
Abstract
Drug target prioritization for new targets and drug repurposing of existing drugs for COVID-19 treatment are urgently needed for the current pandemic. COVID-19 drugs targeting human proteins will potentially relate to different host responses involving multiple molecular pathways. Here we implemented a novel COVID-19 drug target prioritisation protocol by integrating multi-omics Mendelian randomization (MR) of COVID-19 severity with colocalization. By integrating novel MR and existing evidence, we prioritised 353 candidate drug targets with clinical and/or literature evidence of host-coronavirus interaction and one additional target (ENTPD5) with robust MR and colocalization evidence on COVID-19 severity. We further conducted phenome-wide MR of these prioritised targets on 622 complex diseases in 11 SARS-CoV-2 related tissues, which we identified 726 potential causal effects on other diseases, providing information on potential beneficial and adverse effects. This study demonstrated the value of our integrative approach in prioritising drug targets and mechanisms involved in COVID-19 severity. This provides the first steps towards evaluating intervention targets worthy of follow-up for coronavirus and viral infections.
One Sentence Summary Integrating multi-omic evidence to prioritise potential drug targets and identify molecular mechanisms for COVID-19 severity.
Introduction
The outbreak of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the causative agent of novel coronavirus disease 2019 (COVID-19), has been a global pandemic. It is expected to infect a large portion of the world population with a wide spectrum of manifestations, ranging from completely asymptomatic carriers (1) to critical respiratory failure, multi-organ dysfunction, and death (2). This heterogeneity may relate to different host responses involving multiple human proteins and pathways. Considering the relatively high mortality rate of patients with severe disease (3), search for optimal treatments, such as novel drugs to improve survival rate of severe patients is a key objective to reduce the impact of both the current and potential future coronavirus epidemics.
Currently, both clinical trials and experimental studies are ongoing for developing new drugs or repurposing existing drugs for COVID-19. For example, 1,444 clinical trials are currently at status of recruiting or completed (ClinicalTrial.gov searched on December 6th using COVID-19), including remdesivir (4). In parallel with these drug trials, one recent study identified a set of human proteins interacting with 26 SARS-CoV-2 proteins using affinity purification-mass spectrometry in HEK293 cell line by determining the host proteins which interact with SARS-CoV-2 (5). Another study has identified 44 genes associated with SARS-CoV, a closely related coronavirus with SARS-CoV-2, using mouse models (6), which may provide drug targets for general coronavirus interventions. However, in this rapidly developing situation, apart from some early stage trials (7) and observational studies (8), only dexamethasone has been reported to reduce the death rate significantly in severe hospitalized COVID-19 patients in a large randomized controlled trial - RECOVERY (9). Till 2 October 2020, nearly 318 of 405 therapeutic drugs in development for COVID-19 were tested in clinical trials, but their efficacy is as yet unproven (10)(11).
Genetic, transcriptomic and proteomic (“omics”) data could play a role in supporting efficient drug development for various diseases including COVID-19. Recent studies have begun to support the role of genetics in predicting drug trial success (12). Some multi-omics studies have further demonstrated the value of molecular quantitative trait locus (QTL) studies in repurposing existing targets to additional indications as well as prioritizing novel drug targets (13)(14). One approach to utilising molecular QTL data is Mendelian randomization (MR) (15). MR uses genetic variants as instrumental variables to estimate the effect of an exposure (e.g. measured levels of a protein or a gene transcript) on an outcome (e.g. COVID-19 severity) (16), which could prioritise drug targets cost-effectively. MR can also be applied to identify potential mediators (e.g. coagulation), linking the exposure (e.g. a protein) with the outcome (e.g. COVID-19) in a mediation/two-step MR framework (17). In addition, phenome-wide MR could be applied to identify potential drug repurposing opportunities and adverse effects (18).
Recent GWASs (19)(20)(21) and MR studies (22)(23)(24)(25) utilising data from the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/) and GenOMICC consortium have identified a set of genes (e.g. ABO, OAS1, IFNAR2, IL10RB) associated with various COVID-19 phenotypes. However, there are issues that require consideration: i) some genes and proteins (e.g. ABO) are well-known to be pleiotropic, which may violate the “no pleiotropy” (or “exclusion restriction”) assumption of MR; ii) when comparing severe COVID-19 cases with population controls as an outcome, it is not possible to separate the causal effects of becoming infected from any causal effects on disease progression after infection (despite these potentially being separate mechanisms) (26); iii) collider bias (also known as selection bias, sampling bias or ascertainment bias) (27). Each of these could induce spurious associations between the target and COVID-19. Careful instrument and outcome selection are needed for the drug target MR of COVID-19.
To prioritise potential drug targets and molecular mechanisms for COVID-19 severity as well as identify potential beneficial and adverse effects, we combined genetic association information of 1,002 proteins and 15,944 transcripts from eight studies (28)(29)(30)(31)(32)(33)(34)(35) and applied two-sample MR (36) and colocalization analyses (37) to estimate the causal effects of these targets on COVID-19 severity and 622 complex human traits. To enable rapid queries, results of all analyses are available in an open access online platform (https://epigraphdb.org/covid-19/ctda/).
Results
Summary of the study
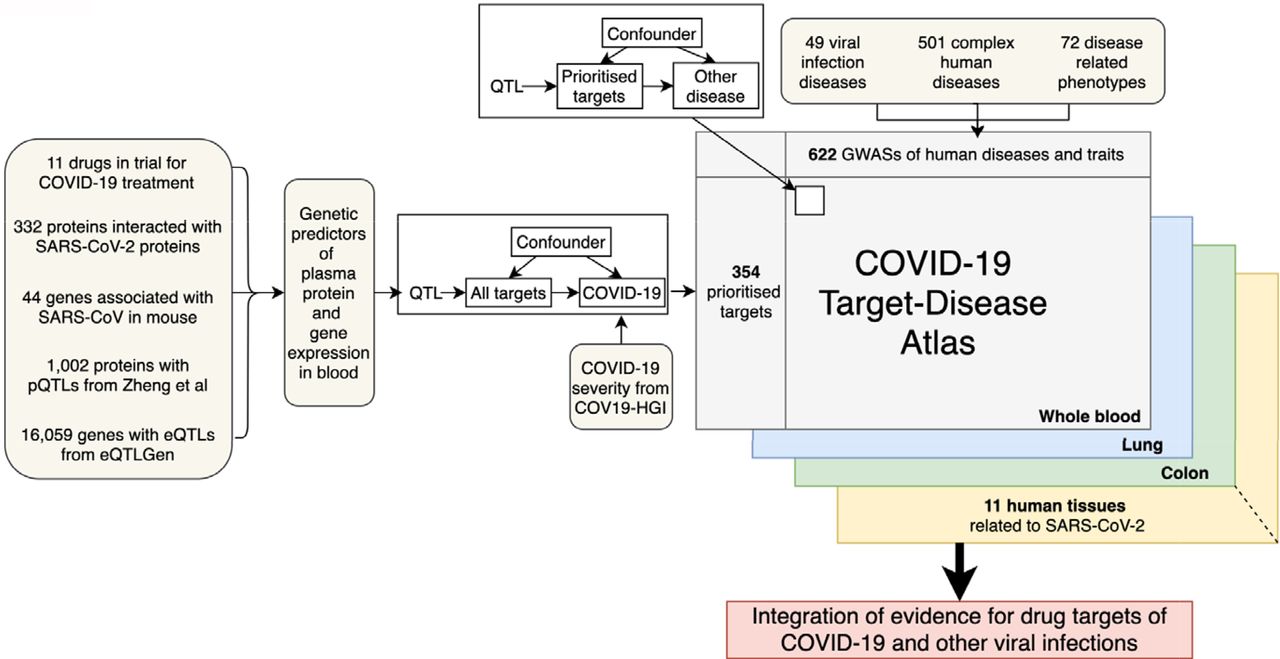
Figure 1 illustrates the design of this study: i) selection of drug targets potentially relevant to COVID-19 from five resources; ii) selection of the COVID-19 severity data and other diseases data; iii) MR analysis of the selected targets on COVID-19 severity; iv) phenome-wide association study of prioritised targets to identify potential beneficial and adverse effects of these targets on 622 complex human traits; v) follow-up mediation/two-step and bidirectional MR analyses of the prioritised targets with MR and colocalization evidence.
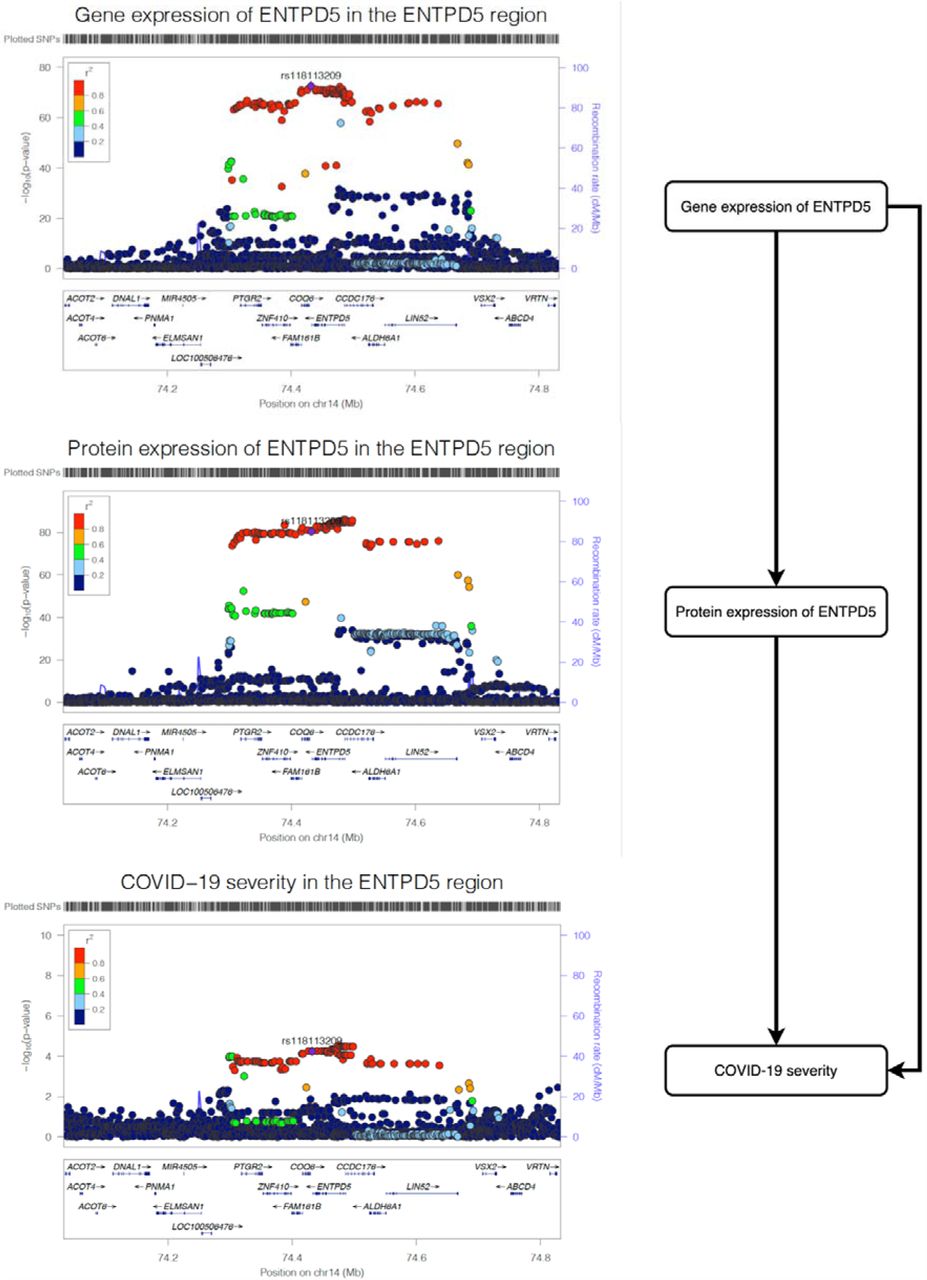
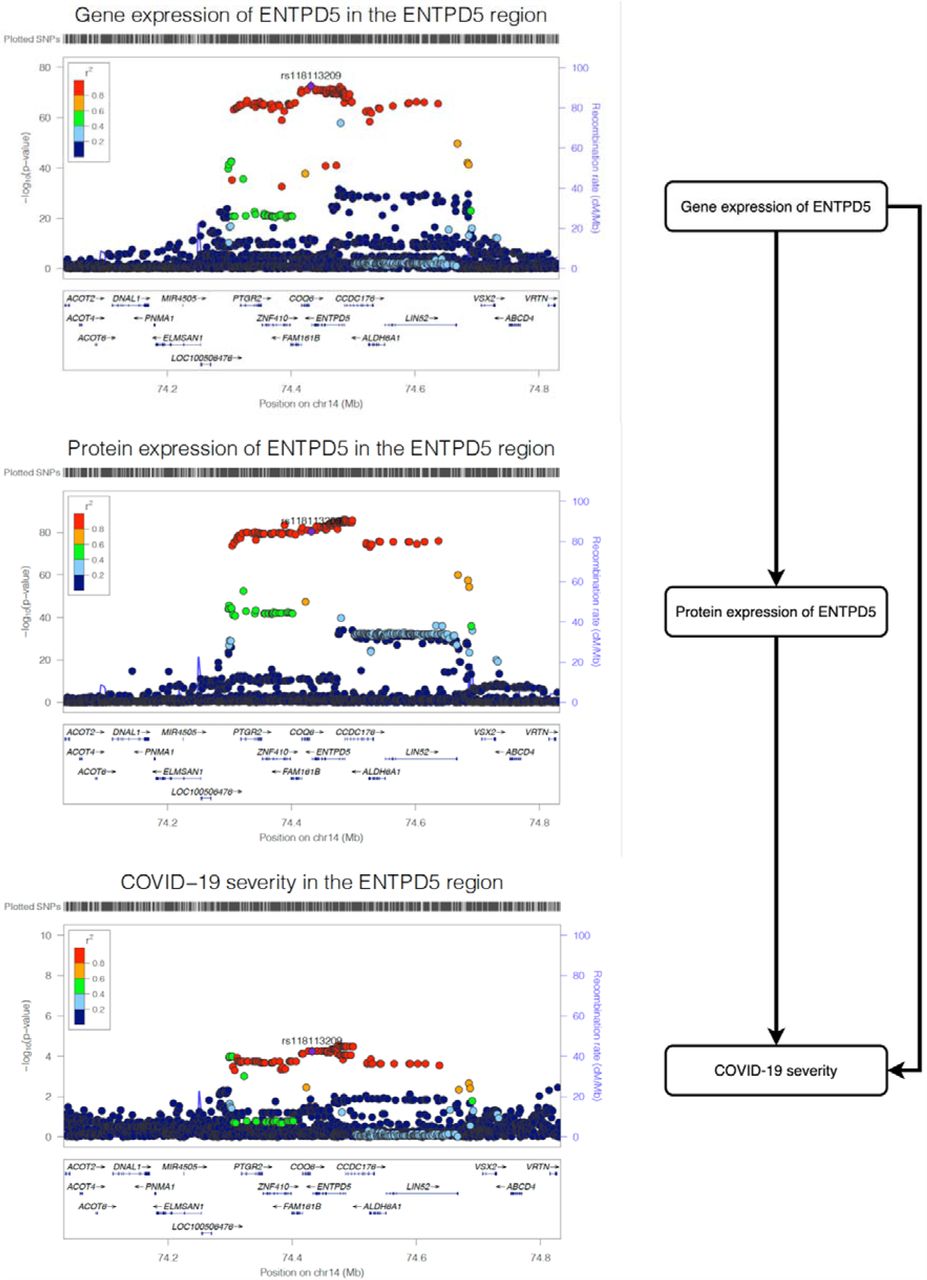
The three regional plots refer to the genetic associations of (A), gene expression of ENTPD5; (B) protein expression of ENTPD5; (C) and COVID-19 severity. The X-axis is the chromosome and position of the ENTPD5 region.
Selection of drug targets for COVID-19
We retrieved drug targets from two resources: i) By using the instrument selection tier system described by Zheng et al (13), we selected 1,002 protein targets with robust genetic predictors and less concerns about pleiotropy (Tier 1 or 2 instruments) from five studies (28)(29)(30)(31)(32) (Supplementary Table 1); ii) we selected 16,059 genes with robust genetic instruments to predict their gene expression levels using data from the eQTLGen consortium (33) (Supplementary Table 2). To further prioritise potential drug targets for COVID-19, we integrated evidence from three resources: i) target genes of 11 drugs under trials for COVID-19 treatment from ClinicalTrials.gov (Supplementary Table 3); ii) 332 human proteins interacting with SARS-CoV-2 proteins in human cell lines (5); iii) 44 genes associated with SARS-CoV in a mouse model (6). After removing duplicates, 380 unique targets were selected (Supplementary Table 4), of which 353 targets have genetic predictors (instruments) in either blood or in any of the 11 SARS-CoV-2 related tissues (data from Zheng et al, eQTLGen and GTEX, Supplementary Table 5). For these targets, both non-pleiotropic cis and trans instruments were considered to maximise the number of targets we included in the MR analyses.
Characteristics of COVID-19 severity
The genetic association data for COVID-19 was obtained from the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/). In total, GWASs of six COVID-19 related phenotypes were provided by the initiative. We excluded three COVID-19 GWASs using population as controls and two additional GWASs using self-reported data as either cases or controls, since these comparisons don’t distinguish between asymptomatic infected, exposed but uninfected and unexposed. Using these data as outcomes for MR, we will not be able to separate the causal effects of exposure to virus, infection and progression of COVID-19 after infection (which may represent separate mechanisms) (Supplementary Figure 1). Even after excluding these studies, we note that collider bias (also known as selection bias, sampling bias or ascertainment bias) could influence our understanding of COVID-19 disease risk and severity (27). Collider bias can induce spurious associations between the exposure (e.g. levels of a target) and the outcome (e.g. COVID-19 severity) (38). For example, COVID-19 testing could be a collider (common consequence) of both asthma and COVID-19 severity, since people with respiratory illnesses may be more likely to seek COVID-19 testing, as are people with more severe symptoms of COVID-19. When we sample individuals based on the collider (COVID-19 testing), that is equivalent to conditioning on the collider (26). This could induce a spurious association between asthma-associated genetic variants (and protein levels) and severity of COVID-19 (Supplementary Figure 2). Despite this limitation, we nevertheless used the GWAS of hospitalized COVID-19 vs. not hospitalized COVID-19 (928 confirmed hospitalized COVID-19 patients, 2,028 confirmed non hospitalised COVID-19 patients) as the dataset to represent COVID-19 severity in our study, since it provides the most meaningful comparison to explore effects on severity. The summary statistics of the genetic associations with COVID-19 severity were used as the outcome dataset for the MR analysis (Supplementary Table 6).
Estimated causal effects of targets on COVID-19 severity
We conducted two-sample MR (36) to comprehensively estimate the causal effects of 1,002 plasma proteins and 16,059 transcripts on COVID-19 severity (Supplementary Figure 1). For the MR analysis of 1,002 plasma proteins, we were able to successfully perform MR on COVID-19 severity for 837 of them due to missingness of genetic variants in the COVID-19 GWAS (Bonferroni corrected P = 5.97×10−5). The protein level of ENTPD5 (represented by a cis protein QTL) showed a positive MR association with COVID-19 severity after multiple testing correction (odds ratio [OR] of COVID-19 severity per standard deviation change of protein level= 2.07, 95%CI=1.47 to 2.92, P=3.29×10−5; Supplementary Table 7). For the MR analysis of 16,059 transcripts, 14,036 of could be analysed with COVID-19 severity using MR (Bonferroni corrected P = 3.56×10−6). None of the transcript MR results showed robust MR evidence after correction for multiple testing (best MR p value= 4.17×10−5, Supplementary Table 8).
For the 353 prioritised targets with experimental or trial evidence, all of them showed no strong MR evidence of a causal effect on COVID-19 severity. Only expression of 10 genes (AATF, NEU1, NUP210, PIGS, PLOD2, PMPCA, RETREG3, SPART, SRP72, TLE3) and levels of one protein (NPC2) showed nominal association on COVID-19 severity (we used a lenient P value threshold of P□<□0.05, maximizing the number of possible genes analysed but also allowing readers to filter out associations should they wish to apply a more stringent threshold, Supplementary Table 9). All of these 11 targets were reported to interact with SARS-CoV-2 proteins in human cell lines (Supplementary Table 4).
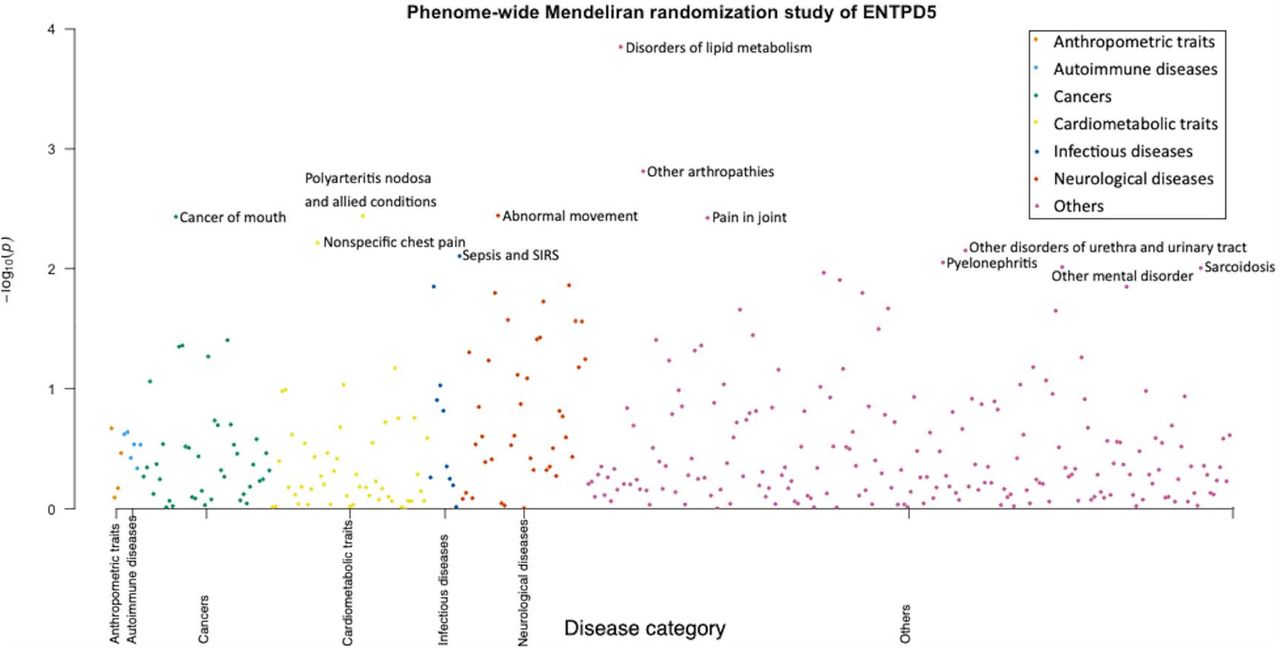
Genetically predicted associations between a protein or gene and COVID-19 severity may arise any of the four scenarios: causality, reverse causality, confounding by linkage disequilibrium (LD) between the leading SNPs for proteins and phenotypes or horizontal pleiotropy (Supplementary Figure 3). For the target, ENPTD5, with strong MR evidence of a causal effect on COVID-19 severity, we conducted single-trait colocalization (37) to distinguish causality from confounding by LD. This analysis showed strong evidence (colocalization probability=99.9%) of a shared genetic effect between ENPTD5 protein levels and COVID-19 severity (Supplementary Table 10A). In addition, we estimated the causal effects of COVID-19 severity on protein expression level of ENTPD5 using bidirectional MR (39) to explore the influence of potential reverse causality (with two conditional independent COVID-19 severity associated SNPs [P<1×10−6] used as instruments and the protein level of ENTPD5 used as outcome). This analysis showed little MR evidence to support reverse causality (OR= 0.94, 95%CI=0.81 to 1.11, P= 0.47; Supplementary Table 10B). In addition, the phenome-wide MR analysis (MR-PheWAS) of ENTPD5 provided no strong MR evidence of protein level of ENTPD5 associated with any of the 622 tested diseases or infection traits at a Bonferroni-corrected threshold of 8.04×10−5 (lowest MR P value=1.41×10−4; Figure 3), which suggested that horizontal pleiotropy is less likely to be an issue for this target.
{kind=link}
{kind=link}
{kind=link}
Different colors refer to different disease areas. For the 622 tested traits, none of them passed the Bonferroni-corrected threshold is 8.04×10−5. Therefore, ENTPD5 are less likely to be influenced by horizontal pleiotropy.
Estimated beneficial and adverse effects of the prioritised targets on other complex diseases
To identify potential beneficial and/or adverse effects of ENTPD5 (prioritised by omics MR of COVID-19 severity) and the 353 additional targets (prioritised by literature evidence), we conducted MR-PheWAS (18) and colocalization (37) of the protein and gene expression level of these targets on 622 traits, which including 49 viral infection phenotypes, 501 human diseases and 72 disease related risk factors (e.g. blood lipids) (Supplementary Table 11). 45,938 target-disease associations were tested in plasma proteome and/or transcriptome in whole blood (P<1.09×10−6 at a Bonferroni-corrected threshold). Where data was available, we also tested the tissue-specific effects of gene expression of the same targets on the outcome phenotypes using data from GTEX. Overall, 372,830 target-disease associations were estimated in the 11 COVID-19 relevant tissues (see the list of tissues in Methods).
As mentioned above, protein level of ENTPD5 was not strongly associated with any of the 622 traits. Considering a more inclusive threshold (a lenient P value threshold of P<0.05), the MR-PheWAS suggested that reducing protein levels of ENTPD5 may increase the risk of lipid metabolism disorders (OR=0.93, 95%CI=0.90 to 0.97, P=1.41×10−4) and may increase the risk of joint disorders such as arthropathies (OR= 0.94, 95%CI=0.91 to 0.98, P= 0.002; Figure 3 and Supplementary Table 12A). The MR-PheWAS also showed that genetically predicted ENTPD5 expression level was not associated with any disease (e.g. asthma) that might influence likelihood of seeking COVID-19 testing (the collider) (see Supplementary Figure 2 for more information). With these analyses we did not find evidence that the MR result of expression level of ENTPD5 on COVID-19 severity was influenced by collider bias, although this does not exclude it as a possibility.
For the 353 targets prioritised by literature evidence, we observed 833 target-disease associations with robust MR evidence in the 11 tested tissues. Using the same omics data as the MR analysis, 726 of the 833 (87.2%) associations also showed strong colocalization evidence (colocalization probability > 70%) (Supplementary Table 12B), making these as more reliable findings of this study. Of these, 366 associations were obtained using a single cis instrument in a Wald ratio model (40), 327 were obtained using a single trans instrument and 33 were estimated using multiple instruments in an inverse variance weighted (IVW) model (36). The remaining 107 (12.8%) associations had evidence from MR but did not have strong evidence of colocalization (probability<70%; Supplementary Table 12C), emphasizing the importance of this approach to address confounding by linkage disequilibrium (LD) in phenome-wide association studies.
Integration of evidence for the prioritised drug targets
We further integrated the 726 target-disease MR associations with clinical trial information from Open Targets (41), ChEMBL (42), DrugBank (43), Drug-gene-interaction (DGI) (44), the druggable genome (45) and ClinicalTrial.gov to prioritise drug targets against COVID-19. Of the 726 target-disease associations, 499 were unique target-disease pairs (due to the same pair appearing in multiple tissues; Supplementary Table 13). Further integrating these trial and literature evidence with the target-COVID-19 severity associations estimated in this study, we found that all targets showed no strong MR evidence of a causal effect on COVID-19 severity. Only 11 of them, including PLOD2, showed nominal effects. PLOD2 gene is the target for three approved drugs: sirolimus, ribavirin and apicidin (Supplementary Table 13). The direction of effect between gene expression of PLOD2 and disease risk was consistent with the therapeutic mechanism of these drugs. The MR-PheWAS of gene expression of PLOD2 suggested that the target may be associated with a few blood cell and lipid traits but is not strongly associated with any disease outcome. For the same reason explained in the ENPTD5 example, the MR-PheWAS did not show clear evidence of collider bias influencing our MR of gene expression of PLOD2 on COVID-19 severity.
Follow-up analyses of the prioritised drug target – ENTPD5
Given the potential causal effect of protein expression level of ENTPD5 on COVID-19 severity was supported by both MR and colocalization analyses, we further investigated this target with some additional follow-up analyses.
Shared causal effect among ENTPD5 gene expression and protein levels on COVID-19 severity
To further confirmed the ENTPD5 finding using plasma proteome data, we estimated the causal effect of the gene expression level of ENTPD5 on COVID-19 severity using data from eQTLGen and observed strong MR (P=5.66×10−5) and colocalization (probability=99.9%) evidence (Supplementary Table 10C). We then estimated the shared causal effect among gene and protein expression of ENTPD5 and COVID-19 severity using multi-trait colocalization analysis (46). We observed robust colocalization evidence to support shared genetic aetiology of the three traits (shared probability=95.4%; Figure 2; Supplementary Table 10D), which further strengthens the evidence level of the association between ENTPD5 and COVID-19 severity.
Causal mediator linking ENTPD5 with COVID-19 severity
We further investigated potential mediators linking protein level of ENTPD5 with COVID-19 using two-step MR (17). We first selected candidate mediators using phenome-wide association study (PheWAS) of the protein (rs57731447) and gene expression (rs118113209) instruments of ENTPD5 (using the IEU OpenGWAS database (47)). Four modifiable traits associated with the protein and/or gene expression instruments of ENTPD5 were selected as candidate mediators (HDL cholesterol, C-reactive protein, coagulation factor VIII and body fat percentage; Supplementary Table 14). We then conducted a two-step MR to link ENTPD5 with the mediators and COVID-19 severity (Supplementary Figure 4). In the first step of the two-step MR, we formally estimated the effect of gene and protein expression of ENPTD5 on mediators, confirmed the causal effects of protein level of ENTPD5 on these four candidate mediators (Supplementary Table 15A). In step 2 of the two-step MR, we estimated the effect of these four candidate mediators on COVID-19 severity but found little evidence to support their causal relationships (Supplementary Table 15B). We thus extended step 2 of the two-step MR by including traits similar to the four candidate mediators, which included lipid traits and lipoproteins, coagulation traits as well as traits related to body weight. We observed good evidence to support a causal role of increased D-dimer on increasing COVID-19 severity (OR=2.36, 95%CI=1.03 to 3.69, P value=0.001) as well as suggestive evidence of increased platelet count on increasing COVID-19 severity (OR=1.46, 95%CI=1.04 to 2.04, P value=0.027; Supplementary Table 15C). Moreover, we observed a protein-protein interaction between coagulation factor VIII (F8) and D-dimer (FGA/FGB/FGG) (data from StringDB implemented in EpiGraphDB). Although we did not identify a single mediator in this analysis, the two-step MR result and protein-protein interaction information suggested that coagulation may be involved in the causal effect of ENPTD5 protein expression on COVID-19 severity.
In addition, to explore the influence of potential reverse causality, we estimated the causal effects of the mediators on protein level of ENTPD5 using bidirectional MR (39) (with the mediators used as exposures and the protein expression level of ENTPD5 used as outcome) and found little evidence to support such reverse relationships, except for triglyceride levels showing evidence of a negative effect on ENTPD5 protein expression levels (Supplementary Table 15D).
Discussion
Genetic, transcriptomic and proteomic data provide a cost-effective approach to prioritise drug targets in the early stages of drug development (13)(28). In this study, we applied multi-omics MR, two-step MR and MR-PheWAS methods in a comprehensive in silico pipeline to prioritise drug targets for COVID-19 severity. Our multi-omics MR and colocalization study of 16,059 transcripts and 1,002 proteins identified putative causal effect of protein level of ENTPD5 on COVID-19 severity. Follow-up two-step MR analyses suggested that coagulation may be involved in the association between ENPTD5 protein levels and COVID-19 severity. Further integrating trial and experimental evidence, we investigated 353 targets for COVID-19. The MR-PheWAS of these 353 targets showed 726 target-disease associations with strong MR and colocalization evidence. However, MR of the gene or protein expression levels of these targets showed no strong evidence of a causal effect on COVID-19 severity. Moreover, the drug targets prioritised in this study were expected to offer therapeutic mechanisms through changing human proteins rather than SARS-CoV-2 itself. We therefore seriously considered the unintended beneficial or adverse effects of these targets on other complex diseases using MR-PheWAS. Our MR-PheWAS analysis suggested that the prioritised targets are unlikely to have a major adverse effect on complex human diseases. To enable the evidence to be widely accessible for COVID-19 drug target research, we constructed an open access browser allowing rapid queries of the drug targets, genetic predictors of the target genes and the target-COVID-19 associations (http://epigraphdb.org/covid-19/ctda/).
The ENTPD5 gene, located on chromosome 14, encodes the NTPase Ectonucleoside Triphosphate Diphosphohydrolase 5, an enzyme mediating extracellular catabolism. A specific role of ENTPD5 is to promote host protein N-glycosylation for proper protein folding (information from Open Targets). SARS-CoV-2 spike protein is extensively N-glycosylated (48) and blocking viral N-glycan biosynthesis was shown to inhibit viral entry (49). Taylor et al. suggested ENTPD5 as a gene associated with severe COVID-19 (50). In this study, our multi-omics MR and colocalization analyses suggested that both gene and protein expression levels of ENTPD5 showed a positive effects on COVID-19 severity. The existing evidence plus our MR findings suggested that future studies are needed to examine the direct role of ENTPD5 inhibition on SARS-CoV-2 viral entry and, subsequently, in the prevention of severe COVID-19. In addition, the follow-up two-step MR analysis did not identify any single mediator but suggested that coagulation dysfunction could be a potential mechanism of ENTPD5 on COVID-19 severity. In the literature, coagulation dysfunction was reported to be observed in hospitalized / severe COVID-19 patients (51)(52)(53)(54), including mild thrombocytopenia (55), increased D-dimer levels (56) and increased thromboembolic events (57)(58). In a candidate-driven genetic association study of severe COVID-19, genetic loci known to be associated with coagulation have been identified, which revealed that SARS-CoV-2 engages activation of coagulation cascades (59). An association between anticoagulation and improved survival was observed in patients with severe COVID-19 even after adjusting for mechanical ventilation (60)(61). Till the 19th of December 2020, 25 randomized controlled trials studying the effect of anticoagulation agents (i.e. Bivalirudin, Enoxaparin, Rivaroxaban and Aspirin) on COVID-19 are under investigation (https://clinicaltrials.gov/).
For the 353 targets with trial or literature evidence, the MR provided no strong evidence of a causal effect of the expressions of these targets showed on COVID-19 severity. Only 11 targets showed nominated associations. This included PLOD2, which is the target for three marketed drugs: sirolimus, ribavirin and apicidin. PLOD2, located on chromosome 3, is found in the endoplasmic reticulum and functions to catalyse the hydroxylation of lysyl residues in collagen-like peptides. Its protein, PLOD2, was revealed to interact with the SARS-CoV-2 protein open reading frame 8 (ORF8) (62), which was identified as containing the most pathogenic variants of all coronavirus proteins (63). Another gene (SRP72) located on chromosome 4, is the only subunit of the signal recognition particle (SRP) that can be phosphorylated. The SRP mediates the transportation of proteins to the plasma membrane. In the context of coronavirus, the SRP mediates the transportation of proteins to the endoplasmic reticulum (64). SRP72 protein also forms part of the viral mRNA translation and gene expression pathways. In the context of COVID-19, SRP72 has been identified as a human protein that interacts with the nonstructural protein 8 (nsp8) of SARS-CoV-2 (65). This SRP-nsp8 interaction may contribute to the reconfiguration of the endoplasmic reticulum during SARS-CoV-2 infection (5).
Some limitations of our analysis are worth noting. Whilst initiatives are underway to collect genetic information for COVID-19 patients (e.g. the COVID-19 host genetics initiative, https://covid-19genehostinitiative.net/), the recent initial GWASs of COVID-19 found few genetic association signals, which highlights the importance of sample size and statistical power. In addition, recent MR studies of COVID-19 identified a few gene targets associated with COVID-19 (19)(20)(21)(22)(23)(24), but these studies downplayed consideration of potential biases in the data. For example, if we use a mixture of population samples as controls (which include unexposed individuals and asymptomatic and untested cases), we will not be able to distinguish the causal effects of being exposed to SARS-CoV2, infected by the virus, or progressing to severe COVID-19 after infection (26). A GWAS using an unbiased population sample screened to detect infection of COVID-19 (e.g. from antibody tests) will be helpful to disentangle this issue, but such a GWAS does not yet exist. Our study excluded the five available COVID-19 GWAS datasets (with populations or self-reported data as controls) to minimise the influence of mixed controls. We only used hospitalised COVID-19 vs. non-hospitalised COVID-19 as the outcome for MR, which had the most reliable definition of cases and controls. However, given the GWAS of hospitalized COVID-19 vs. not hospitalized COVID-19 was still selected on the basis of COVID-19 status, collider bias could still be an issue (see Supplementary Figure 2). To estimate the influence of potential collider bias, we conducted MR-PheWAS of the 353 prioritised targets and ENTPD5 on 622 human diseases and phenotypes. The prioritised targets such as ENTPD5 and PLOD2 did not show much evidence of a causal effect on major diseases (e.g. asthma), which suggested that gene/protein level change of these targets are not obviously acting through these disease on selection (e.g. COVID-19 testing). However, the targets prioritised by our study should still be carefully reviewed in future trials before clinical application. A second limitation is that the drug targets evaluated in this study were proxied using a limited number of instruments, which means the putative causal effects rely on one or two genetic instruments. Thus, these associations support causality but do not prove it, as horizontal pleiotropy remains an alternative possibility. In addition, even though our results suggest some biological links between the target and diseases, these only provide evidence for the very first step of the drug development process. Finally, whilst these are plausible targets for COVID-19 severity, we cannot predict whether successful intervention would impact on risk of infection or other disease characteristics relevant to public health (e.g. viral shedding).
In conclusion, this study prioritised ENTPD5 as a drug target for COVID-19 severity using an integrated analytical protocol including MR and colocalization approaches. ENTPD5 is related to anti-coagulation, which suggests a potential molecular pathway for COVID-19 treatment. This study evidences the value of genetic approaches in drug target validation and drug repurposing. This provides the very first step towards evaluating intervention targets that are worth following-up for coronavirus and viral infections.
Materials and Methods
Selection of genetic instruments for COVID-19 drug targets
First, genetic association information of gene and protein expression levels were looked up from two resources: i) protein expression levels in plasma from five studies (28)(29)(30)(31)(32) implemented in Zheng et al. (13); ii) gene expression levels in whole blood from eQTLGen consortium (33). The genetic variants, genes and proteins were further mapped to genome build GRCh37.p13 coordinates. We used the following criteria to select genetic instruments:
We selected SNPs that were associated with any protein or gene expression (using a P-value threshold ≤5⨯10−8) in at least one of the four GWASs, including both cis and trans instruments.
We then conducted LD clumping for the instruments using the 1000 Genome European samples as reference panel, which was implemented in the TwoSampleMR R package (14) to identify independent instruments for each protein/gene. We used r2 < 0.001 as the threshold to exclude correlated instruments in the cis (or trans) gene region.
After instrument selection, 1,674 conditioning distinguished pQTL signals of 1,002 proteins (Supplementary Table 1) and 39,630 conditioning distinguished eQTLs signals for 16,059 transcripts (Supplementary Table 2) were kept as instruments for the genetic analyses of this study. To identify cis and trans instruments, we split instruments into two groups: 1) 29,610 cis-acting instruments within a 500Kb window from each side of the leading QTL of the protein/transcript; 2) 10,020 instruments outside the 500Kb window of the leading QTL were designated as trans instruments. Whilst trans instruments may be more prone to pleiotropy, their inclusion could increase statistical power as well as the scale of the study. Therefore, for the proteins and genes with cis instruments or trans instruments, we conducted MR analyses using both sets of instruments (Supplementary Table 1 and 2).
Second, we selected additional drug targets that were prioritised by evidence from the following three resources: i) a list of 11 drugs under trials for COVID-19 treatment were extracted from ClinicalTrials.gov (Supplementary Table 3). These drugs were mapped to their target genes using Drug-Gene-Interaction (DGI) database (http://dgidb.org/) (44) and CHEMBL database (42); ii) human proteins interacting with SARS-CoV-2 proteins from Gordon et al. (5); iii) genes associated with SARS-CoV from Gralinski et al. (6). After de-duplicating these, 380 unique drug targets were selected for our study (Supplementary Table 4). Next, gene and protein expression levels of these targets were looked up from four resources: protein expression levels in plasma from four studies (28)(29)(30)(31)(32) implemented in Zheng et al. (13), gene expression levels in whole blood from eQTLGen consortium (33), tissue specific gene expression levels in 7 tissues from the GTEx consortium (34) and gene expression levels in two kidney tissues from Gilles et al. (35). After the mapping step, 353 drug targets with genetic variants robustly associated with the transcripts and/or proteins were included as the start point of the instrument selection (Supplementary Table 5).
For any of the 353 targets, we mapped the drug targets with related drug names using four platforms: OpenTargets (41), ChEMBL (42), DrugBank (43) and DGI platforms (44). After this analysis, we mapped one target, PLOD2, to the drugs which target it (Supplementary Table 9). We further mapped the targets to the previously reported “druggable genome” (45). This study stratified the potential drug targets from across the genome into three tiers. Tier 1 (1427 genes) included efficacy targets of approved small molecules and biotherapeutic drugs, as well as targets modulated by clinical-phase drug candidates; tier 2 was composed of 682 genes encoding proteins closely related to drug targets, or with associated drug-like compounds; and tier 3 contained 2370 genes encoding secreted or extracellular proteins, distantly related proteins to approved drug targets. For the 11 additional prioritised COVID-19 drug targets, one target was mapped to tier 1, 2 targets to tier 3 (Supplementary Table 9).
Selection of genetic association information for COVID-19 severity
The genetic association information of COVID-19 severity were obtained from the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/). Among the six available genetic association data listed in Supplementary Table 6, we considered the hospitalized COVID-19 vs. not hospitalized COVID-19 as the most suitable dataset to represent COVID-19 severity. The major advantage of this data compared to other datasets (e.g. COVID-19 vs. population) is that all participants of this genetic analysis were confirmed COVID-19 cases, therefore this analysis will be less bias by data selection (e.g. there will be some unconfirmed COVID-19 cases in the population so it is not a perfect control dataset). We used the GWAS summary statistics of the hospitalized COVID-19 vs. not hospitalized COVID-19 as the outcome for the MR analysis.
This GWAS combined European samples from three large scale biobanks: UK Biobank, DECODE and FinnGen (928 confirmed hospitalized COVID-19 patients, 2,028 confirmed non hospitalised COVID-19 patients; Supplementary Table 6).
Causal inference and sensitivity analyses
Mendelian randomization analysis
In the initial MR analysis, 1,674 instruments of 1,002 plasma proteins and 39,630 instruments of 16,059 gene transcripts in whole blood were used as the exposures to proxy the effect of these targets on COVID-19, which maximized the statistical power of the study and supported us to obtain an overall view of the target-COVID-19 associations. The hospitalized COVID-19 vs. not hospitalized COVID-19 were used as the outcome to proxy COVID-19 severity. Due to missingness of available exposure or outcome data, the causal relations of expression of 837 proteins and 14,036 transcripts on COVID-19 severity were tested in this study. We selected a P-value threshold of 0.05, corrected for the number of tests, as our threshold for prioritising MR results for follow up analyses (N_protein= 837, P_protein< 5.97×10−5; N_transcript= 14,036, P_transcript <3.56×10−6) The Wald ratio (40) method was used to obtain MR effect estimates for protein and/or transcript targets with only one instrument. For protein and/or transcript targets with two or more instruments, the inverse variance weighted (IVW) method (36) was used to estimate the causal effects.
Colocalization analysis
For the drug targets - COVID-19 associations, results that survived the Bonferroni corrected threshold (P_protein< 5.97×10−5 or P_transcript< 3.56×10−6) in the MR analysis were evaluated using colocalization analysis (the “coloc” R package), which estimates the posterior probability of each genomic locus containing a single variant affecting both the target and the phenotype (37). We used the default prior probabilities that a variant is equally associated with each phenotype (p1=1×10−4; p2=1×10−4) and both phenotypes jointly (p12=1×10−5). A posterior probability of > 70% for the colocalization hypothesis in this analysis would suggest that the two association signals are likely to colocalize within the test region (noted as “Colocalised”). The rest of the target-phenotype associations were noted as “Not colocalised”.
Tissue Specificity analysis
The functional receptor of SARS-CoV-2, ACE2 (66), is highly expressed in multiple organs, including gastrointestinal tract, gallbladder, testis, and kidney. This is consistent with the fact that whilst SARS-CoV-2 infection primarily manifests with acute respiratory illness, SARS-CoV-2 can also be detected in faeces (67) and kidney tubules (68). The presence of SARS-CoV-2 in the alimentary tract for longer than in the respiratory system (69) suggests that the intestine may be a hidden reservoir of SARS-CoV-2. We therefore set out to explore differences in potential target effects in different tissues. In order to understand the tissue specific effects of the candidate targets of COVID-19 on human phenotypes, we selected the 9 tissues in which ACE2 is highly expressed, including testis, lung, kidney cortex, kidney glomerular, kidney tubulointerstitial, stomach, colon transverse, small intestine terminal ileum and colon sigmoid.
Tissue specific gene expression data of the 353 targets in each selected tissue were obtained from two studies: GTEx V8 and Gillies et al (34)(35). After selection, 580 instruments of 218 gene transcripts were selected in the 9 tissues, which included 141 instruments for 132 gene transcripts in testis, 125 instruments for 115 transcripts in lung, 20 instruments for 20 transcripts in kidney cortex, 6 instruments for 6 in kidney glomerular, 8 instruments for 8 transcripts in kidney tubulointerstitial, 71 instruments for 67 transcripts in stomach, 84 instruments for 81 transcripts in colon sigmoid, 98 instruments for 96 transcripts in colon transverse and 41 instruments for 39 transcripts in small intestine terminal ileum (Supplementary Table 6). The same MR and colocalization analysis pipeline were applied for the tissue specific analysis.
Follow-up analyses of prioritised targets for COVID-19 severity
For protein and/or transcript targets with robust MR (Bonferroni corrected threshold for tested proteins=5.97×10−5; Bonferroni corrected threshold for tested transcripts=3.56×10−6) and colocalization (probability >80%) evidence, we conducted a set of follow-up analyses to further confirm the association between targets and COVID-19 severity as well as to identify potential mediators/mechanisms linking the targets with COVID-19 severity.
Multi-trait colocalization analysis of prioritised targets
We first explore whether the causal variants were shared across transcriptome, proteome and COVID-19 severity for top MR findings. We applied multi-trait colocalization implemented in the moloc R package (46). The default prior probabilities of 1×10−4 for any one layer of association, 1×10−6 for any two layers of associations and 1×10−7 for colocalization of all three layers of associations were used in the moloc analysis. An overall colocalization probability of three traits (Pa,bc+Pab,c+Pac,b+Pabc) > 80% would suggest that the three association signals are likely to colocalize within the test region.
Mediation/two-step Mendelian randomization analysis of prioritised targets
For targets showing robust evidence from either protein or transcript MR, we further identified the potential causal mediators linking the prioritised targets with COVID-19 severity using two-step MR (17). In first step, we used the protein / expression QTLs as instruments to estimate the effect of the proteins/transcripts on the potential mediator (e.g. an intermediate trait such as coagulation). In second step, we used the SNPs associated with the mediator as instruments to estimate the effect of the mediator on COVID-19 severity (Supplementary Figure 3). To make a comprehensive selection of candidate mediators (intermediate traits), we conducted a phenome-wide scan of the protein and transcript instruments (QTLs) in all 34,494 available human traits in the IEU Open GWAS database (47). For four traits showing suggestive association with the QTLs (P<0.001), we conducted mediation/two-step MR for these mediators.
Bidirectional MR to identify potential reverse causality
We further estimated the potential reverse causality of the mediators on ENTPD5 using bi-directional MR (39). The prioritised mediators were treated as the exposures and ENTPD5 protein level were selected as the outcome of this MR analysis.
Analysis software
The MR analyses (including Wald ratio, IVW, two-step MR and bidirectional MR) were conducted using the TwoSampleMR R package (github.com/MRCIEU/TwoSampleMR) (70). The MR results were plotted as Manhattan plots and forest plots using code derived from the ggplot2 package in R (https://cran.r-project.org/web/packages/ggplot2/index.html).
MR PheWAS of prioritised targets for COVID-19 severity
For the 353 prioritised drug targets with trial or experimental evidence and targets with robust MR evidence of association with COVID-19 severity, we further conducted a phenome-wide MR analysis (MR-PheWAS) to identify potential beneficial and/or adverse effects of these targets on other human diseases. The QTLs for the prioritised targets were chosen as the exposures for the MR-PheWAS. For the outcomes, 49 viral infection phenotypes from the GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics) (71), 504 human diseases and 72 disease related traits (e.g. blood lipids) selected from the IEU Open GWAS database (47) were selected as outcomes for this analysis (Supplementary Table 11). These 573 outcomes were selected using the following inclusion criteria:
The GWAS with the greatest expected statistical power (e.g. largest sample size / number of cases) when multiple GWAS records of the same disease / risk factor were available in the Open GWAS database.
GWAS with betas, standard errors and effect alleles for all tested variants (i.e. full GWAS summary statistics available)
For the MR-PheWAS results that survived the Bonferroni corrected threshold (P< 1.1⨯10−6), we further evaluated them using colocalization analysis, which estimates the posterior probability of each genomic locus containing a single variant affecting both the target and the phenotype (37). The same prior probabilities applied for the previous colocalization were applied here. A posterior probability of > 70% for the colocalization hypothesis in this analysis would suggest that the two association signals are likely to colocalize within the test region (noted as “Colocalised”). In addition, we conducted an approximate colocalization analysis (called LD check), in which the LD between the sentinel variant of each target and the 30 strongest SNPs in the region associated with the phenotype were checked. LD r2 > 0.7 between the sentinel variant and any of the 30 SNPs associated with the phenotype was used as evidence of colocalization (noted as “LD Check”). The rest of the target-phenotype associations were noted as “Not colocalised”.
COVID-19 Target-Disease Atlas (CTDA) browser of the EpiGraphDB platform
We have made all MR results openly available to browse or download at the COVID-19 Target-Disease Atlas (CTDA) browser within the EpiGraphDB platform (http://epigraphdb.org/covid-19/ctda/). This includes 14,873 unique target-COVID-19 severity associations evidence for the omics MR as well as 372,830 unique target-disease associations evidence for 353 targets on 622 diseases/phenotypes in 11 SARS-CoV-2 related tissues. Users are able to query the study results by the targeted gene/protein name and QTL SNPs via the online platform, and the results are presented in searchable tables as well as volcano plots. In addition, users can programmatically access the results using the /covid-19/ctda endpoints in the application programming interface (API) of EpiGraphDB via http://api.epigraphdb.org/.
Data Availability
The data needed for the analysis were available via MR-Base platform. The results is available via the EpiGraphDB platform http://epigraphdb.org/covid-19/ctda/
Supplementary Materials
TableS1. The available pQTL instruments of 1,002 plasma proteins.
TableS2. The available eQTL instruments of 16,059 blood transcripts.
TableS3. Drugs in trials for COVID-19 treatment and their target genes.
TableS4. The available genetic information for SARS-CoV-2 target genes from pQTL and tissue specific eQTL resources.
TableS5. The genetic predictors and association information of the SARS-CoV-2 target genes.
TableS6. The genome-wide association studies of the 6 COVID-19 phenotypes.
TableS7. The plasma protein-COVID-19 severity associations with suggestive Mendelian randomization evidence (P<0.05).
Note: The target-COVID-19 severity associations with Mendelian randomization p value < 10−2 were included in this table.
TableS8. The blood transcription-COVID-19 severity associations with suggestive Mendelian randomization evidence (P<0.05).
Note: The target-COVID-19 severity associations with Mendelian randomization p value < 10−2 were included in this table.
TableS9. The target-COVID-19 severity associations with suggestive Mendelian randomization evidence (P<0.05).
TableS10. The colocalization results for proteins, expression levels of ENTPD5 and COVID-19 severity.
Note: The target-disease associations passed Bonferroni corrected threshold (6.0×10−5) were tested further by colocalization analysis.
TableS11. The list of 622 human phenotypes been used as outcomes for the phenome-wide mendelian randomization analysis.
TableS12. The phenome-wide Mendelian randomization results for the 12 prioritised drug targets.
TableS13. The triangulation table mapping drug target gene with drug name, the drug trial and druggability information.
Note: The target genes were mapped to the drugs using DGI, OpenTargets, ChEMBL and/or Drugbank databases. The drug trial information for each drug was identified from DGI, OpenTargets, ChEMBL, Drugbank and/or ClinicalTrial.gov.
TableS14. Phenome-wide association scan of the pQTL and eQTL instruments of ENTPD5.
TableS15. Two-step Mendelian randomization analysis linking ENTPD5 with the potential mediators and COVID-19 severity
Note: Four modifiable traits showed association signals with the pQTL and eQTL of ENTPD5 were selected as candidate mediators (HDL cholesterol, C-reactive protein, coagulation factor VIII and body fat percentage).
Funding
JZ is funded by a Vice-Chancellor Fellowship from the University of Bristol. This research was also funded by the UK Medical Research Council Integrative Epidemiology Unit (MC_UU_00011/1, MC_UU_00011/4). This study was funded/supported by the NIHR Biomedical Research Centre at University Hospitals Bristol NHS Foundation Trust and the University of Bristol (TRG). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health and Social Care. YMZ is supported by National Natural Science Foundation of China (81800636). HZ is supported by the University of Michigan Health System–Peking University Health Science Center Joint Institute for Translational and Clinical Research (BMU2017JI007). This work was supported by a Wellcome Trust Institutional Translational Partnership Award (209739/Z/17/Z).
Competing interests
No competing interests
Availability of data
The data used in this study is available via the IEU Open GWAS platform (https://gwas.mrcieu.ac.uk/). The results of this study is available via the EpiGraphDB platform http://epigraphdb.org/covid-19/ctda/
Code availability
The code to conduct Mendelian randomization analysis was available via the TwoSampleMR R package (https://mrcieu.github.io/TwoSampleMR/).
Author contributions
JZ and YMZ selected the drug targets; JZ performed the Mendelian randomization analysis; JZ and DB performed the colocalization analysis; JZ conducted the triangulation between MR and drug trials; JZ and YMZ conducted the drug target prioritisation; YL developed the database and web browser; JZ and YMZ wrote the manuscript; DB, YL, LW, XZL, HZ and TRG reviewed the paper and provided key comments; JZ, YMZ and TRG conceived and designed the study and oversaw all analyses.
Acknowledgments
This research was conducted using the UK Biobank resource under application number 15825. The UK Biobank received ethnical approval from the research ethnic committee (REC reference for UK biobank 11/NW/0382) and participants provided written informed consent.
Footnotes
Added the MR results of proteins, genes on COVID-19 severity and integrated these results with literature evidence
References




