https://clades.nextstrain.org/).
Maximum likelihood phylogenetic trees were produced using the above curated dataset using IQ-TREE v2.1.2 30. Evolutionary model selection for trees were inferred using ModelFinder 31 and trees were estimated using the GTR+F+I model with 1000 ultrafast bootstrap replicates32. All trees were visualised with Figtree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/), rooted on the SARS-CoV-2 reference sequence and nodes arranged in descending order. Nodes with bootstraps values of <50 were collapsed using an in-house script.
A time tree indicating temporal divergence was inferred using TreeTime v0.75 33. For this a further subsample of 100 sequences from the multiple sequence alignment used to make the maximum likelihood tree were selected, which represented sequences belonging to NextStrain clade 20B. This included the 23 patient genomes, all genomes from two of the three long-term shedders (one was excluded as it belonged to NextStrain clade 19A), 75 subsampled Cambridge sequences, and the SARS-CoV-2 reference strain MN908947.3. All sequences were re-aligned using MAFFT v7.473 with automatic flavour selection 29 and used as input.
Molecular substitution (clock) rates for the index patient, as well as three long-term shedders and two recently described immunocompromised patients from literature, were estimated using BEAST v2.6.3 34 using a HKY substitution model with 4 rate categories drawn from a gamma distribution, a strict clock and a coalescent exponential population tree prior. MCMC was run for 100 million iterations excluding a 15% burn-in. Tracer v1.7.1 was used to analyse the BEAST trace in order extract the clock rate and ensure convergence had occurred.
In-depth allele frequency variant calling
The SAMFIRE package35 was used to call allele frequency trajectories from BAM file data. Reads were included in this analysis if they had a median PHRED score of at least 30, trimming the ends of reads to achieve this if necessary. Nucleotides were then filtered to have a PHRED score of at least 30; reads with fewer than 30 such reads were discarded. Distances between sequences, accounting for low-frequency variant information, was also conducted using SAMFIRE. The sequence distance metric, described in an earlier paper 11, combines allele frequencies across the whole genome. Where L is the length of the genome, we define q(t) as a 4 x L element vector describing the frequencies of each of the nucleotides A, C, G, and T at each locus in the viral genome sampled at time t. For any given locus i in the genome we calculate the change in allele frequencies between the times t1 and t2 via a generalisation of the Hamming distance
 where the vertical lines indicate the absolute value of the difference. These statistics were then combined across the genome to generate the pairwise sequence distance metric
where the vertical lines indicate the absolute value of the difference. These statistics were then combined across the genome to generate the pairwise sequence distance metric

The Mathematica software package was to conduct a regression analysis of pairwise sequence distances against time, leading to an estimate of a mean rate of within-host sequence evolution. In contrast to the phylogenetic analysis, this approach assumed the samples collected on days 93 and 95 to arise via stochastic emission from a spatially separated subpopulation within the host, leading to a lower inferred rate of viral evolution for the bulk of the viral population.
Inference of selection
Under the assumption of a large effective population size, a deterministic one-locus model of selection was fitted to genome sequence data describing changes in the frequency of the variant T39I. Where q(t) represents the frequency of a single variant at time t, we used a maximum likelihood method to infer the initial variant frequency at time t=1, and the selection coefficient s, for times from the initial time point to the disappearance of the variant from the population. Specifically, where n(ti) and N(ti) were the number of observations of the variant and the total read depth at that locus at time ti, we fitted the model
 so as to maximise the binomial likelihood
so as to maximise the binomial likelihood

A similar calculation was performed to estimate the mean effective selection (incorporating intrinsic selection for the variant plus linkage with other selected alleles) acting upon the variant D796H during the final period of CP therapy. In this case selection was modelled as being time-dependent, kicking in with the administration of therapy. We fitted the model:
 to the data, setting τ = 95.
to the data, setting τ = 95.
Recombination Detection
All sequences were tested for potential recombination, as this would impact on evolutionary estimates. Potential recombination events were explored with nine algorithms (RDP, MaxChi, SisScan, GeneConv, Bootscan, PhylPro, Chimera, LARD and 3SEQ), implemented in RDP5 with default settings 36. To corroborate any findings, ClonalFrameML v1.12 37 was also used to infer recombination breakpoints. Neither programs indicated evidence of recombination in our data.
Structural Viewing
The Pymol Molecular Graphics System v2.4.0 (https://github.com/schrodinger/pymol-open-source/releases) was used to map the location of the four spike mutations of interested onto a SARS-CoV-2 spike structure visualised by Wrobel et al (PDB: 6ZGE) 38.
Generation of Spike mutants
Amino acid substitutions were introduced into the D614G pCDNA_SARS-CoV-2_Spike plasmid as previously described39 using the QuikChange Lightening Site-Directed Mutagenesis kit, following the manufacturer’s instructions (Agilent Technologies, Inc., Santa Clara, CA).
Pseudotype virus preparation
Viral vectors were prepared by transfection of 293T cells by using Fugene HD transfection reagent (Promega). 293T cells were transfected with a mixture of 11ul of Fugene HD, 1µg of pCDNAΔ19Spike, 1ug of p8.91 HIV-1 gag-pol expression vector40,41, and 1.5µg of pCSFLW (expressing the firefly luciferase reporter gene with the HIV-1 packaging signal). Viral supernatant was collected at 48 and 72h after transfection, filtered through 0.45um filter and stored at −80°C. The 50% tissue culture infectious dose (TCID50) of SARS-CoV-2 pseudovirus was determined using Steady-Glo Luciferase assay system (Promega).
RT activity in virion containing supernatant was measured as previously described.
Serum/plasma pseudotype neutralisation assay
Spike pseudotype assays have been shown to have similar characteristics as neutralisation testing using fully infectious wild type SARS-CoV-212.Virus neutralisation assays were performed on 293T cell transiently transfected with ACE2 and TMPRSS2 using SARS-CoV-2 Spike pseudotyped virus expressing luciferase42. Pseudotyped virus was incubated with serial dilution of heat inactivated human serum samples or convalescent plasma in duplicate for 1h at 37°C. Virus and cell only controls were also included. Then, freshly trypsinized 293T ACE2/TMPRSS2 expressing cells were added to each well. Following 48h incubation in a 5% CO2 environment at 37°C, the luminescence was measured using Steady-Glo Luciferase assay system (Promega).
mAb pseudotype neutralisation assay
Virus neutralisation assays were performed on HeLa cells stably expressing ACE2 and using SARS-CoV-2 Spike pseudotyped virus expressing luciferase as previously described43. Pseudoyped virus was incubated with serial dilution of purified mAbs13 in duplicate for 1h at 37°C. Then, freshly trypsinized HeLa ACE2-expressing cells were added to each well. Following 48h incubation in a 5% CO2 environment at 37°C, the luminescence was measured using Bright-Glo Luciferase assay system (Promega) and neutralization calculated relative to virus only controls. IC50 values were calculated in Graphpad Prism.
Acknowledgements
We are immensely grateful to the patient and family. We would also like to thank the staff at CUH and the NIHR Cambridge Clinical Research Facility. We would like to thank Dr Ruthiran Kugathasan and Professor Wendy Barclay for helpful discussions and Dr Martin Curran, Dr William Hamilton and Dr. Dominic Sparkes. We would like to thank Prof Andres Floto and Prof Ferdia Gallagher. We thank Dr James Voss for the kind gift of HeLa cells stably expressing ACE2. COG-UK is supported by funding from the Medical Research Council (MRC) part of UK Research & Innovation (UKRI), the National Institute of Health Research (NIHR) and Genome Research Limited, operating as the Wellcome Sanger Institute. RKG is supported by a Wellcome Trust Senior Fellowship in Clinical Science (WT108082AIA). LEM is supported by a Medical Research Council Career Development Award (MR/R008698/1). SAK is supported by the Bill and Melinda Gates Foundation via PANGEA grant: OPP1175094. DAC is supported by a Wellcome Trust Clinical PhD Research Fellowship. CJRI acknowledges MRC funding (ref: MC_UU_00002/11). This research was supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre, the Cambridge Clinical Trials Unit (CCTU) and by the UCL Coronavirus Response Fund and made possible through generous donations from UCL’s supporters, alumni and friends (LEM). JAGB is supported by the Medical Research Council (MC_UP_1201/16).
References




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Subject Area
Reviews and Context
1
Comment
0
TRIP Peer Reviews
0
Community Reviews
0
Automated Services
198
Blogs/Media
Author Videos