
ABSTRACT
Background In the field of immuno-oncology, predicting treatment response or survival of cancer patients remains a challenge. Efforts to overcome these challenges focus mainly on the discovery of new biomarkers. Owing to the complexity of cancers and their tumor microenvironment, only a limited number of candidate biomarkers eventually enters clinical practice, despite advances in cellular and molecular approaches.
Methods A computational modeling approach based on ordinary differential equations was used to simulate the fundamental mechanisms that dictate tumor-immune dynamics and show its implications on responses to immune checkpoint inhibition (ICI) and patient survival. Using in silico biomarker discovery trials, we extracted fundamental principles underlying the success rates of biomarker discovery programs.
Results Our main finding is the prediction of a tipping point – a sharp state transition between immune control and immune evasion – that follows a strongly non-linear relationship between patient survival and both immunological and tumor-related parameters. In patients close to the tipping point, ICI therapy may lead to long-lasting survival benefits, whereas patients far from the tipping point may fail to benefit from these potent treatments.
Conclusion These findings have two important implications for clinical oncology. First, the apparent conundrum that ICI induces substantial benefits in some patients yet completely fails in others could be, to a large extent, explained by the presence of a tipping point. Second, predictive biomarkers for immunotherapy should ideally combine both immunological and tumor-related markers, as the distance of a patient’ status from the tipping point cannot, in general, be reliably determined from solely one of these. The notion of a tipping point in cancer-immune dynamics could help to optimize strategies in biomarker discovery to ensure accurate selection of the right patient for the right treatment.
INTRODUCTION
Immunotherapies are revolutionizing clinical care for cancer patients. The most widely used approach, immune checkpoint inhibition (ICI), can lead to long-term survival benefits in patients with advanced melanoma (1), lung cancer (2), and renal cell carcinoma (3). However, not all patients benefit from ICI therapy, and adequate predictions of treatment response have proven elusive so far (4, 5). Efforts to improve these predictions focus mainly on discovering biomarkers in aberrant molecular pathways within the tumor microenvironment that drive immunosuppression and therapeutic resistance (6, 7). These include genomic alterations in oncogenic drivers, the absence of tumor-specific antigens, and the presence of immunosuppressive molecules or cells (8, 9). Despite substantial efforts, only a limited fraction (according to one estimate, <1% (10)) of proposed cancer biomarkers find their way into the clinical practice. These apparent challenges in identifying biomarkers for immunotherapy and translating them into clinical practice could be a consequence of the inherent complexity of cancers and their interaction with the immune system.
To unravel the complexities of cancers and their treatments, researchers have adopted mathematical and computational approaches to complement laboratory research. A plethora of modeling approaches are available, ranging from simple one-variable equations to complex spatial agent-based simulation models. In silico modeling has contributed to fundamental insights into tumor growth and cancer progression (11-13), tumor-immune control (e.g., neoantigen prediction as targets for immunotherapy) (14), identification of tumor-associated genes (15), verification of treatment-related safety concerns such as hematological toxicity (16), prediction of treatment responses to chemo- and immunotherapy (17-19), investigation of drug-induced resistance (20), and timing of anti-cancer treatments (21-23). In the context of disease course dynamics, ordinary differential equation (ODE) models have proven useful over the years. ODE models follow the principle that a model should be “as simple as possible but not simpler”. Based on plausible biological assumptions, they aim to reduce the complex reality of the modeled system to its bare essentials to enable investigation of critical underlying dynamics. For instance, Fassoni et al. used ODE models to predict that dose de-escalation of tyrosine kinase inhibitors targeting the oncogenic protein BCR-ABL1 in patients with chronic myeloid leukemia (chronic phase) does not lead to worse long-term outcomes (24). The recent results of the DESTINY trial support this prediction (25).
In this study, we investigate the consequences of tumor-immune dynamics on patients’ responses to ICI and survival with an ODE model. Our model reveals a tipping point within tumor-immune dynamics – a critical threshold for survival culminating in an all-or-nothing principle – that has profound implications for a patient’s disease course and outcome. We show how the presence of such a tipping point alone robustly induces heterogeneous immunotherapy treatment outcomes, and how this complicates the search for both prognostic and predictive biomarkers.
METHODS
Capturing core mechanisms of the tumor microenvironment in a mathematical model
We constructed a mathematical model based on a system of ordinary differential equations (ODE) to capture essential interactions between cancerous cells and lymphocytes in the tumor microenvironment. Our model represents tumorigenesis in patients, starting with the malignant transformation of a single cell.
The model describes the following processes: (1) conversion of naive T cells into antigen-specific effector T cells in lymph nodes; (2) the clonal expansion of effector T cells in the lymph nodes; (3) tumor growth; (4) subsequent attraction of tumor-infiltrating lymphocytes; and (5) formation of tumor-immune cell complexes to facilitate tumor cell killing. Below we describe the model; a mathematical formulation containing the model equations is provided as Supplementary Information. Tumor growth is represented by the generalized exponential model proposed by Mendelsohn (26), meaning that at each time interval, a fraction of tumor cells divide. The dividing fraction decreases as the tumor burden increases since substantial parts of a larger tumor mass, such as the necrotic core, are no longer able to proliferate. During tumor development, T cells are produced in the lymph nodes. This process starts with the conversion of naive T cells (i.e., not activated antigen-specific) into antigen-specific effector T cells at a rate proportional to the tumor burden. Effector T cells expand clonally and migrate into the tumor microenvironment, where they form tumor-immune complexes (i.e., an immune cell attached to a tumor cell). Within these complexes, T cells kill the tumor cells.
The killing rate expression is derived from the Michaelis-Menten kinetics for enzyme-substrate interaction with a quasi-steady-state approximation, as described by Borghans et al. (27). Given that complex formation and dissociation occur on a time scale several orders of magnitude smaller than the time scale for tumor growth, this approximation states that this process can be regarded as a constant process. Further simplification of the killing rate expression using the Padé approximation yields the double saturation killing rate of T cells (27). T cells follow a ‘monogamous killing’ strategy, meaning that one T cell interacts with one tumor cell at a time (27, 28). In addition, T cells die and exit the system at a fixed rate.
Simulation parameters
The simulation parameters are listed in Table 1.
The parameters were chosen to mimic realistic in vivo intercellular behavior. The rationale for the choice of each parameter is explained below.
In a human adult, an estimated repertoire of approximately 1010 - 1011 naive CD8+ T cells is present (30, 31). Naive CD8+ T cells need to be primed to become activated effector T cells. The CD8+ T cell precursor frequency – the frequency at which any given peptide-MHC complex is recognized by naive antigen-specific CD8+ T cells – is on the order of 1 : 100.000 (30). Priming should be limited primarily to naive CD8+ T cells in one of the tumor areas draining lymph nodes. A human body contains ±600 lymph nodes. At steady states, roughly 40% of all lymphocytes reside in lymph nodes, meaning that 40.000 naive T cells (≈ 70 naive CD8+ T cells per lymph node) can be primed (32, 33). We assume that priming occurs primarily in the tumor-draining lymph node station (per station harboring around 20 lymph nodes (34)), leading to an optimal priming rate of 1400 T cells per day. Considering that DCs might present multiple epitopes and antigens, and that T cell priming in vivo might occur suboptimally, we set a priming rate of at most 2500 cells per day. The order of magnitude of these priming rates corresponds to priming rates found in chronic infectious diseases (35). Due to evasive mechanisms, anti-tumor immunity is a more dormant process than an immune response to infections (36). Therefore, we scaled the priming rate with tumor size (Supplementary Information), which translates into a maximum production of 106 antigen-specific CD8+ T cells per day via clonal expansion. Next, we assume that all antigen-specific effector T cells migrate into the tumor microenvironment to interact with tumor cells (i.e., complex formation).
Complex formation and dissociation rates are described by ‘the Michaelis constant’, which we derived from literature (28). The Michaelis constant describes the ratio between complex formation and dissociation.
The killing rate of effector T cells has been investigated mainly in the context of infectious disease. In their review (37), Halle et al. discuss discrepancies between in vitro and in vivo killing rates of effector T cells. Dependent on the context, killing rates of effector T cells vary from 1 target per 5 minutes to 0-10 targets per day (37), but tumor cells are considered difficult to kill. Extensive variation in experimental in vivo per capita killing rates (i.e., the number of cells killed by an effector T cell per unit of time) complicates the selection of a default fixed killing parameter. Therefore, we investigated T cell dynamics over a range of overall killing rates as described using the monogamous killing regime in a double saturation model by Gadhamsetty et al. (28). The double saturation model ensures that the killing rate saturates with respect to the tumor cell and the effector cell densities. Consequently, our model’s maximum per capita killing rate is 2.5 cells-1 day-1: one T cell can kill at most 2.5 tumor cells per day, provided there are abundant target cells available and there is no competition with other T cells. The default tumor growth rate is one cell day-1, but we varied this parameter extensively in our simulations. Taken together, our default parameter values led to simulations of disease courses with realistic survival times in patients with malignancies and matched the order of magnitude of tumor growth rates as reported by others (38).
Patient simulations
We simulated tumor development in patients up to a maximum of 5 years. Note that depending on emergent tumor-immune dynamics, simulated patients may not reach the overall survival endpoint during this interval. Each time step in the simulation corresponded to one day. At baseline, one tumor cell and a pool of 106 naïve tumor-specific T cells are present in a patient. Activated effector T cells are absent. We defined the time of diagnosis as the time at which the tumor burden exceeded 65 * 108 cells and became clinically apparent. This cut-off corresponds to the assumption that a tumor with a volume of 1 cm3 contains 108 tumor cells (39) and that several primary tumors (e.g., lung cancer, colon carcinoma, and renal cell carcinoma) are diagnosed as spherical structures with a median diameter of approximately 5 cm (40-42). The ‘lethal’ tumor burden of patients in these simulations is estimated at 1012 cells, corresponding to a total tumor mass of approximately 22 * 22 * 22 cm.
Model implementation
We implemented our ODE model in C++. The Boost library ‘odeint’ was used to solve the system of ordinary differential equations (43). The code is available at GitHub: https://github.com/jeroencreemers/tipping-point-cancer-immune-dynamics. Analyses and visualizations were performed in R.
RESULTS
Modeling tumor-immune dynamics yields realistic disease trajectories
To investigate the consequences of tumor-immune dynamics on the survival kinetics of patients, we used a computational modeling approach. We aimed to capture the interplay between tumor- and immune cells in the tumor microenvironment and simulate tumor growth in patients (see Methods). Our ODE model captured four essential processes in anti-tumor immunity: priming of naive antigen-specific CD8+ T cells, clonal expansion of effector T cells in lymph nodes, tumor growth leading to effector T cell attraction into the tumor microenvironment, and formation of tumor-immune cell complexes to enable tumor cell killing (Fig. 1A).
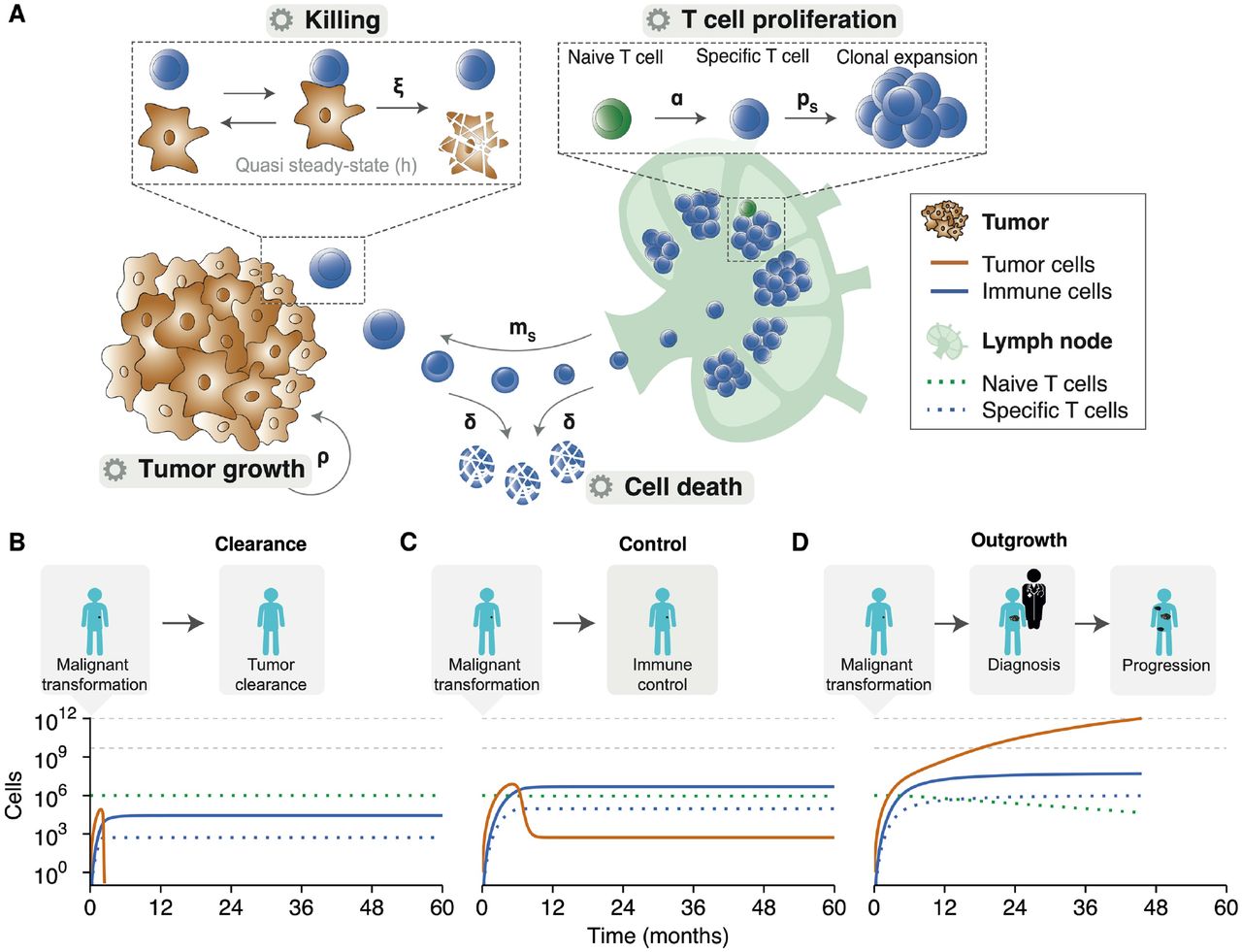
(A) The ODE model describes fundamental processes in the tumor microenvironment. Parameters: α = naive T cell priming rate, δ = effector T cell death rate, ξ = effector T cell killing rate, ρ = tumor growth rate, ps = effector T cell proliferation rate, and ms = effector T cell migration rate. (B) An effective anti-tumor immune response can eradicate tumor cells before the clinical manifestation of a tumor. (C) After an initial state in which the tumor outpaces the immune system, the immune system can suppress tumor growth and controls it in a subclinical state. (D) The natural course of disease for a clinically apparent tumor. An initial malignant transformation is followed by tumor growth until clinical diagnosis. Despite the activation of adaptive immunity, the tumor prevails. A stage of progressive disease follows, ultimately culminating in cancer-related death. The horizontal grey lines indicate (from bottom to top): the tumor burden at diagnosis and the tumor burden at death, respectively. Simulation parameters are added in Supplementary Table 1.
We simulated tumor development from malignant transformation of a single cell, via clinical detection of a tumor, to advanced disease and possibly death. Depending on the tumor growth and the cytotoxic capacity of effector T cells, the ‘time to clinical manifestation’ and overall survival varied. Despite this variation, our simulations consistently showed three possible outcomes: 1) effector T cells inhibited tumor cell outgrowth and eradicated the tumor before clinical manifestation (Figure 1B); 2) effector T cells were initially unable to inhibit tumor cell outgrowth, but caught up and suppressed tumor growth to a balanced subclinical state (Figure 1C); or 3) exponential tumor growth outpaced the immune system’s control and gave rise to a clinically detectable tumor (Figure 1D). These three scenarios only led to two clinically different outcomes in patients: either a tumor became clinically evident, or the immune system could suppress/eradicate a tumor at an early stage. A balanced equilibrium state, in which the immune system keeps a clinically evident tumor under persistent control, does not exist in this deterministic version of our model.
Patient survival depends on a tipping point in tumor-immune dynamics
To better characterize these dichotomous survival kinetics, we examined how tumor-immune dynamics influenced patient survival by varying the tumor growth rate and the T cell killing rate over a broad range of possible values.
First, we focused solely on the tumor-component by varying the tumor growth rate. An increase in tumor growth did not gradually shorten overall survival in patients (Figure 2A). On the contrary, a critical threshold was present. Once the threshold was exceeded, the kinetics ‘flip’ from a state of immune control (Figure 2A, inset 1) to a state in which the tumor could evade immune control (Figure 2A, inset 2).

(A) A gradual increase in tumor growth reveals a tipping point, where long-term survival (immune control; inset 1) abruptly changes to short-term survival (immune evasion; inset 2). (B) A similar analysis reveals a tipping point along the immune axis, again differentiating short-term survival (immune evasion; inset 1) from long-term benefit (immune control; inset 2). (C) The tipping point is present across the entire range of parameters examined. Cure and progressive disease are the dominant states, whereas subclinical tumor control only occurs within a limited parameter range (inset). Simulation parameters are shown in Supplementary Table 2.
Second, we investigated the influence of the T cell killing rate on overall survival. As for the death rate, a gradual increase in the cytotoxic capacity of effector T cells did not induce a gradual change in survival times. Instead, a sharp state transition was again observed that differentiated short from long survival (Figure 2B). This coincided with the phenotypes ‘immune evasion’ and ‘immune control’.
To visualize this sudden state transition or ‘tipping point’ in tumor-immune dynamics as a function of both tumor proliferation and cytotoxic killing at the same time, we visualized the joint influence of the tumor growth rate and T cell killing rate on survival in a heatmap (Figure 2C). This ‘phase diagram’ shows that the tipping point is not only present for specific parameter values but is a fundamental property in our model. By contrast, the state of subclinical tumor control was not universally present around the tipping point (Figure 2C, inset) but manifested itself only in a narrow range of parameters. Nevertheless, the general presence of a tipping point indicates that small perturbations in either tumor growth rate or T cell killing rate in the vicinity of a tipping point may result in substantial overall survival differences in patients, whereas much larger perturbations far away from the tipping point would have far less effect.
Immune checkpoint inhibitors induce a survival benefit by shifting patients over a tipping point
So far, we have described tumor-immune interactions during the natural course of malignant disease. In a clinical setting, however, therapeutic interventions are available to steer disease courses. Dependent on the treatment of choice, a specific effect is exerted on the tumor microenvironment. Treatment effects vary from constraining the proliferative capacity of tumor cells (e.g., chemotherapy or targeted therapy) to increasing the T cell pool (e.g., CAR T cells) or expanding the proliferative capacity of T cells (e.g., cancer vaccines; Figure 3A). Given the unparalleled responses of advanced malignancies to immunotherapy, we focused on the consequences of a tipping point for responses to immune checkpoint inhibitors (ICI), but these findings could be extended to other therapies as well. In this study, we limited the treatment effect of ICI to their primary mode of action: the augmentation of the T cell killing rate (Figure 3A).

(A) Treatments target processes or cell populations in the tumor microenvironment. (B-C) Two criteria need to be met to induce long-term survival: (B) ICI need to augment T cell killing sufficiently, and (C) the treatment effect needs to be retained for a prolonged time. An inadequate treatment effect or limited treatment duration led at maximum to a temporary survival benefit. (D-E) In patient populations with variation in only (D) the tumor (i.e., growth rate), or (E) the immune system (i.e., T cell killing rate), the distance to a tipping point determines the clinical benefit. Without treatment, survival was limited (grey bars). In contrast, ICI induced long-term survival solely in patients close to a tipping point (green bars). See also Supplementary Table 3.
In the presence of a tipping point, ICI could induce a long-term survival benefit under two conditions: 1) the effect of treatment needs to be potent enough to shift a patient over a tipping point (Figure 3B), and 2) the treatment effect needs to be sustained long enough for a patient to benefit from the treatment (Figure 3C). The treatment effect is defined as the multiplication factor of the T cell killing rate. When both criteria were satisfied, ICI were able to induce a long-term survival benefit. However, if the treatment effect (anti-PD1 effect < 12.6) or duration (less than ±5 months) proved inadequate, any survival benefit was only temporary, and inevitable tumor progression would ultimately limit overall survival (Figure 3B/C; insets). These survival kinetics depend not solely on therapeutic features of ICI but rather on the interplay between patient and ICI characteristics. To illustrate this, we simulated twenty patients with identical immune systems (i.e., identical T cell killing rates). In the absence of ICI therapy, variation in the tumor growth rate – that is, variation in the distance to a tipping point – led to a limited variation in survival (Figure 3D; grey bars). When these same patients were treated with ICI, a survival benefit is induced in all patients. However, the extent of this benefit differs and is dependent on the distance to a tipping point. Following clinical observations, long-term survival is only induced in the subset of patients close to a tipping point (Figure 3D; green bars). Similar findings were obtained in a population of patients with identical tumors but different immune systems. Without treatment, hardly any survival variation is present (Figure 3E; grey bars). Again, treatment with ICI induced dichotomous clinical outcomes: a small survival benefit in most patients, with long-term survival in a subset (Figure 3E; green bars). Hence, the mere presence of a tipping point yields heterogeneity in treatment outcomes.
Tipping points determine patient outcomes in dynamic patient trajectories
Thus far, our simulations considered tipping points generated in patients with fixed characteristics. However, disease courses in patients are certainly not fixed and are, to a certain extent, subject to (random) variation. We hypothesized that interpatient variability in clinical outcomes could be (partially) attributable to this dynamic behavior of cancers and the interaction with the immune system. Such variation might reflect biological processes (e.g., accumulating mutations, the expression of checkpoint molecules, and the availability of nutrients) that alter anti-tumor immunity and promote or hamper tumor development. We reasoned that the subsequent dynamics could drive patients towards and ultimately over a tipping point – or move patients away from it, which would limit the survival benefit of these treatments. To verify this hypothesis, we simulated the effect of dynamically evolving tumors (Figure 4A) or immune systems (Figure 4B) in identical patients compared to a static reference patient. Specifically, we varied the tumor growth rate and the T cell killing rate randomly over time (parameter values are included in Supplementary Table 4). Upon reaching a diagnosable tumor volume, all patients in these examples were treated with ICI. As expected, stochastic dynamics prompted survival differences and induced a survival benefit in a subset of patients. In a heterogeneous patient population, this led to an interesting finding: the initial distance to a tipping point, along with the dynamics itself, determined the clinical outcome of patients treated with ICI (Figure 4C, Supplementary Figure 1). At population level, this led to a distinction between three subsets of patients: (1) patients far away from a tipping point with an unmodifiable bad prognosis (non-responders), (2) patients close to a tipping point with a favorable prognosis (responders), and, most importantly, (3) patients in between these groups (potential responders). In the last subset, tumor dynamics ultimately determined the treatment response, and thereby the clinical outcome (Figure 4C; grey box). A clinically important ramification of dynamic trajectories is that even if the subset to which a patient belongs is known at baseline, dynamics could alter the distance to a tipping point and, thereby, the prognosis of a patient. Therefore, it might be impossible to predict the prognosis solely based on characteristics measured upon diagnosis. Dynamic trajectories can significantly diversify patient outcomes, meaning that continuous variation in the tumor growth rate (Figure 4D) or T cell killing rate (Figure 4E) leads to an entire spectrum of patient outcomes.

(A-B) Examples of dynamic disease courses in patients with identical tumors and immune systems at baseline, respectively. (A) Evolving tumors (i.e., random variation in tumor growth rate over time) and (B) continuous variation in the potency of the immune system (i.e., killing rate) lead to divergent survival outcomes. The grey dotted line indicates the baseline values for the growth rate and killing rate, respectively. (C) Dynamic trajectories in a heterogeneous patient population can move patients towards or away from a tipping point. The grey box indicates patients in which dynamic trajectories strongly alter survival outcomes. See also Supplementary Figure 1. In dynamic trajectories, (D) baseline tumor growth and (E) baseline T cell killing rates cannot accurately predict overall survival. Note: all patients in these examples are treated with ICI. The red and black dotted lines indicate the 25% and 75% quantiles, respectively. See also Supplementary Table 4.
Implications of tipping points for biomarker discovery studies
Biomarker discovery studies aim to improve the prediction of patient survival upon treatment. We observed that tipping points are crucial in shaping survival kinetics. Therefore, accurate survival predictions would require the consideration of tipping points. Ideally, a prognostic biomarker (or biomarker panel) would consistently distinguish long-term survivors from their counterparts. Since the non-linear survival dynamics following a tipping point weaken the correlation between a single biomarker and survival, the question is: how can we screen for biomarkers in a more efficient manner that takes this tipping point into account?
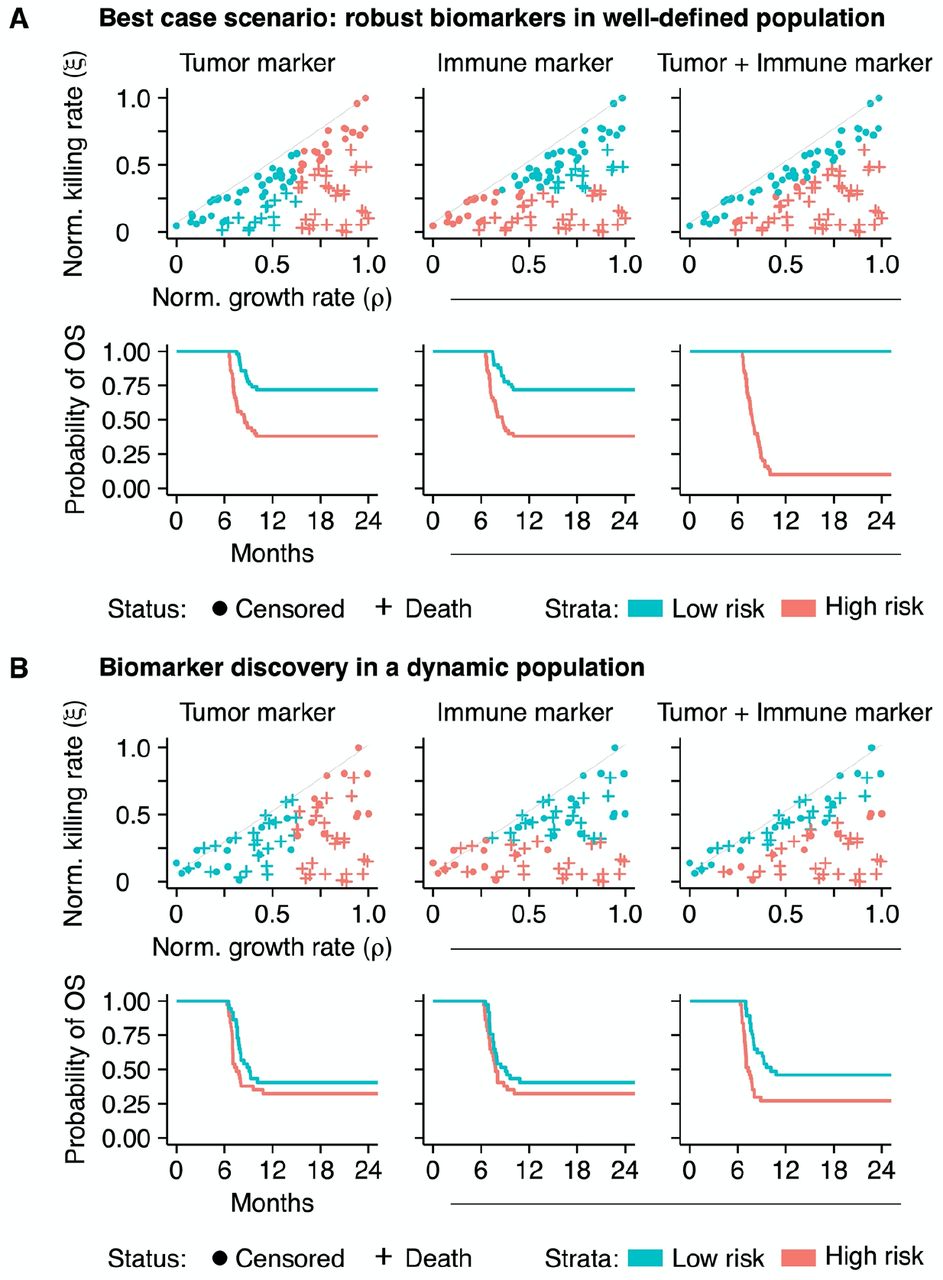
We approached this question with an in silico biomarker discovery study. We measured the value of two potential biomarkers at baseline in patients (n=100) that were subsequently treated with ICI. We simplified the cohort by fixing the tumor and immune characteristics of these patients over time and assumed to have access to an entirely accurate biomarker (i.e., no measurement error; Figure 5A). Within this cohort, we predicted the prognosis of patients based on either the tumor or immune marker (the first and second columns of Figure 5A, respectively). As is common in practice (though from a statistical point of view far from ideal), we dichotomized the biomarker using its median as a cut-off. Although survival differentiation based on these biomarkers alone was partially possible, it remained far from optimal. However, when we constructed a biomarker panel including both biomarkers, it highly accurately discriminated short-term from long-term survivors (third column of Figure 5A, Supplementary information). Note that despite variability in time from diagnosis, the initial plateau in the survival curves was caused by the fact that all tumors were diagnosed with identical sizes and immediately treated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) An in silico biomarker discovery study in a ‘fixed’ patient cohort: while a single biomarker – either a tumor or an immune marker – can predict survival to some extent (the first and second columns), information from both markers in a biomarker panel enhances the predictive capacity greatly (third column). (B) Dynamic disease trajectories challenge survival prediction with ‘baseline’ biomarkers and stress the need for continuous monitoring. Disease dynamics challenge the predictive value of single ‘baseline’ biomarkers (the first and second columns; compare to Figure 5A). A biomarker panel improves survival predictions in this cohort (the third column) but is still defied by evolving dynamics. See also Supplementary figure 5.
In clinical practice, the assumption of a ‘fixed’ patient trajectory does not hold. Therefore, we simulated this cohort again with dynamic trajectories. Due to the dynamics, a subgroup of patients did not develop clinical tumors and was excluded from the analysis. The prediction of a patient’s prognosis with a single biomarker, either from the tumor or the immune system, in a dynamic cohort became increasingly challenging (the first and second columns of Figure 5B). The combination of both markers in a biomarker panel increased the predictive capacity slightly, enabling the prediction of prognosis to some extent. However, in line with the notion of personalized medicine, the accurate and individualized prediction of prognosis based on baseline characteristics was not feasible in a significant subgroup of patients due to dynamic tumor-immune interactions (third column of Figure 5B). Two important findings are derived from these observations: First, due to the non-linear tumor-immune dynamics with respect to survival, it is highly unlikely that a single biomarker can accurately predict a patients’ prognosis. Since survival kinetics emerge from the interplay between a cancer and the immune system, biomarkers from both systems need to be incorporated simultaneously into a biomarker panel to improve the predictive value. Second, biomarker measurements at baseline are merely a situational snapshot of the disease conditions at a specific point in time. Depending on the magnitude of the dynamics, it might become challenging or even impossible to correctly predict the prognosis of patients from these biomarkers, stressing the need for continuous monitoring.
DISCUSSION
This study investigated how tumor-immune dynamics relate to ICI-induced treatment responses and survival kinetics of patients. We predict that a tipping point is present in the tumor-immune interaction. This finding implies that at the basis of the intricate interplay between a developing malignancy and the immune system contrasting disease states determine disease outcome: a state where the immune system controls tumor outgrowth and a state in which a tumor escapes immune defense. A stable “steady state” in which tumor growth and the immune response perfectly balance each other for extended periods seems only plausible in a subclinical setting. We show that treatment with ICI can induce a survival benefit by shifting a patient over a tipping point, thereby tipping the balance in tumor-immune dynamics in favor of survival. In line with clinical observations of interpatient variability in disease courses, we found that dynamics in patient trajectories might contribute to unpredictability in treatment responses. Moreover, we showed how the tipping point in dynamic patient trajectories defies simple strategies for outcome prediction in biomarker discovery studies, stressing the need for advanced mechanism-based monitoring.
Tipping points are well-known in complex systems such as financial markets and ecosystems but are also present in medicine (44, 45). State transitions might progress gradually or abruptly. If a system balances around a critical threshold, small perturbations might induce an abrupt transition to a contrasting state. In oncology, phenomena as partial or complete radiologic responses during treatment or (hyper)progression after discontinuation of treatment suggest the presence of state transitions (46, 47). Based on these observations, a tipping point in cancer immunotherapy had been speculated upon (48). Experimentally, tipping points are most clearly represented by early preclinical work in the PD-1/PD-L1 axis. Consistent with our findings, dichotomous treatment responses arise in syngeneic DBA/2 mice inoculated with P815/PD-L1 cells (49). While genetically identical with similar tumor characteristics, anti-PD-L1 antibodies could prolong survival in only a subset of the mice, likely due to stochastic differences in immune responses and TCR repertoire. Additional in vivo data supporting the theory of tipping points in oncology is derived from studies on dynamic network biomarkers, showing its relevance during the onset of metastasis in hepatocellular carcinoma (50) and the development of treatment resistance in breast cancer (51). This study provides a potential mechanistic explanation for this phenomenon in immuno-oncology and shows its implications on the induction of long-term survival in clinical practice and biomarker discovery. From a biomechanistic perspective, such state transitions in cancer immunotherapy arise due to fundamental differences in proliferation kinetics between tumors and the immune system. While tumor cell proliferation is virtually unrestricted, immune cell proliferation is much more limited and tightly controlled. Our finding that tipping points affect not only natural disease courses but also treatment responses underlines the importance of these kinetics.
An important implication of tipping points within tumor-immune dynamics involves biomarker discovery. Biomarkers are developed to predict prognosis and steer clinical decision making. Disease outcomes in cancer patients are essentially determined by the interplay between two complex systems: the tumor and the immune system. Our model predicts that factors from both systems should be considered to improve the predictive power of biomarkers. However, in contrast with this seemingly straightforward prediction, current research mainly focuses on factors derived from one of the two complex systems. Expression of programmed death ligand-1 (PD-L1) on tumor tissue illustrates this properly: while 45% of patients with PD-L1 positive tumors show objective responses to anti-PD(L)1 immunotherapy, 15% of patients with PD-L1 negative tumors also show objective responses (52). Other explanations for this difference include heterogeneous intratumoral and inter-metastases expression patterns, positivity-threshold selection, and differences in immunohistochemical staining protocols. In that respect, tumor mutational burden (TMB) might prove to be a highly relevant biomarker. The mutation rate is a tumor-intrinsic factor associated with the phenotypical aggressiveness of tumors (53). Simultaneously, a high mutational burden might induce a plethora of neoantigens, linking this tumor-intrinsic factor directly to adaptive immunity. Clinical observations of a stronger association between TMB and response rates to anti-PDL1 immunotherapy compared to PD-L1 expression in patients with urothelial carcinoma support this hypothesis (54). Thus, reinforcing common calls to integrate multiple existing biomarkers for immunotherapy prediction outcomes (55, 56), our research indicates that a combination of both immunological and tumor-related parameters should be the basis of any biomarker discovery effort. The strongly non-linear dynamics resulting from the tipping point mean that a one-dimensional approach will likely be insufficient.
Our approach has to be interpreted in light of some limitations. Although the ‘coarse-grained’ nature of ODE models allows focusing on the major common underlying mechanisms in many cancers, it is also a potential pitfall. For example, metabolic processes such as hypoxia, immune-suppressive characteristics of the tumor microenvironment such as the presence of FoxP3+ regulatory T cells or expression of transforming growth factor β, the presence of additional relevant effector cells such as natural killer cells, and the availability of nutrients are only implicitly represented by our model in a single killing efficacy parameter. This simplification also holds for treatments. In this study, ICI was limited to its main mode of action: the augmentation of the T cell killing rate. While the ‘true’ mechanistic effects might be more widespread, sufficient data to correctly parameterize more complex models remains scarce. Furthermore, it should be emphasized that an ODE model contains limited spatial information; while we distinguish between lymphatic tissue and the tumor microenvironment, all cells within the microenvironment are identical, and all processes affect cells in the same manner. We do not expect that explicit incorporation of these processes or translation of the model into a spatial variant alters our findings of a tipping point; however, it might be of interest to verify these hypotheses in future research in spatial agent-based models.
In conclusion, we used a computational modeling approach to show that the clinical outcome of cancer patients is determined by tipping points in tumor-immune dynamics. A tipping point influences not only response to treatment but also the prognosis of patients and has major implications for future biomarker research.
Data Availability
The code of the ODE model is available at GitHub.
https://github.com/jeroencreemers/tipping-point-cancer-immune-dynamics.
DECLARATIONS
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
The code of the ODE model is available at GitHub: https://github.com/jeroencreemers/tipping-point-cancer-immune-dynamics.
Competing interests
WJL reports consultancy activities for Douglas Pharmaceuticals and MSD; research funding from Douglas Pharmaceuticals, AstraZeneca, and ENA therapeutics; patents PCT/AU2019/050259 and PCT/AU2015/000458 (all outside this work). NM reports personal fees from Bayer and Merck Sharp & Dohme; grants and personal fees from Jansen-Cilag, Roche, Astellas, and Sanofi (all outside this work). WRG reports consultancy activities for Bristol-Myers Squibb, IMS Health, Janssen-Cilag, Sanofi, and MSD; speaker fees from ESMO and MSD; and research funding from Bayer, Astellas, Janssen-Cilag, and Sanofi (all outside this work).
Funding
JC was funded by the Radboudumc. WJL was supported by Fellowships from the NHMRC, the Simon Lee Foundation, and the Cancer Council Western Australia. CF received an ERC Adv Grant ARTimmune (834618) and an NWO Spinoza grant. IV received an NWO-Vici grant (918.14.655). JT was supported by a Young Investigator Grant (10620) from the Dutch Cancer Society and an NWO grant (VI.Vidi.192.084).
Authors’ contribution
JHAC and JT conceived this study. JHAC performed the experiments and wrote the manuscript under the supervision of JT. All authors provided feedback on the manuscript and reviewed the manuscript prior to submission.
Acknowledgements
Not applicable.
LIST OF ABBREVIATIONS
- ICI
- Immune Checkpoint Inhibition
- ODE
- Ordinary Differential Equation
- PD-(L)1
- Programmed Death-(Ligand) 1
- TMB
- Tumor Mutational Burden
REFERENCES



