
Abstract
Proteins detectable in peripheral blood may influence COVID-19 susceptibility or severity. However, understanding which circulating proteins are etiologically involved is difficult because their levels may be influenced by COVID-19 itself and also subject to confounding factors. To identify circulating proteins influencing COVID-19 susceptibility and severity we undertook a large-scale two-sample Mendelian randomization (MR) study, since this study design can rapidly scan hundreds of circulating proteins and reduces bias due to confounding and reverse causation. We began by identifying the genetic determinants of 955 circulating proteins in up to 10,708 SARS-CoV-2 uninfected individuals, retaining only single nucleotide polymorphisms near the gene encoded by the circulating protein. We then undertook an MR study to estimate the effect of these proteins on COVID-19 susceptibility and severity using the Host Genetics Initiative. We found that a standard deviation increase in OAS1 levels was associated with reduced COVID-19 death or ventilation (N = 2,972 cases / 284,472 controls; OR = 0.48, P = 7×10−8), COVID-19 hospitalization (N = 6,492 / 1,012,809; OR = 0.60, P = 2×10−7) and COVID-19 susceptibility (N = 17,607 / 1,345,334; OR = 0.81, P = 6×10−5). Results were consistent despite multiple sensitivity analyses probing MR assumptions. OAS1 is an interferon-stimulated gene that promotes viral RNA degradation. Other potentially implicated proteins included IL10RB. Available medicines, such as interferon-beta-1b, increase OAS1 and could be explored for their effect on COVID-19 susceptibility and severity.
Introduction
Since its onset in late 2019, the COVID-19 pandemic has caused more than 1 million deaths worldwide, and infected over 34 million individuals.1 Despite the scale of the epidemic, there are at present no disease-specific therapies that can reduce the morbidity and mortality of SARS-CoV-2 infection, and apart from dexamethasone therapy in oxygen dependent patients,2 most clinical trials have shown at most mild or inconsistent benefits in disease outcome.3–5 Further, vaccines are at least several months away, and whether they elicit protective immunity is unclear. Therefore, validated targets are needed for COVID-19 therapeutic development.
One source of such targets is circulating proteins. Recent advances in large-scale proteomics have enabled the measurement of thousands of circulating proteins at once and when combined with evidence from human genetics, such targets greatly improve the probability of drug development success.6–8 While de novo drug development will take time—even in the accelerated arena of COVID-19 therapies— repositioning of currently available molecules targeting those proteins, if they exist, could also provide an accelerated opportunity to deliver new therapies to patients.
Nevertheless, since confounding and reverse causation often bias traditional circulating protein epidemiological studies, disentangling the causal relationship between circulating proteins and COVID-19 susceptibility or severity is challenging. Confounding happens when both the circulating protein and COVID-19 share a common cause, and reverse causation occurs when COVID-19 itself influences the level of a circulating protein. Even with sophisticated statistical methods, both are difficult to control for in observational studies. One way to address these limitations is by using Mendelian randomization (MR), a genetic epidemiology method that uses genetic variants as instrumental variables to test the effect of an exposure (here protein levels) on an outcome (here COVID-19 outcomes). Given that each person’s genotype is essentially randomly assigned at conception, due to Mendel’s second law, this greatly reduces bias due to confounding. Since genotypes are always assigned prior to disease onset, MR studies are not influenced by reverse causation. However, MR rests on three core assumptions.9 First, the genetic variants must be associated with the exposure of interest. Second, they do not affect the outcome, except through the exposure of interest (i.e. a lack of horizontal pleiotropy) and third, they do not associate with the confounders of the exposure-outcome relationship.
Of these, the most problematic is the second assumption—a lack of horizontal pleiotropy. One way to help avoid bias due to horizontal pleiotropy is to use genetic variants that influence circulating protein levels which are adjacent to the gene which encodes the circulating protein through the use of cis-protein quantitative trait loci (cis-pQTLs).8 Given their close proximity to the target gene, cis-pQTLs are likely to influence the level of the circulating protein by directly influencing its transcription or translation, and therefore less likely to affect the outcome of interest (COVID-19) through pleiotropic pathways. The choice of cis-pQTLs also supports the first and third assumptions of MR, as it uses a biologically plausible pathway from the genetic variant to the exposure.
Recently developed MR methods, using two-sample MR,10 allow for the rapid scanning of hundreds of exposures if genome-wide association studies (GWASs) have been done for both the biomarker and the outcome.8 Nevertheless, given that MR only requires an association between the genetic instrument and the exposure, a causal association between the two may be confounded by linkage disequilibrium (LD, the non-random association of genetic variants assigned at conception). This would happen if the selected genetic variants associated with both protein levels and COVID-19 are a part of separate causal mechanisms but are also related through LD.11 To probe this potential problem, colocalization tests can assess for the presence of bias from LD.
Understanding the etiologic role of circulating proteins in infectious diseases can be challenging because the infection itself often leads to large changes in circulating proteins, which may act against the infectious agent. Thus, it may appear that an increase in a circulating protein, such as a cytokine, is associated with a worsened outcome, when in fact, the cytokine may be the host’s response to this infection and help to mitigate this outcome. It is therefore important to identify genetic determinants of the protein levels in the non-infected state, which would reflect a person’s baseline predisposition to the level of a protein. Further, circulating proteins may execute their biological roles inside the cell, but then translocate to the peripheral blood, and thus their circulating levels can, in some circumstances, offer insights into their intracellular roles.
In this study, we therefore undertook two-sample MR and colocalization analyses to combine results from large-scale GWASs of circulating protein levels and COVID-19 outcomes12 in order to prioritize proteins likely influencing COVID-19 outcomes. We began by identifying the genetic determinants of circulating protein levels in large-scale protein level GWASs. Then, after selecting the SNPs associated with circulating proteins, which were proximal to their encoded genes (cis-pQTLs), we assessed whether these cis-pQTLs were associated with COVID-19 outcomes in the ICDA Host Genetics Initiative COVID-19 outcomes GWASs and then undertook MR analyses to estimate the effect of the circulating protein level of COVID-19 outcomes.
Results
MR using cis-pQTLs, and pleiotropy assessment
We began by obtaining the genetic determinants of circulating protein levels from six large proteomic GWAS of European individuals (Sun et al13 N=3,301; Emilsson et al14 N=3,200; Pietzner et al15 N=10,708; Folkersen et al16 N=3,394; Yao et al17 N=6,861 and Suhre et al18 N=997). A total of 955 proteins from these six studies had cis-pQTLs associated at a genome-wide significant level (P < 5×10−8) with protein levels, or highly correlated proxies (LD R2 > 0.8), in the meta-analyses of data the from COVID-19 Host Genetics Initiative (https://www.covid19hg.org/results/) which included results from the GenOMICC program (https://genomicc.org/)19. We then undertook MR analyses using 1,388 directly matched cis-pQTLs and 50 proxies as genetic instruments for their associated circulating proteins on three separate COVID-19 outcomes: 1) Very severe COVID-19 disease, (defined as individuals experiencing death, mechanical ventilation, non-invasive ventilation, high-flow oxygen, or use of extracorporeal membrane oxygenation) using 2,972 cases and 284,472 controls; 2) COVID-19 disease requiring hospitalization using 6,492 cases and 1,012,809 controls and 3) COVID-19 susceptibility using 17,607 cases and 1,345,334 controls. These case-control phenotype definitions are referred to as A2, B2, and C2 by the COVID-19 Host Genetics Initiative, respectively. In all outcomes, cases required evidence of SARS-CoV-2 infection. For the very severe COVID-19 and hospitalization outcomes, COVID-19 cases were defined as laboratory confirmed SARS-CoV-2 infection based on nucleic acid amplification or serology tests. For the COVID-19 susceptibility outcome, cases were also identified by review of health records (using International Classification of Disease codes or physician notes).
MR analyses revealed that the levels of four circulating proteins, 2’-5’-oligoadenylate synthetase 1 (OAS1), interleukin-10 receptor beta subunit (IL10RB), ABO, and liposaccharide binding protein (LBP) were associated with one or more COVID-19 outcomes after Bonferroni correction for the number of proteins tested (P<5.2×10−5) (Table 1, Table S1-3). We note that Bonferroni correction is overly conservative given the non-independence of the circulating protein levels. Notably, increased OAS1 levels were strongly associated with protection from all three COVID-19 outcomes. Further, these effect sizes became more pronounced with more severe outcomes, such that each standard deviation increase in OAS1 levels was associated with a decreased odds of COVID-19 susceptibility of 0.81 (95% CI: 0.73-0.90, P=5.63×10−5), decreasing further for hospitalized COVID-19 (OR=0.60; 95% CI: 0.50-0.73, P=1.69×10−7) and very severe COVID-19 (OR=0.48; 95% CI: 0.37-0.63, P=6.77×10−8) (Figure 1).

We next assessed whether the cis-pQTL associated with OAS1 levels (rs4767027) was associated with any other phenotypes across more than 5,000 outcomes, as catalogued in PhenoScanner,20 which catalogues associations of SNPs with outcomes from all available GWASs. We found that the OAS1 cis- pQTL was strongly associated with circulating OAS1 levels (P=6.20×10−26) in serum, whereas it was not associated with any other traits or protein levels (Bonferroni-corrected P<5.0×10−5). These findings reduce the possibility that the MR estimate of the effect of OAS1 on COVID-19 outcomes is due to horizontal pleiotropy. Finally, except for the susceptibility outcome, the effect of rs4767027 did not demonstrate evidence of heterogeneity across COVID-19 Host Genetics Initiative GWAS meta-analyses (Table 1).
Next we identified an independent SNP associated with OAS1 circulating protein levels, which was not at the OAS1 locus and is thus as trans-SNP (rs62143197, P value for association with OAS1 levels =7.10 x 10−21). Due to strong association with many other proteins, such as annexin A2 (P=5.62 x 10−237) and small ubiquitin-related modifier 3 (P=9.12 x 10−178), including this trans-SNP could introduce bias from potential horizontal pleiotropy effects and was thus not considered in the MR analyses.
Similarly, using the cis-pQTL for IL10RB (rs2834167), we found that a one standard deviation increase in circulating IL10RB level was associated with a decreased odds for hospitalized COVID-19 (OR = 0.51; 95% CI: 0.37-0.70, P=3.61×10−5) and very severe COVID-19 (OR=0.48; 95% CI: 0.31-0.75, P=1.08×10−3). However, circulating IL10RB protein level was not associated with COVID-19 susceptibility. Using PhenoScanner, we could not find evidence of pleiotropic effects of the cis-pQTL for IL10RB. The IL10RB cis-pQTL also showed a homogeneous effect across cohorts for the three COVID-19 outcomes (Table 1).
We found that cis-pQTLs for ABO (rs505922) and LBP (rs2232613) were strongly associated with several other proteins, suggesting potential pleiotropic effects (Table S4). Given their known involvement in multiple physiological processes, these results were expected, but highlights that these MR analyses may suffer from significant bias from horizontal pleiotropy.
Protein-Altering Variants
cis-pQTLs may be, or may be in LD with, protein altering variants (PAVs)13 and thus influence MR-prioritized proteins if these PAVs alter the affinity and binding of the molecules used to quantify their level. We thus assessed if the cis-pQTLs for the MR-prioritized proteins were PAVs, or in LD (R2>0.8) with PAVs, and if so, conditional analyses for cis-pQTLs, conditioning on their PAVs in LD, were undertaken to test if the cis-pQTL remained associated with the protein level. rs2834167 (IL10RB) and rs2232613 (LBP) are nonsense and missense variants, respectively, therefore both could be subject to potential binding effects. rs4767027 (OAS1) is an intronic variant, which is in LD with missense variants, however, the missense variants in LD with rs4767027 were absent in the original imputed dataset from Sun et al, therefore, no assessment for aptamer binding effects could be made. rs505922 (ABO) is not in LD with known missense variants.
Colocalization Studies
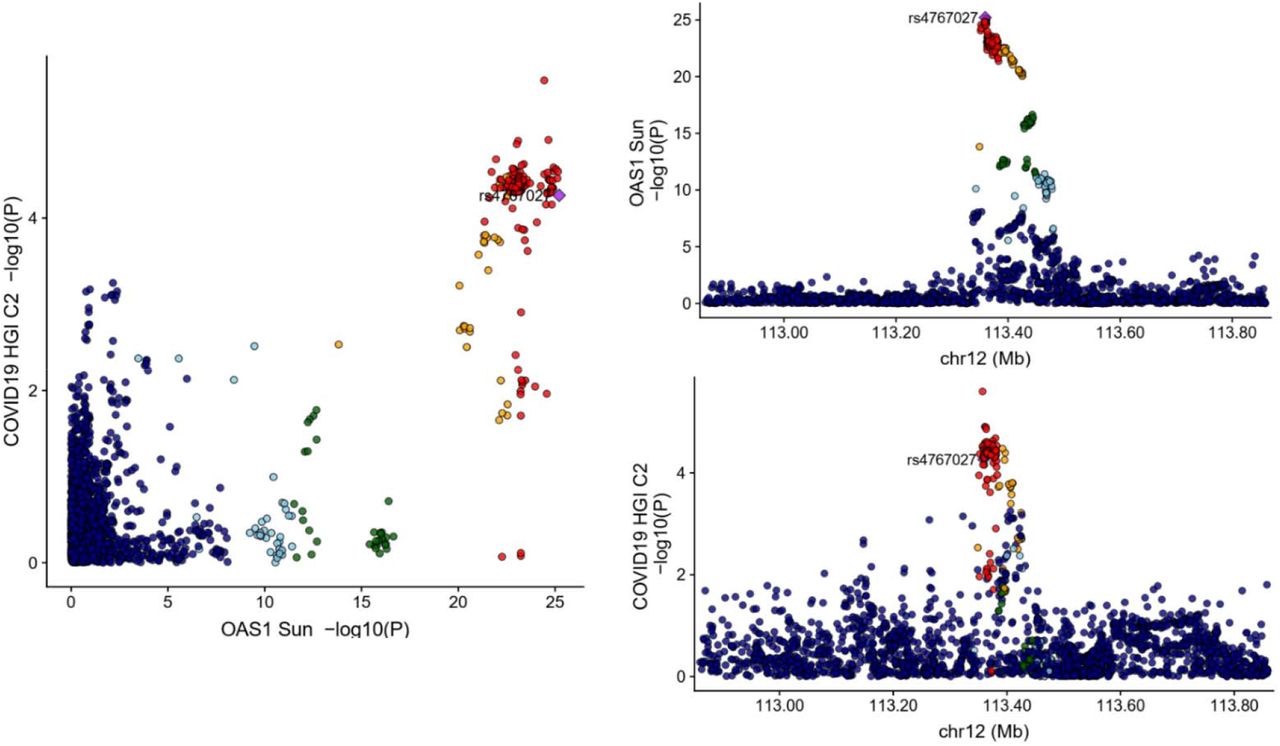
To test whether confounding due to LD may have influenced the estimated effect of circulating OAS1 on the three different COVID-19 outcomes, we tested the probability that the genetic determinants of OAS1 circulating protein level were shared with the three COVID-19 outcomes using colocalization analyses. These were performed using coloc, a Bayesian statistical test implemented in the coloc R package.11 We found that the posterior probability that OAS1 levels and COVID-19 outcomes shared a causal signal (the posterior probability for hypothesis 4 in coloc, PP4) in the 1Mb locus around the cis-pQTL rs4767027 was 0.75 for very severe COVID-19 (Figure 2), 0.72 for hospitalization due to COVID-19 (Figure 3), and 0.88 for COVID-19 susceptibility (Figure 4). This suggests that there is likely one shared causal signal for OAS1 circulating protein levels and COVID-19 outcomes.
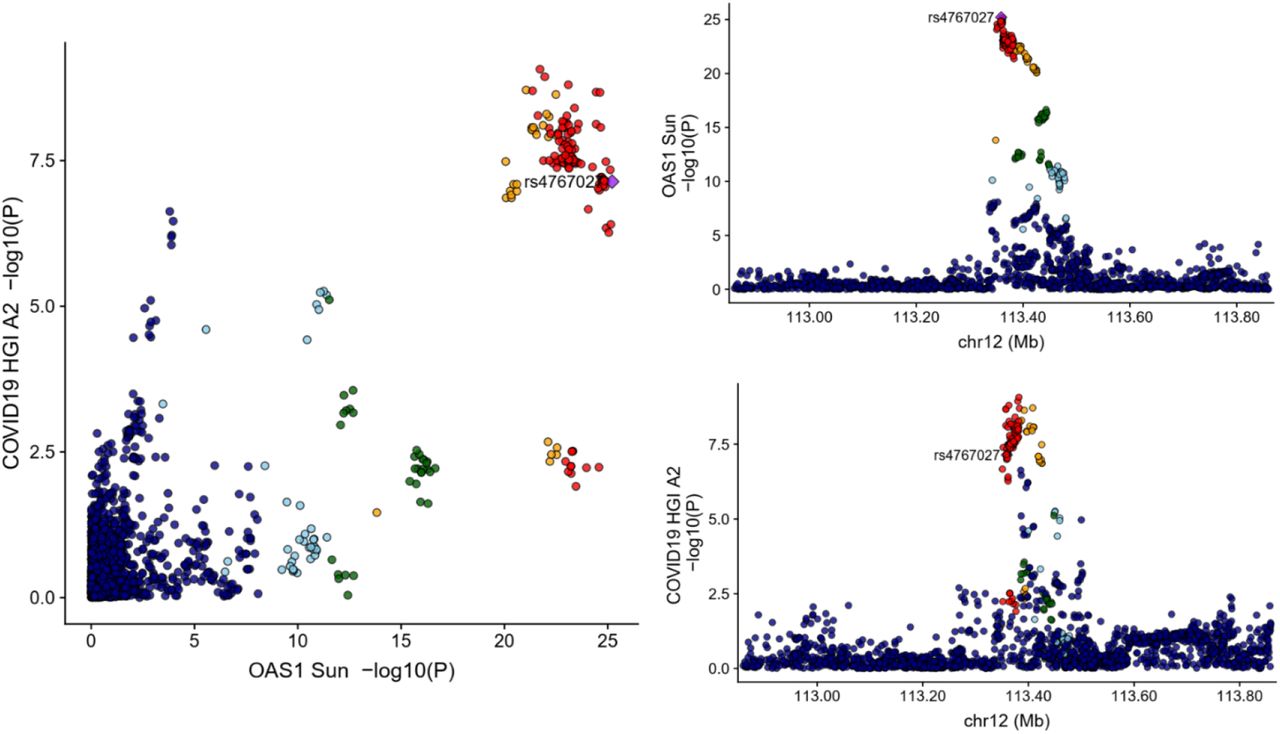
Created using locuscomparer R package,51 the cis-pQTL rs4767027 is shown as a purple diamond. The GWAS used for OAS1 level was Sun et al. LD is based on the 1KG European panel, red dots represent SNPs having LD R2>0.8 with the cis-pQTL. Yellow dots represent SNPs having LD R2>0.6 with the cis-pQTL. Coloc results were PP4=0.75; PP3=0.25.

Created using locuscomparer R package, the cis-pQTL rs4767027 is shown as a purple diamond. The GWAS used for OAS1 level was Sun et al. LD is based on the 1KG European panel, red dots represent SNPs having LD R2>0.8 with the cis-pQTL. Yellow dots represent SNPs having LD R2>0.6 with the cis-pQTL. Coloc results were PP4=0.72; PP3=0.28.

Created using locuscomparer R package, the cis-pQTL rs4767027 is shown as a purple diamond. LD is based on the 1KG European panel, red dots represent SNPs having LD R2>0.8 with the cis-pQTL. Yellow dots represent SNPs having LD R2>0.6 with the cis-pQTL. Coloc results were PP4=0.88; PP3=0.10.
Colocalization of circulating LBP levels and the three COVID-19 outcomes showed strong colocalization between LBP levels and hospitalization due to COVID-19 (PP4 = 0.98, Figure S2), but lack of colocalization between LBP level and COVID-19 susceptibility (PP4 = 0.48, Figure S1) and very severe COVID-19 (PP4 = 0.07, Figure S3). Colocalization of ABO levels and different COVID-19 outcomes showed no colocalization between ABO level and different COVID-19 outcomes (posterior probability of single shared signal = 0.03, 0.02 and 0.005 for ABO level and COVID-19 susceptibility, hospitalization due to COVID-19 and very severe COVID-19, respectively). This was likely due to the multiple independent causal signals at the ABO locus that could not be assessed by coloc (Figures S4-6). We were unable to perform colocalization analysis for IL10RB due to a lack of genome-wide summary level data from the original proteomic GWAS.
eQTL studies
We next assessed whether the cis-pQTL, rs4767027 for circulating OAS1 levels, influenced expression of OAS1 levels across different tissues using the data from the GTEX consortium.21 We found that rs4767027 was an eQTL for OAS1 in whole blood (P=2.3 x 10−173), skin (P=4.2 x 10−149), adipose (P=1.4 x 10−147) and, critically, in lung (P=4.5 x 10−139). It is also an eQTL for the nearby genes OAS3 (P=1.1 x 10− 78) and OAS2 (P=6.9 x 10−21) in cultured fibroblasts, spleen, pancreas and whole blood. OAS1, OAS2 and OAS3 all encode enzymes in the same 2’, 5’ oligoadenylate synthase family that bind to and activate RNase L, leading to viral RNA degradation and inhibit viral replication.22 Investigating IL10RB, we found that its cis-pQTL is an eQTL for IL10RB in the brain (P=7.9×10−34), heart (P=4.9×10−22) and lung (P=1.2×10−13).21 Figure 5 summarizes data supporting the role of OAS1 in COVID-19 outcomes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Note that this Figure is not intended to be a directed acyclic graph.
Discussion
Disease-specific therapies are needed to reduce the morbidity and mortality associated with COVID-19 outcomes. In this large-scale two-sample MR study of 955 proteins assessed for three COVID-19 outcomes in up to 17,600 cases and 1.3 million controls, we provide evidence that increased OAS1 and IL10RB levels are strongly associated with reduced COVID-19 susceptibility, hospitalization and severe outcomes. For OAS1 the protective effect size was particularly large, such that a 50% decrease in the odds of very severe COVID-19 was observed per standard deviation increase in OAS1 circulating levels. Since therapies exist that activate OAS1, repositioning them as potential COVID-19 treatments should be prioritized.
Each of the four MR-identified proteins has orthogonal evidence implicating their role in viral infections. For example, the OAS gene cluster on chromosome 12 includes OAS1, OAS2 and OAS3, which share significant homology and differ only in their number of OAS units. These genes all encode similar enzymes that are essential to host responses to viral infections. They are induced by interferons and activate latent RNase L, resulting in direct viral and endogenous RNA destruction, as demonstrated in in-vitro studies.23 They also increase expression of both IRF3 and IRF7, both genes involved in interferon gene expression. As an interferon stimulated gene24, OAS1 polymorphisms have been involved in host immune response to several classes of viral infection including influenza25, herpes simplex26, hepatitis C, West Nile27 Dengue28, and SARS-CoV29 viruses. Given that OAS1 is an intracellular enzyme leading to viral RNA degradation, it is probable that the circulating levels of this enzyme reflect intracellular levels of this protein. While further experiments are required to better understand the role of OAS1 in SARS-CoV-2 infection, it is likely that the site of action of this enzyme upon the virus would be in the intracellular compartment.
Molecules currently exist which can increase OAS1 activity. Interferon beta-1b, which is an OAS1 agonist,30 is currently used to treat multiple sclerosis and has been shown to induce OAS1 expression in blood.31 A recent randomized controlled trial showed that interferon beta-1b combined with lopinavir-ritonavir reduced mortality in MERS-CoV infections, another human coronavirus.32 While lopinavir-ritonavir, rather than interferon beta-1b, may have been responsible for this mortality reduction a recent COVID-19 randomized controlled trial from the RECOVERY group showed no benefit from lopinavir-ritonavir in multiple COVID-19 outcomes.33 Indeed, according to clinicaltrials.gov, there are multiple on-going phase II randomized controlled trials testing interferon beta-1b for COVID-19 outcomes. In-vitro evidence also exists demonstrating that pharmacological inhibition of phosphodiesterase-12, which normally degrades the OAS enzymes, potentiates this OAS-mediated antiviral activity.22 PDE-12 inhibitors potentiate the action of OAS1, 2 and 3.34 Interestingly other coronaviruses in the same betacoronavirus family as SARS-CoV-2 have been shown to produce viral proteins that degrade the OAS family of proteins, and antagonize RNase-L activity, leading to evasion of the host immune response.35,36 Thus classes of medications currently exist that lead to increased OAS1 levels and could be explored for their effect upon COVID-19 outcomes.
Importantly OAS2 and OAS3 circulating levels were not assessed by any of the protein level GWASs and the lead cis-pQTL for OAS1 at this locus likely influences OAS2 and OAS3 expression levels. Thus, it remains possible that, in addition to influencing OAS1 levels, the lead cis-pQTL may also influence the abundance of OAS2 and OAS3. In a recent transcription-wide association study pre-print from the GenOMICC program19, OAS3 demonstrated differences in predicted expression across tissues in critically ill COVID-19 patients. Nevertheless, all three genes share similar functions, structure and activity, highlighting that increases in the activity, or level of this family of proteins are likely to protect against COVID-19 susceptibility and severity.37
IL10RB encodes for the beta subunit of the IL10 receptor, and is part of a cluster of immunologically important genes including IFNAR1 and IFNAR2, both recently implicated in severe COVID-19 pathophysiology.38 IFNAR1 and 2 encode the interferon alpha/beta receptor subunits 1 and 2, respectively. Interestingly, while there exists a cis-pQTL strongly associated with IFNAR1 levels, it was not associated with any of the COVID-19 outcomes (P ∼ 0.5). Further, IFNAR1 had no trans-pQTLs identified, which means that the IL10RB cis-pQTL does not likely reflect IFNAR1 levels. However, since IFNAR2 was not measured in any proteomic studies, we could not test the effect of its circulating levels on COVID-19 outcomes. IL10RB mediates IL10 anti-inflammatory activity through its downstream inhibitory effect on many well-known pro-inflammatory cytokines such as janus kinases and STAT1.39 While overexpression of IL10 has been involved in the persistence of multiple chronic bacterial infections such as tuberculosis,40 its role remains poorly understood in acute infections. In sepsis, a disease state characterized by high levels of cytokine activity and a rise in multiple biomarkers associated with inflammation, there is also a well-established paradoxical increase in anti-inflammatory IL10 production by leukocytes, especially in the early stage of the disease.41 Most importantly, while in a normal physiological state, IL10 is usually only produced at a low level by neutrophils, its production is strongly upregulated by IL4, itself upregulated by lipopolysaccharides (LPS) when they bind LBPs.42,43 While LPS’s are well-known for their role in triggering gram-negative bacterial sepsis, their role in other acute infections and respiratory diseases is likely broader, and involves complex sequences of cytokine signaling.44–47 Nevertheless, as our MR studies showed that LBP and IL10RB protein levels affected COVID-19 outcome with a concordant effect direction, and given the known role of overt inflammation in COVID-19 morbidity, this pathway likely deserves more investigation.
This study has limitations. First, we used MR to test the effect of circulating protein levels measured in a non-infected state. This is because the effect of the cis-pQTLs upon circulating proteins was estimated in individuals who had not been exposed to SARS-CoV-2. Once a person contracts SARS-CoV-2 infection, levels of circulating proteins could be altered and this may be especially relevant for cytokines such as IL10 (which binds to IL10RB), whose levels may reflect host response to the viral infection. Thus, the results presented in this paper should be interpreted as an estimation of the effect of circulating protein levels, when measured prior to infection. On-going studies will help to clarify if the same cis-pQTLs influence circulating protein levels during infection. As emphasized above, these circulating protein levels may also reflect intracellular biological processes, such as may be the case for OAS1. Second, this study suffers a high false-negative rate. Our goal was not to identify every circulating protein influencing COVID-19 outcomes, but rather to provide evidence for few proteins with strong cis-pQTLs since these proteins are more likely to be robust to the assumptions of MR studies. Future large-scale proteomic studies with more circulating proteins properly assayed should help to overcome these limitations. Third, the colocalization analyses in this study are also subject to potential bias from mixed ethnicity from the COVID-19 outcome GWAS. Previous studies have already established statistical colocalization methods may be overly sensitive to the accurate estimation of LD structure, and thus fail to colocalize48 due to small differences in LD, or the presence of more than one causal signal at a locus. Nevertheless, the locus zoom plots (Figures 2-4) show similar signals arising from the same genomic regions for the protein and COVID-19 outcomes. Fourth, most MR studies assume a linear relationship between the exposure and the outcome. Thus, our findings would not identify proteins whose effect upon COVID-19 outcomes has a clear threshold effect.
In conclusion, we have used genetic determinants of circulating protein levels and COVID-19 outcomes obtained from large-scale studies and found compelling evidence that OAS1 has a protective effect on COVID-19 susceptibility and severity. Known pharmacological agents that increase OAS1 levels (such as interferon beta-1b and PDE12 inhibitors) could be explored for their effect on COVID-19 outcomes.
Methods
Cohorts
pQTL GWAS
We systematically identified pQTL associations from six large proteomic GWASs.13–18 Each of these studies undertook proteomic profiling using either SomaLogic somamer technology, or O-link proximal extension assays.
COVID GWAS and COVID-19 Outcomes
To assess the association of cis-pQTLs with COVID-19 outcomes, we used the largest COVID-19 meta-analytic GWAS to date from the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/results/). The participating cohorts provided GWAS summary statistics data from either large-scale biobanks (N = 18), hospital-based cohorts (N = 13), or direct-to-consumer genotyping companies (N = 1), which were meta-analyzed across ancestries and made publicly available. For our study, we used three of these GWAS meta-analyses, based on sample size and clinical relevance. These outcomes were very severe COVID-19, hospitalized patients with COVID-19, and susceptibility to COVID-19 (named A2, B2, and C2, respectively in the COVID-19 Host Genetics Initiative).
Very severe COVID-19 cases were defined as hospitalized individuals with COVID-19 as the primary reason for hospital admission with laboratory confirmed SARS-CoV-2 infection (nucleic acid amplification tests or serology based), and death or respiratory support (intubation, continuous positive airway pressure, Bilevel Positive Airway Pressure, or continuous external negative pressure, Optiflow/very high flow Positive End Expiratory Pressure Oxygen). Simple supplementary oxygen (e.g. 2 liters/minute via nasal cannula) did not qualify for case status. Controls were all individuals in the participating cohorts who did not meet this case definition.
Hospitalized COVID-19 cases were defined as individuals hospitalized with laboratory confirmed SARS-CoV-2 infection (using the same microbiology methods as for the very severe phenotype), where hospitalization was due to COVID-19 related symptoms. Controls were all individuals in the participating cohorts who did not meet this case definition.
Susceptibility to COVID-19 cases were defined as individuals with laboratory confirmed SARS-CoV-2 infection, health record evidence of COVID-10 (international classification of disease coding or physician confirmation), or with self-reported infections (e.g. by questionnaire). Controls were all individuals in the participating cohorts who did not meet this case definition.
Two-sample Mendelian randomization
We used two-sample MR analyses to screen and test potential circulating proteins for their role influencing COVID-19 outcomes. In two-sample MR, the effect of SNPs on the exposure and outcome are taken from separate GWASs. This method often improves statistical power, because it allows for larger sample sizes for the exposure and outcome GWAS.49
Exposure definitions: We conducted MR using six large proteomic GWAS studies.13–18 Circulating proteins from Sun et al, Emilsson et al and Pietzner et al were measured on the SOMAlogic platform, Suhre et al, Yao et al and Folkersen et al used protein measurements on the O-link platform. We selected proteins with only cis-pQTLs to test their effects on COVID-19 outcomes, because they are less likely to be affected by potential horizontal pleiotropy. The cis-pQTLs were defined as the genome-wide significant SNPs (P < 5 × 10−8) with the lowest P value within 1 Mb of the transcription start site (TSS) of the gene encoding the measured protein.8 We selected one cis-pQTL per protein per each study. We included the same proteins represented by different cis-pQTLs from different studies in order to cross examine the findings. For cis-pQTLs that were not present in the COVID-19 GWAS, SNPs with LD R2>0.8 and with minor allele frequency (MAF) < 0.42 were selected as proxies, but MAF > 0.3 was used for palindromic alignment for proxy SNPs. cis-pQTLs with palindromic effects and with minor allele frequency (MAF) > 0.42 were removed prior to MR to prevent allele-mismatches. Bonferroni correction was used to control for the total number of proteins tested using MR. We recognize that this is an overly conservative correction, given the non-independence of the circulating proteins, but such stringency should reduce false positive associations. MR analyses were performed using the TwoSampleMR package in R,50 using Wald ratio to estimate the effect of each circulating protein on each of the three COVID-19 outcomes. After matching of the cis-pQTLs of proteins with COVID-19 GWAS and the removal of palindromic SNPs, a total of 537 proteins (543 directly matched IVs and 22 proxies) from Sun et al, 750 proteins (731 directly matched IVs and 20 proxies) from Emilsson et al, 94 proteins (85 directly matched IVs and 7 proxies) from Pietzner et al, 74 proteins (72 directly matched IVs) from Suhre et al, 24 proteins (24 directly matched IVs) from Yao et al and 13 proteins (12 directly matched IVs and 1 proxy) from Folkersen et al were used as instruments for the MR analyses across the three COVID-19 outcomes (Table S5-7).13–18
Pleiotropy assessments
A common pitfall of MR is horizontal pleiotropy, which occurs when the genetic variant affects the outcome via pathways independent of circulating proteins. The use of circulating protein cis-pQTLs greatly reduces the possibility of pleiotropy, for reasons described above. We also searched in the PhenoScanner database, a large catalogue of observed SNP-outcome relationships involving > 5,000 GWAS done to date to assess potentially pleiotropic effects of the cis-pQTLs of MR prioritized proteins, by testing the association of cis-pQTLs with other circulating proteins (i.e. if they were trans-pQTLs to other proteins or traits). For cis-pQTLs of MR prioritized proteins, if they were measured on SOMAlogic platform, we assessed the possibility of potential aptamer-binding effects (where the presence of protein altering variants may affect protein measurements). We also checked if cis-pQTLs of MR prioritized proteins had significantly heterogeneous associations across COVID-19 populations in each COVID-19 outcome GWAS.
Colocalization analysis
Finally, we tested colocalization of the genetic signal for the circulating protein and each of the three COVID-19 outcomes using colocalization analyses, which assess potential confounding by LD. Specifically, for each of these MR significant proteins with genome-wide summary data available, for the proteomic GWASs, a stringent Bayesian analysis was implemented in coloc R package to analyze all single nucleotide variants (SNV) with MAF > 0.01 in 1MB genomic locus centered on the cis-pQTL. Colocalizations with posterior probability for hypothesis 4 (PP4, that there is an association for both protein level and COVID-19 status and they are driven by the same causal variant) > 0.5 were considered likely to colocalize (which means the highest posterior probability for all 5 coloc hypotheses), and PP4 > 0.8 was considered to be highly likely to colocalize.
Ethics declarations
All cohorts contributing data to this study received ethics approval from their respective ethics review board.
Data Availability
Data from proteomics studies are available from the referenced peer-reviewed studies or their corresponding authors, as applicable. Summary statistics for the COVID-19 outcomes are publicly available for download on the COVID-19 Host Genetics Initiative website (www.covid19hg.org).
Data availability
Data from proteomics studies are available from the referenced peer-reviewed studies or their corresponding authors, as applicable. Summary statistics for the COVID-19 outcomes are publicly available for download on the COVID-19 Host Genetics Initiative website (www.covid19hg.org).
Author contributions
Conception and design: SZ, GBL, TN and JBR. Data analyses: SZ and TN. Manuscript writing: SZ, GBL, TN and JBR. Data acquisition: TN, GBL, DM, DEK, JA, MA, NK, ZA, NR, MB, CG, XX, CT, BV, VF and JBR. Interpretation of data: SZ, GBL, TN, YC, DEK, VF and JBR. Intellectual contribution to the manuscript: SZ, GBL, TN, YC, VF, VM, DEK and JBR. All authors were involved in preparation of the further draft of the manuscript and revising it critically for content. All authors gave final approval of the version to be published. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Footnotes
Funding: The Richards research group is supported by the Canadian Institutes of Health Research (CIHR: 365825; 409511), the Lady Davis Institute of the Jewish General Hospital, the Canadian Foundation for Innovation (CFI), the NIH Foundation, Cancer Research UK, Genome Québec, the Public Health Agency of Canada and the Fonds de Recherche Québec Santé (FRQS). TN is supported by Research Fellowships of Japan Society for the Promotion of Science (JSPS) for Young Scientists. JBR is supported by a FRQS Clinical Research Scholarship. SZ is supported by a CIHR fellowship and a FRQS fellowship. GBL is supported by the a CIHR scholarship, and a joint FRQS and Québec Ministry of Health and Social Services scholarship. Support from Calcul Québec and Compute Canada is acknowledged. TwinsUK is funded by the Welcome Trust, Medical Research Council, European Union, the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. These funding agencies had no role in the design, implementation or interpretation of this study. VM is supported by a Canada Excellence Research Chair. The Kaufmann lab is supported by the CIHR, NIH, CFI, AmFAR and FRQS.
Disclosures: JBR has served as an advisor to GlaxoSmithKline and Deerfield Capital.
Reference updated
References



