
Abstract
Molecular testing and surveillance of the spread and mutation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are critical public health measures to combat the pandemic. There is an urgent need for methods that can rapidly detect and sequence SARS-CoV-2 simultaneously. Here we describe a method for multiplex isothermal amplification of the SARS-CoV-2 genome in 20 minutes. Based on this, we developed NIRVANA (Nanopore sequencing of Isothermal Rapid Viral Amplification for Near real-time Analysis) to detect viral sequences and monitor mutations in multiple regions of SARS-CoV-2 genome for up to 96 patients at a time. NIRVANA uses a newly developed algorithm for on-the-fly data analysis during Nanopore sequencing. The whole workflow can be completed in as short as 3.5 hours, and all reactions can be done in a simple heating block. NIRVANA provides a rapid field-deployable solution of SARS-CoV-2 detection and surveillance of pandemic strains.
Main
The novel coronavirus disease (COVID-19) pandemic is one of the most serious challenges to public health and global economy in modern history. SARS-CoV-2 is a positive-sense RNA betacoronavirus that causes COVID-191. It was identified as the pathogenic cause of an outbreak of viral pneumonia of unknown etiology in Wuhan, China, by the Chinese Center for Disease Control and Prevention (CCDC) on Jan 7, 20202. Three days later, the first SARS-CoV-2 genome was released online through a collaborative effort by scientists in universities in China and Australia and Chinese public health agencies (GenBank: MN908947.3)3. One week after the publication of the genome the first diagnostic detection of SARS-CoV-2 using real-time reverse transcription polymerase chain reaction (rRT-PCR) was released by a group in Germany4.
To date, rRT-PCR assays of various designs, including one approved by the US Centers for Disease Control and Prevention (US CDC) under emergency use authorization (EUA)5, have remained the predominant diagnostic method for SARS-CoV-2. Although proven sensitive and specific for providing a positive or negative answer, rRT-PCR provides little information on the genomic sequence of the virus, knowledge of which is crucial for monitoring how SARS-CoV-2 is evolving and spreading and ensuring successful development of new diagnostic tests and vaccines. To this end, samples need to go through a separate workflow–typically Illumina shotgun metagenomics or targeted next-generation sequencing (NGS)6. Because NGS requires complicated molecular biology procedures and high-value instruments in centralized laboratories, it is performed in < 1% as many cases as rRT-PCR, evidenced by the number of genomes in the GISAID database (39,954) and confirmed global cases tallied by the WHO (6,535,354) as of June 5, 2020.
Both rRT-PCR and NGS are sophisticated techniques whose implementation is contingent on the availability of highly-specialized facilities, personnel and reagents. These limitations could translate into long turn-over time or inadequate access to tests even in developed countries. To overcome these issues and accelerate COVID-19 testing, several PCR-free nucleic acid detection assays have been proposed as point-of-care replacements of rRT-PCR. Chief among them is reverse transcription coupled loop-mediated isothermal amplification (RT-LAMP7), which has been used for rapid detection of SARS-CoV-2 RNA using colorimetric readout8,9. RT-LAMP can also be coupled to CRISPR-Cas1210 to increase specificity using lateral flow readout11,12. On the sequencing front, the pocket-sized Oxford Nanopore MinION sequencer has been used for rapid pathogen identification in the field13,14. Because MinION offers base calling on the fly, it is an attractive platform for consolidating viral nucleic acid detection by PCR-free rapid isothermal amplification and viral mutation monitoring by sequencing.
However, there are several challenges for an integrated point-of-care solution based on RT-LAMP and Nanopore sequencing. RT-LAMP requires a complex mixture of primers that increases the chance of non-specific amplification and makes it difficult to multiplex. Additionally, LAMP amplicons used for SARS-Cov-2 detection are short (<180 bp)9,11. Sequencing singleplex short amplicons not only fails to take advantage of the long-read and sequencing throughput (∼10 Gb) of the minION flow cell, it is also prone to false negative reporting due to amplification failure. To the best of our knowledge, no multiplexed isothermal amplification of SARS-CoV-2 has been reported. Nanopore sequencing has its own caveats too. Due to its relatively high basecalling error, sophisticated algorithms are needed for accurate variant detection15,16. New bioinformatics tools are also needed to accurate call the presence of viral sequences (substituting for rRT-PCR) and analyze virus mutations (substituting for NGS).
Here we developed isothermal recombinase polymerase amplification (RPA) assays to simultaneously amplify multiple regions (up to 1365 bp) of the SARS-CoV-2 genome. This forms the basis for an integrated workflow to detect the presence of viral sequences and monitor mutations in multiple regions of the genome in up to 96 patients at a time (Fig. 1a). We developed a bioinformatics pipeline for on-the-fly analysis to reduce the time to answer and sequencing cost by stopping the sequencing run when data are sufficient to provide confident answers.
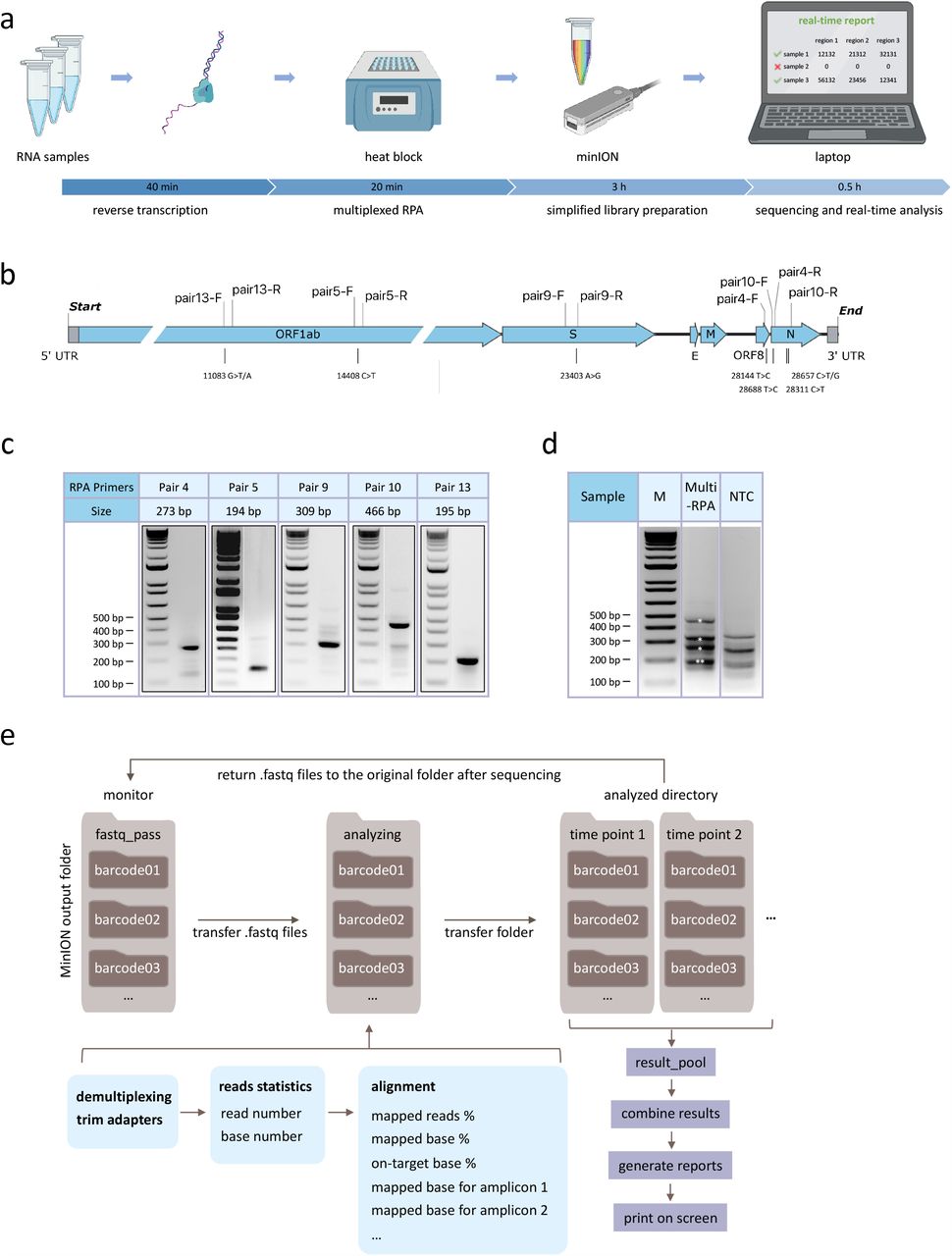
a, Schematic representation of NIRVANA. RNA samples were subjected to reverse transcription, followed by multiplex RPA to amplify multiple regions of the SARS-CoV-2 genome. The amplicons were
b, purified and prepared to the Nanopore library using an optimized barcoding library preparation protocol. In the end, the sequencing was performed in the pocket-sized Nanopore MinION sequencer and sequencing results were analyzed by our algorithm termed RTNano on the fly.
c, The RPA primers used in this study were plotted in the SARS-CoV-2 genome. The corresponding prevalent variants were labeled under the genome.
d, Agarose gel electrophoresis results of singleplex RPA with selected primers shown next a molecular size marker. The amplicons range from 194 bp to 466 bp.
e, Agarose gel electrophoresis results of multiplex RPA. All of the five amplicons were shown in the gel with correct size (asterisks, note that pair 5 and 13 have similar sizes). The no template control (NTC) showed a different pattern of non-specific amplicons. M: molecular size marker.
f, Pipeline of RTNano real-time analysis. RTNano monitors the Nanopore MinION sequencing output folder. Once newly generated fastq files are detected, it moves the files to the analyzing folder and makes a new folder for each sample. If the Nanopore demultiplexing tool guppy is provided, RTNano will do additional demultiplexing to make sure reads are correctly classified. The analysis will align reads to the SARS-CoV-2 reference genome and collect key information, including percentage of mapped reads, percentage of mapped bases, percentage of on-target bases, base number that aligned to each targeted region. As sequencing proceeds, RTNano will merge the newly analyzed results with existing ones to update the current sequencing statistics.
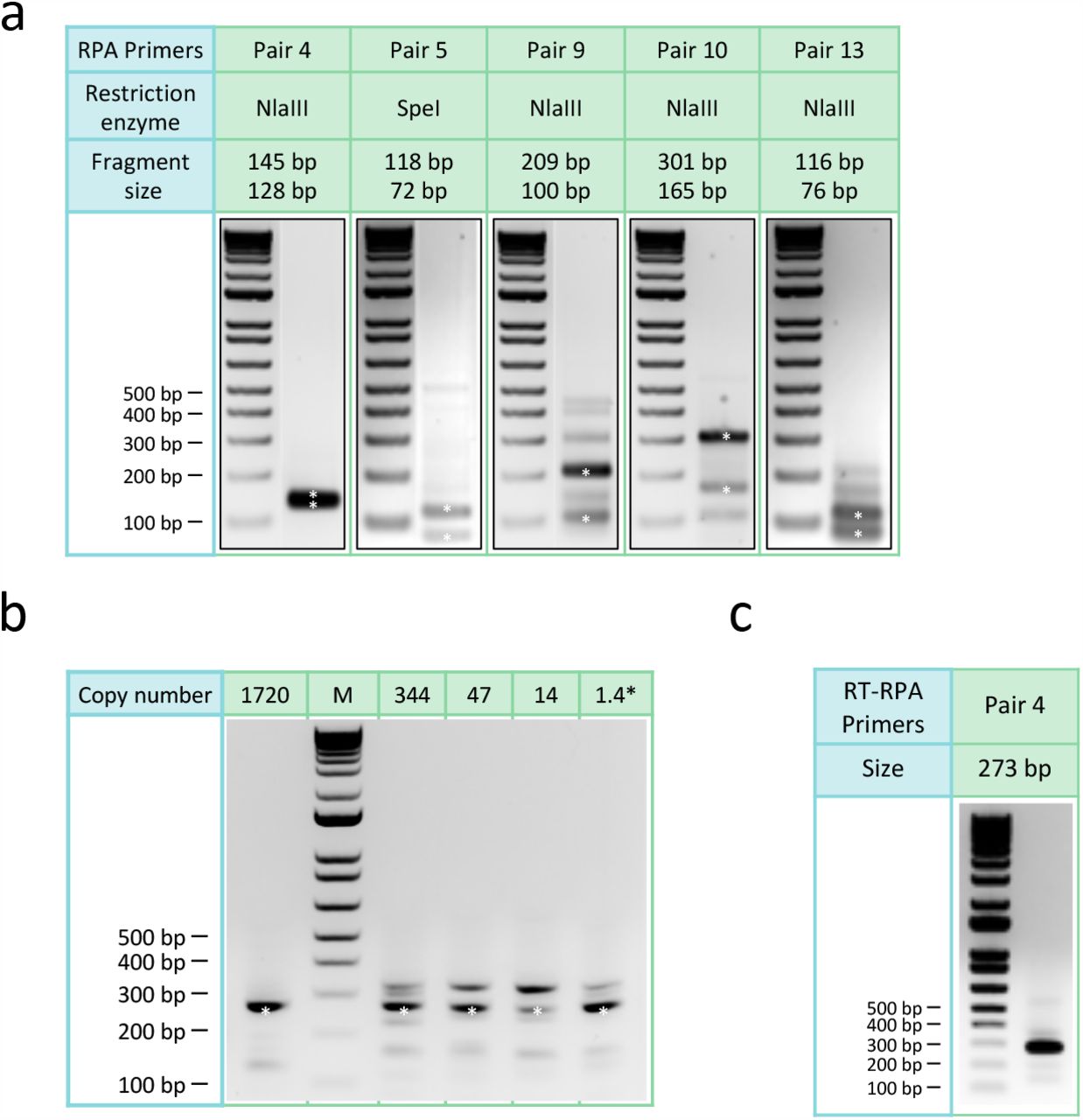
Singleplex RPA was first performed to test its ability to amplify the SARS-CoV-2 genome from a nasopharyngeal swab sample tested positive for the virus using the US CDC assays5 (CT value=21). Sixteen primers were tested in 12 combinations to amplify 5 regions harboring either reported signature mutations17,18 useful for strain classification or mutation hotspots (GISAID, as of March 15, 2020). Five pairs of primers showed robust amplification of DNA of predicted size (range: 194-466 bp) in a 20-min isothermal reaction at 39 °C (Fig. 1b-c). The specificity of all 5 RPA products was verified by restriction enzyme digestion (Supplementary Fig. 1a). The limit of detection of RPA reaches below 10 copies (Supplementary Fig. 1b). Furthermore, we showed that these RPA reactions could be multiplexed to amplify the five regions of the SARS-CoV-2 genome in a single reaction (Fig. 1d), thus significantly simplifying the workflow.
We next performed multiplex RPA using ten COVID-19 positive samples (US CDC assays5, CT value range: 15 to 27.9). Two control samples were also included to test cross-reactivity and specificity. One control sample contained 21 common respiratory pathogens such as influenza virus, coronaviruses (HKU1, 229E, NL63 and OC43), and respiratory syncytial virus, but not SARS-CoV, SARS-CoV-2, or MERSV (see methods), while the other one was a blank control without microorganisms. Multiplex RPA products of the 12 samples were individually barcoded, pooled and prepared into a Nanopore sequencing library using an optimized protocol to save time. The whole workflow from RNA to sequencing takes approximately 4 hours (Fig. 1a).
The barcoded library was sequenced on a Nanopore MinION using a R9.4.1 flow cell. Besides its excellent portability, the MinION also provides real-time base-calling results. To take advantage of this, we developed a bioinformatics pipeline for real-time analysis of reads, termed Real-Time Nanopore sequencing monitor (RTNano) (Fig. 1e). It continuously monitors the output folder during the sequencing run and generates analysis reports in a matter of seconds. To reduce barcode misclassification during demultiplexing of error-prone Nanopore reads, RTNano included an additional round of demultiplexing using stringent parameters (see methods). After detecting new fastq files, RTNano generated a summary report for each demultiplexed sample, including current read number, base number, and alignment details (Fig. 1e). The alignment statistics were used to determine the presence of the virus. Because base calling and demultiplexing may lead to a brief delay, we refer to the workflow as Nanopore sequencing of Isothermal Rapid Viral Amplification for Near real-time Analysis (NIRVANA) (Fig. 1a).
In the Nanopore sequencing run of patient samples, RTNano identified the first positive sample at 15 minutes, and accurately called all 12 samples at 30 mins (Fig. 2a-b). The 10 positive samples had a deep read coverage of all five amplified regions (Fig. 2a-b, d). We found that the two control samples contained reads mapped to some but not all regions (Fig. 2a-b, d). This could be due to either a small chance of barcode hopping in the one-pot adapter ligation step or fortuitous carryover of positive samples despite our best efforts (see methods). Because of the extraordinary sensitivity of RPA, even a few molecules of viral RNA in aerosols could result in productive amplification. Nonetheless, the lack of any read in 2 out of the 5 target regions even after 12 hours of sequencing clearly distinguish control negative samples from positive ones (Fig. 2c). These data show the importance of using multiple independent RPA amplicons to rule out false positives.

a, Real-time analysis results by RTNano collected at 15 min after sequencing started. A control sample bc17 was classified as negative because lack of reads in two targeted regions (the last region is a combination of primer pair 4 and 10). Bc18 had not been analyzed at this time. The rest of the samples were classified as positive for SARS-CoV-2.
b, Real-time analysis results by RTNano collected at 30 min after sequencing started. The other blank control sample bc18 was classified as negative, while classification of the others remained the same.
c, Similar to (b) but the results were collected after 12 hours of sequencing run. The classification of all samples remained the same.
d, IGV plots showing Nanopore sequencing read coverage on the SARS-CoV-2 genome. Bc17 and 18 showed no read mapped to two targeted regions (red boxes), while other samples showed reads covering all of the targeted regions.
e, The sequencing throughput of barcoded samples. Twenty percent of reads were binned to unclassified due to the stringent demultiplexing algorithm.
f, The SNVs detected in multiplex RPA sequencing and their position as shown in the Nextstrain data portal (Nextstrain.org). A total of 16 SNVs were detected from 10 SARS-CoV-2 positive samples.
g, Equipment used in NIRVANA. The whole workflow can be done with one laptop, one Nanopore MinION sequencer, two pipettes, two boxes of pipette tips, and a heating block (using a miniPCR™ mini16 here).
We acquired a total of 1.7 million reads from this barcoded library (Fig. 2e, Supplementary Fig. 2a). After demultiplexing, the reads were distributed relatively evenly among the barcodes (samples). Due to the stringent demultiplexing procedure, 20.3% of the reads were unclassified. RTNano had an integrated function to quickly analyze variants in each sample during sequencing. It detected 16 single nucleotide variants (SNVs) in the ten positive samples and all of them have been reported in GISAID (Fig. 2f, as of June 7, 2020). The reported SNVs suggested that the strains in samples bc13, bc14, and bc15 are close to clade 19B (nt28144 T/C), first identified in Wuhan, China, while the strains in bc16, and bc19-24 are close to clade 20 (nt14408 C/T, 23403 A/G), first becoming endemic in Europe. Given the fact that bc13, bc14, and bc15 were collected early in the pandemic, while the others were more recent, the SNV signature of the strains revealed by NIRVANA is consistent with the pattern of COVID-19 case importation, which initially came from China and shifted to Europe after a ban of flight from China. Prospectively collecting such data regularly could guide public health policy making to better control the pandemic.
To validate the SNVs and compare NIRVANA with conventional RT-PCR amplicon sequencing19, we chose three samples (bc13, bc14 and bc15) to perform multiplex RT-PCR amplicon sequencing on the Nanopore MinION. Variant calling was done by RTNano using the same parameters and the results showed that RT-PCR sequencing confirmed all 3 SNVs detected by NIRVANA (Supplementary Fig. 2b).
We further compared the SNVs of bc13, bc14 and bc15 with their corresponding assembled genome from Illumina sequencing published in GISAID (EPI_ISL_437459 for bc13, EPI_ISL_437460 for bc14, and EPI_ISL_437461 for bc15). All of the three SNVs existed in the assembled genome.
Taken together, NIRVANA provides accurate calling of positive and negative samples on the fly. The whole workflow can be even shorter by performing one-pot RT-RPA (Supplementary Fig. 1c) so that the time from RNA to answer can be as short as 3.5 hours. All molecular biology reactions in the workflow can be done in a simple heating block, and all necessary supplies fit into a briefcase (Fig. 2g, Supplementary Fig. 2c). We expect it to provide a rapid field-deployable solution of pathogen (including but not limited to SARS-CoV-2) detection and surveillance of pandemic strains.
METHODS
RNA samples and primers
Anonymized RNA samples were obtained from Ministry of Health (MOH) hospitals in the western region in Saudi Arabia. The use of clinical samples in this study is approved by the institutional review board (IRB# H-02-K-076-0320-279) of MOH and KAUST Institutional Biosafety and Bioethics Committee (IBEC). Oropharyngeal and nasopharyngeal swabs were carried out by physicians and samples were steeped in 1 mL of TRIzol (Invitrogen Cat. No 15596018) to inactivate virus during transportation. Total RNA extraction of the samples was performed following instructions as described in the CDC EUA-approved protocol using the Direct-Zol RNA Miniprep kit (Zymo Research Cat. No R2070) or TRIzol reagent (Invitrogen Cat. No 15596026) following the manufacturers’ instructions. The Respiratory (21 targets) control panel (Microbiologics Cat. No 8217) was used as controls in multiplex RPA.
Reverse transcription
Reverse transcription of RNA samples was done using either NEB ProtoScript II reverse transcriptase (NEB Cat. No M0368) or Invitrogen SuperScript IV reverse transcriptase (Thermo Fisher Scientific Cat. No 18090010), following protocols provided by the manufacturers. After reverse transcription, 5 units of thermostable RNase H (New England Biolabs Cat. No M0523S) was added to the reaction, which was incubated at 37 °C for 20 min to remove RNA. The final reaction was diluted 10-fold to be used as templates in RPA. All of the web-lab experiments in this study were conducted in a horizontal flow clean bench to prevent contaminations. The bench was decontaminated with 70% ethanol, DNAZap (Invitrogen, Cat no. AM9890) and RNase AWAY (Invitrogen, Cat no. 10328011) before and after use. The filtered pipette tips (Eppendorf epT.I.P.S.® LoRetention series) and centrifuge tubes (Eppendorf DNA LoBind Tubes, Cat. No 0030108051) used in this study were PCR-clean grade. All of the operations were performed carefully following standard laboratory operating procedures.
Singleplex RPA
Singleplex RPA was performed using TwistAmp® Basic kit following the standard protocol. Thirteen pairs of primers covering N gene, S gene, ORF1ab and ORF8 were tested and the corresponding amplicons were purified by 0.8X Beckman Coulter AMPure XP beads (Cat. No A63882) and eluted in 40 µl H2O. The purified amplicons were first analyzed by running DNA agarose gel to check the specificity and efficiency. The most robust five pairs of primers with correct size were further analyzed by NlaIII (NEB Cat. No R0125L) and SpeI (NEB Cat. No R0133L) digestion following standard protocols. For one-pot RT-RPA, 10 U of AMV Reverse Transcriptase (NEB Cat no. M0277S) and 20U of SUPERase•In™ RNase Inhibitor (Invitrogen Cat no. AM2694) were added to a regular RPA reaction mix. The RT-RPA was carried out per the manufacturer protocol.
Multiplex RPA
Multiplexed RPA was done by add all of the five pairs of primers in the same reaction. The total final primer concentration is limited to 2 μM. To achieve an even and robust amplification, we empirically determined the final concentration of the primers as follows: 0.166 µM for each pair-4 primer, 0.166 µM for each pair-5 primer, 0.242 µM for each pair-9 primer, 0.26 µM for each pair-10 primer, 0.166 µM for each pair-13 primer, 29.5 µl of primer free rehydration buffer, 1 µl of 10-fold diluted cDNA, 7µl H2O. The reaction was incubated at 39 °C for 4 min, then vortexed and spun down briefly, followed by a 16-min incubation at 39 °C. The RPA reaction was purified using Qiagen QIAquick PCR purification kit (Qiagen Cat No. 28106).
Library preparation and sequencing
The RPA library preparation was done using Native barcoding expansion kit (Oxford Nanopore Technologies EXP-NBD114) following Nanopore PCR tiling of COVID-19 Virus protocol (Ver: PTC_9096_v109_revE_06Feb2020) with a few changes. We determined the concentration of each RPA samples using a Qubit™ 4 fluorometer with the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific Q32851). The end-prep reaction was done separately in 15 µl volume using 40 ng of each multiplex RPA samples. After that, we followed the same procedures as described in the official protocol. The RT-PCR library preparation was done using Native barcoding expansion kit (Oxford Nanopore Technologies EXP-NBD104) according to the standard native barcoding amplicons protocol. The sequencing runs were performed on an Oxford Nanopore MinION sequencer using R9.4.1 flow cells.
Bioinformatics
RTNano scanned the sequencing folder repeatedly based on user defined interval time. Once newly generated fastq files were detected, it moved the files to the analysis folder and made a new folder for each sample. If the Nanopore demultiplexing tool guppy is provided, RTNano will do additional demultiplexing to make sure reads are correctly classified. The analysis part utilized minimap220 to quickly align the reads to the SARS-CoV-2 reference genome (GenBank: NC_045512). After alignment, RTNano will collect key information, including percentage of mapped reads, percentage of mapped bases, percentage of on-target bases, base number that aligned to every targeted region. With the sequencing continuing, RTNano will merge the newly analyzed result with completed ones to update the current sequencing statistics. Users can use these numbers to determine virus-positive samples in real time. A positive sample is characterized by presence of reads covering all of the targeted regions. RTNano is ultra-fast, a typical analysis with additional guppy demultiplexing of 5 fastq files (containing 4000 reads each) will take ∼10s using one thread in a MacBook Pro 2016 15-inch laptop. Variant calling was performed using samtools (v1.9) and bcftools (v1.9)21. The detected variants were filtered by position (within the targeted regions) and compared with the data in Nextstrain.org as of Jun 2, 2020.
Data Availability
RTNano and sequencing data in this study are available upon request.
Data and materials availability
RTNano and sequencing data in this study are available upon request.
Author contributions
ML and CB performed majority of the experiments related to Nanopore sequencing. CB wrote the code and performed the bioinformatics analysis. CB and ML analyzed the data and wrote the manuscript. ML GM and JX performed molecular biology experiments. FSA, AK, AMH, and NAMA collected clinical samples. SM extracted RNA and performed molecular assays. SH and AP coordinated the clinical samples and molecular testing. ML and CB conceived the study. ML supervised the study.
Funding
The research of the Li laboratory was supported by KAUST Office of Sponsored Research (OSR), under award numbers BAS/1/1080-01 and URF/1/3412-01-01, and special support for KAUST R3T from the office of Vice President of Research and OSR.
Figure legend

a, Agarose gel electrophoresis results of restriction enzyme digestion. The amplicon of pair 5 was digested by SpeI while the others were digested by NlaIII. The digested DNA bands (asterisks) were of expected sizes.
b, Agarose gel electrophoresis results showing the sensitivity of RPA in amplifying the SARS-CoV-2 genome. Primer pair 4 was used in the experiment. Reliable amplification can be achieved with 1.4 copies (calculated from dilution) of the SARS-CoV-2 genome.
c, Agarose gel electrophoresis result of one-pot reverse transcription and RPA reaction using primer pair 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
a, The length distribution and percent reference identity of all samples sequenced. The unclassified reads were short in size and their overall reference identity was lower than the others.
b, IGV plots showing the nt28144 T/C SNV in bc13, 14 and 15 from RPA and RT-PCR Nanopore sequencing. The blue bar represents the C base while the red bar represents the T base. All of the 3 SNVs detected in RPA sequencing were confirmed by RT-PCR sequencing.
c, Equipment used in the NIRVANA workflow. All equipment can be packed into a suitcase.
Acknowledgements
We thank KAUST Rapid Research Response Team (R3T) for supporting our research during the COVID-19 crisis. We thank members of the KAUST R3T for generously sharing materials and advices. We thank members of the Li laboratory, Yeteng Tian, Baolei Yuan, Xuan Zhou, Yingzi Zhang, and Samhan Alsolami for helpful discussions; Marie Krenz Y. Sicat for administrative support. We thank members of the Pain lab, Rahul Salunke and Amit Subudhi for technical assistance.
REFERENCE



