Article Text

Abstract
Aim: Chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) is a multisystem disease, the pathogenesis of which remains undetermined. The authors have recently reported a study of gene expression that identified differential expression of 88 human genes in patients with CFS/ME. Clustering of quantitative PCR (qPCR) data from patients with CFS/ME revealed seven distinct subtypes with distinct differences in Medical Outcomes Survey Short Form-36 scores, clinical phenotypes and severity.
Methods: In this study, for each CFS/ME subtype, those genes whose expression differed significantly from that of normal blood donors were identified, and then gene interactions, disease associations and molecular and cellular functions of those gene sets were determined. Genomic analysis was then related to clinical data for each CFS/ME subtype.
Results: Genomic analysis revealed some common (neurological, haematological, cancer) and some distinct (metabolic, endocrine, cardiovascular, immunological, inflammatory) disease associations among the subtypes. Subtypes 1, 2 and 7 were the most severe, and subtype 3 was the mildest. Clinical features of each subtype were as follows: subtype 1 (cognitive, musculoskeletal, sleep, anxiety/depression); subtype 2 (musculoskeletal, pain, anxiety/depression); subtype 3 (mild); subtype 4 (cognitive); subtype 5 (musculoskeletal, gastrointestinal); subtype 6 (postexertional); subtype 7 (pain, infectious, musculoskeletal, sleep, neurological, gastrointestinal, neurocognitive, anxiety/depression).
Conclusion: It was particularly interesting that in the seven genomically derived subtypes there were distinct clinical syndromes, and that those which were most severe were also those with anxiety/depression, as would be expected in a disease with a biological basis.
Statistics from Altmetric.com
Take-home messages
Expression patterns of 88 chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) associated genes revealed seven distinct CFS/ME subtypes with distinct differences in clinical symptoms and severity.
It was particularly interesting that those CFS/ME subtypes that were most severe were also those with anxiety/depression, as would be expected in a disease with a biological basis.
Chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) is a disease characterised by severe and debilitating fatigue, sleep abnormalities, impaired memory and concentration, and musculoskeletal pain.1 In the western world, the population prevalence is estimated to be of the order of 0.5%.2 3 Research studies have identified various features relevant to the pathogenesis of CFS/ME such as viral infection, immune abnormalities and immune activation, exposure to toxins, chemicals and pesticides, stress, hypotension, lymphocyte abnormalities and neuroendocrine dysfunction. However, the precise underlying disease mechanisms and means by which these abnormalities inter-relate in patients with CFS/ME remain to be clarified.4 5
We have previously described a study of gene expression in peripheral blood from 25 patients with CFS/ME, diagnosed according to the Centers for Disease Control (CDC) diagnostic criteria, and 50 normal blood donors using the Affymetrix U133+2 microarray. Genes showing differential expression were further analysed using quantitative PCR (qPCR) in 55 patients with CFS/ME and 75 normal blood donors. Differential expression was confirmed for 88 genes, 85 of which were upregulated and three downregulated. Highly represented functions were haematological disease and function, immunological disease and function, cancer, cell death, immune response and infection. Clustering of qPCR data from patients with CFS/ME revealed seven distinct subtypes with distinct differences in Medical Outcomes Survey Short Form-36 (SF-36) scores, clinical phenotypes and severity.6
In this study, we determined for each CFS subtype, the fold difference of each of the 88 CFS-associated genes compared with normal persons. Using a fold-difference cut-off of ⩾1.5, we then determined those genes that are differentially expressed in each CFS subtype. For each subtype, we report respective gene functions/pathways, gene interactions and disease associations, and relate these to the clinical phenotype details.
METHODS
Subjects and clinical characterisation
Analyses in this paper are based upon clinical and genomic data from patients with CFS/ME whose blood was used for qPCR confirmation of microarray data, as previously reported.6 In total, 55 such patients were enrolled from clinics in Dorset, Bristol and London, UK, and New York City, USA (one patient from Leicester, UK, was managed by a clinic in London). These cases were diagnosed according to the Centers for Disease Control (CDC) diagnostic criteria for CFS/ME.1 Patients with psychiatric disease were excluded using the Minnesota International Neuropsychiatric Interview, thus ensuring that none of our patients was suffering from major psychiatric disease or abuse of alcohol or other drugs. In addition, patients who smoked in the previous year, or were currently taking (or were within 3 months of taking) antibiotics, steroids or antidepressants were excluded from the study.
For all enrolled subjects, according to the recommendations of the International CFS Study Group,7 severity of physical and mental fatigue was assessed using the Chalder Fatigue Scale,8 level of disability was assessed using the Medical Outcomes Survey SF-36, accompanying symptoms were characterised using the Somatic and Psychological Health Report, sleep abnormalities were assessed using the Pittsburgh sleep questionnaire, and assessment of type and severity of pain was performed using the McGill pain questionnaire. For the patients with CFS/ME, neurocognitive testing was performed using the spatial span (SSP) and verbal recognition memory (VRM) modules of the Cantab software (Cambridge Cognition, Cambridge, UK), which showed abnormal results in CFS/ME.6 9
For each patient with CFS, the severity of particular symptoms and level of function were taken from the questionnaires described above. Then for each CFS subtype, which was derived by clustering of qPCR data as previously described,6 mean values for each symptom and score were calculated and compared between the subtypes. Analysis of variance (ANOVA) was used to determine the significance of differences in individual SF-36 domain scores between CFS subtypes.
Patients and controls gave written consent according to guidance of the Wandsworth Research Ethics Committee (approval number 05/Q0803/137). For the New York patients, approval of the local institutional review board was obtained. The human experimentation guidelines of the US Department of Health and Human Services were followed in this study.
Determination of differential expression of human genes in each CFS/ME subtype
The threshold cycle (Ct) for each test gene in each sample was compared with a calibrator sample to calculate a ΔCt value. ΔCt values were then normalised to the Ct value for an endogenous control gene, glyceraldehyde-3-phosphate dehydrogenase, in respective samples to give the ΔΔCt values. Relative quantity (RQ) values (2−ΔΔCt) for each mRNA of interest were then calculated. Samples showing a difference between minimum and maximum RQ values of ⩾100 (indicating poor replicate concordance) were excluded. The t test was used to compare the RQ values for patients with CFS/ME with the RQ values of the controls. Genes whose mean RQ values differed between the groups (at p⩽0.05) were included in our CFS/ME-associated gene signature.6 RQ values for all 88 CFS/ME-associated genes were normalised and clustered using Genesis software.10
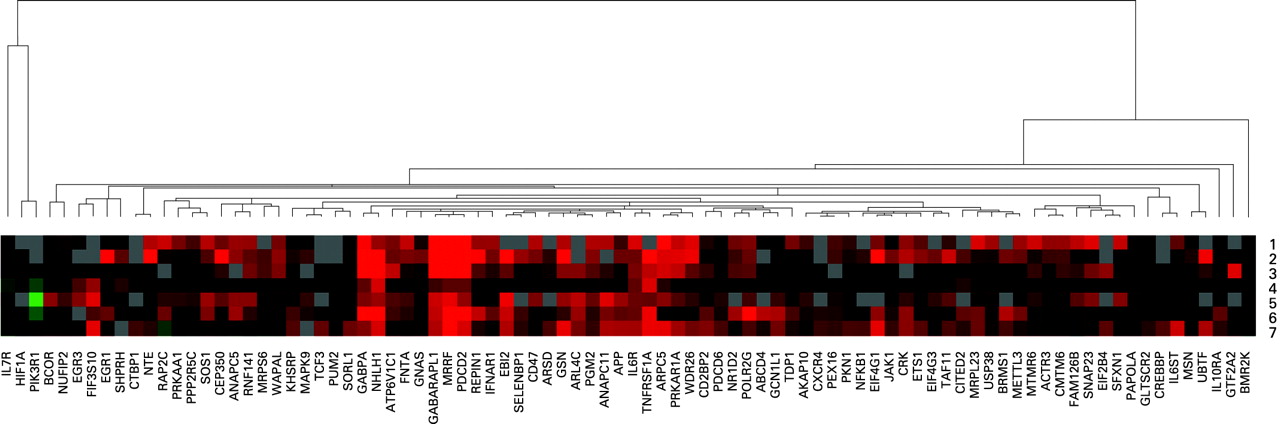
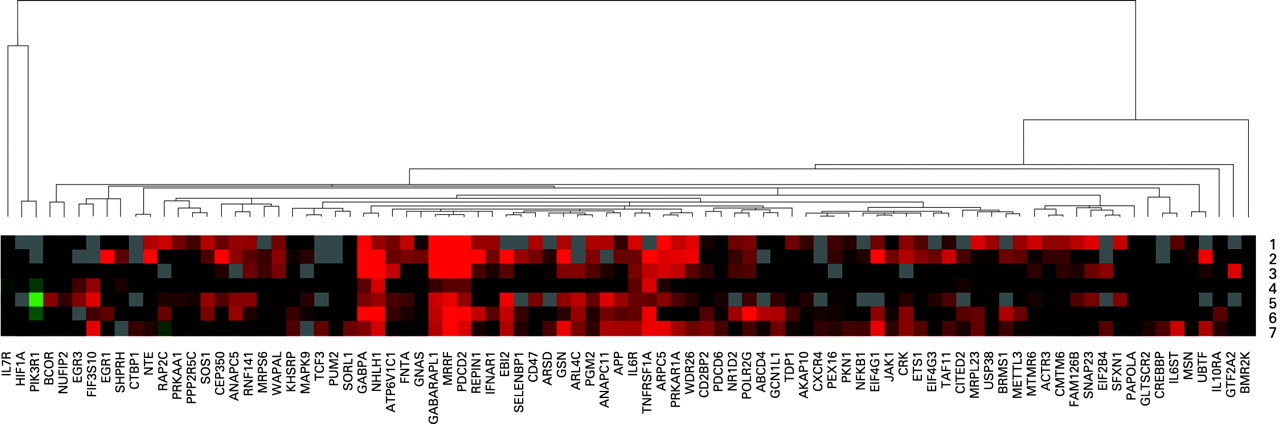
For each CFS subtype, mean RQ values were calculated. Then, for each gene, the mean RQ value for each CFS subtype was divided by the mean RQ value of the normal blood donors, to provide fold-difference values for each CFS subtype. For each subtype, genes were included for analysis assuming they showed fold-difference values (mean RQ in CFS subtype/mean RQ in normal) in qPCR experiments of ⩾1.5. Thus an individual gene list was generated for each CFS subtype within the 88-gene signature for CFS. Mean fold-difference values were clustered using Cluster version 2.11 software (without normalisation) and visualised using Treeview version 1.60 software.11
Analysis of gene function and interaction in each CFS/ME subtype
Each of these subtype-specific gene lists was analysed for gene function and interaction using Ingenuity Pathways Analysis (IPA) software (Ingenuity, Redwood City, California, USA) in order to link CFS/ME-subtype-associated genes into networks based on recognised interactions, and to discern the top associated diseases and disorders, molecular and cellular functions, associated physiological system development and function and canonical pathways.
RESULTS
Subjects and clinical characterisation
Clinical and genomic data from a total of 55 patients with CFS/ME fulfilling CDC diagnostic criteria were used for this study. Of these, 19 were male, and 36 were female, with an overall mean age of 41.6 years and a mean duration of disease of 3.2 years. Additional clinical details are provided elsewhere.6 This study included several patients with CFS/ME whose disease was severe and necessitated bed rest for much of the day, and patients who were able to attend an outpatient clinic. Normal blood donors were used as a comparison group and clinical data for these are available elsewhere.6
Genomic CFS/ME subtypes
As previously reported, clustering of qPCR data revealed the presence of seven genomic CFS subtypes with distinct profiles of gene expression within the 88-gene CFS gene signature.6 Fold-difference values (mean RQ in CFS/mean RQ in normal) for all patients with CFS and for each CFS subtype are shown in table 1. For each subtype, genes with fold-difference values of ⩾1.5 were noted and used in further analysis. This resulted in the following numbers of differentially-expressed genes in each subtype: 58 (CFS subtype 1), 70 (CFS subtype 2), 48 (CFS subtype 3), 27 (CFS subtype 4), 66 (CFS subtype 5), 69 (CFS subtype 6) and 71 (CFS subtype 7). In table 1, genes without values are those for which there were missing data for particular subtypes.
Analysis of gene function in each CFS/ME subtype
Using IPA software, the gene list for each CFS subtype was analysed to determine the most important associated diseases and disorders, molecular and cellular functions, associated physiological system development and function and canonical pathways. The results of this analysis are shown in table 2. As regards disease associations, analysis revealed some common (neurological, haematological, cancer) and some distinct (metabolic, endocrine, cardiovascular, immunological, inflammatory) disease associations among the subtypes. This was also true for cellular and molecular functions, and physiological system development and function analyses (table 2). As regards the canonical pathways implicated in each subtype, there was more variation between subtypes than for the previous analyses, probably because these assignments are based on fewer genes per pathway. Interleukin (IL)6 signalling was implicated in subtypes 1, 2, 5 and 6; B cell receptor signalling was implicated in subtypes 4 and 6; oestrogen receptor signalling was implicated in subtype 7; ephrin receptor signalling was implicated in subtypes 1, 2 and 7; and insulin receptor signalling was implicated in subtypes 3, 4 and 6 (table 2).
Analysis of gene interaction in each CFS/ME subtype
Gene interaction was assessed for each subtype using IPA software. For each subtype, this analysis generated between two and five large networks (arbitrarily defined as containing eight or more CFS-associated genes) based on published gene interactions (data not shown) and a variable number of smaller networks and single genes for which interactions were not known. For each subtype, all networks, large and small, were combined into a single network, indicating genes found to be upregulated and downregulated and then stratified to show the subcellular location of each (fig 1A–G).

Clinical features of each CFS subtype
Number of patients, mean age and male:female ratio for each subtype were as follows: subtype 1 (2, 27 years, 1:1), subtype 2 (5, 49 years, 4:1), subtype 3 (2, 32 years, 0:2), subtype 4 (19, 44.3 years, 8:11), subtype 5 (7, 51 years, 0:7), subtype 6 (14, 41.1 years, 6:8), subtype 7 (3, 47 years, 0:3). Subtypes 3, 5 and 7 were made up of females only, subtype 2 was predominantly male, and the remainder were mixed; age differences were less clearly demarcated.
Mean questionnaire scores for each subtype are shown in fig 2(A,B). Clinical symptom severity for each subtype is shown in fig 2(C). CFS subtypes 1 and 7 were the most severe, followed sequentially by subtypes 2, 4, 5 and 6/3. ANOVA testing revealed significant differences between groups for the SF-36 total score (p = 0.016), social functioning (p = 0.03), and emotional role (p = 0.003), while the difference between groups approached significance for general health (p = 0.08) and mental health (p = 0.08). After adjusting for multiple comparisons, significant associations were found between specific groups and clinical phenotypes. Subtype 7 had most pain, lowest SF-36 scores (along with subtype 1), and most severe individual symptoms including swollen glands, sore throat and headaches. Subtype 1 had the worst cognition and mental health score, and poor sleep despite having the least pain. Subtype 4 had moderate neurocognitive function and cognitive defects, combined with moderate levels of bodily pain and sleep problems. Subtype 5 had the best mental health but poor neurocognitive function, gastrointestinal complaints and the most marked muscle weakness and postexertional malaise. Subtype 2 had marked postexertional malaise, muscle pain and joint pain but poor mental health (fig 2A–C).

Summary clinical features of each subtype were as follows: subtype 1 (cognitive, musculoskeletal, sleep, anxiety/depression); subtype 2 (musculoskeletal, pain, anxiety/depression); subtype 3 (mild); subtype 4 (cognitive); subtype 5 (musculoskeletal, gastrointestinal); subtype 6 (postexertional); subtype 7 (pain, infectious, musculoskeletal, sleep, neurological, gastrointestinal, neurocognitive, anxiety/depression). It is particularly interesting that in these genomically derived subtypes, there were distinct clinical syndromes and that those that were most severe were also those with anxiety/depression, as would be expected in a disease with a biological basis.
As regards subtype associations with geographical location, subtypes 4 and 6 were predominant in Dorset, subtype 4 was predominant in London and New York, and subtype 5 was predominant in Bristol (fig 2D).
CFS/ME associated genes that are specifically targeted by existing drugs
Within the CFS gene signature, there were five human genes that are known to be targeted by one or more existing drugs designed or intended for use in other diseases. Based on the expression levels of these five genes, these drugs may be predicted to be beneficial for particular CFS subtypes. These genes, corresponding drugs and CFS subtypes are as follows: APP (AAB-001; subtypes 1, 2, 3, 4, 5, 6 and 7); CXCR4 (JM1300; subtypes 5 and 6); FNTA (lonafarnib, tipifarnib; subtypes 1, 2, 3, 5 and 6); IL6ST (tocilizumab; subtypes 1, 2, 5, 6 and 7); TNF (golimumab, adalimumab, etanercept, certolizumab pegol, infliximab; subtype 2).
DISCUSSION
This study follows on from our paper describing differential expression of 88 human genes in patients with CFS6 and its purpose is to expand upon the brief description of the genomic and phenotypic aspects of the CFS subtypes given in the earlier paper.
It has long been recognised that subtypes of CFS/ME exist, and it has been believed that these subtypes may, at least in part, reflect particular aetiological factors.12 A symptom-based approach has had some success in identifying musculoskeletal, inflammatory and neurological subtypes13; however, those groups had only minor differences in overall functional severity in contrast to those of the present study.
It is intriguing that within our 88-gene signature, there are several genes with links to various aetiological triggering factors. For example, virus infection (EIF4G1, EBI2) and organophosphate exposure (neuropathy target esterase (NTE)). EIF4G1 is an eukaryotic translation initiation factor that is bound and cleaved by a range of viruses, including enteroviruses, which both trigger and persistently infect patients with CFS.14 15 Whistler and colleagues have also reported upregulation of EIF4G1 transcript variant 5 (the same variant as we report) in patients with CFS who have rapid (?triggered by virus infection) as compared with insidious onset.16 EIF4G1 is a component of the protein complex EIF4F, which is crucial in translation.17 These viruses divert EIF4G1 from its utilisation by the cellular machinery to facilitate production of viral proteins.17 EIF4G1 is upregulated in CFS subtypes 1, 2, 3, 4, 6 and 7 (table 1; fig 3 and fig 1A,B,C,D,F,G).
{kind=link}
{kind=link}
{kind=link}
Various CFS-associated genes identified have been previously shown to be upregulated in Epstein-Barr virus (EBV) infection, namely NFKB1, EGR1, ETS1, GABPA, CREBBP, CXCR4, EBI2, HIF1A, JAK1, IL6R, IL7R and PIK3R1. This is very interesting as EBV is a recognised trigger of CFS and is known to reactivate upon stress.18 However, it is difficult to draw conclusions as to the inter-relationship of these genes in the different subtypes (fig 3). The EBV transcription factor BRLF1 was found to be over-represented in the original CFS gene signature; however, this was not tested by PCR.6 The EBV genes BRLF1 and BZLF1 mediate the switch from latent to lytic phases of EBV infection and during this process they transactivate many human genes. It is interesting that the BRLF1 gene has been identified as being over-represented in the transcription factor analysis, and that specific IgG to the Zebra protein (BZLF1 gene product) has been reported previously in patients with CFS/ME.19
EBI2 is a gene that is upregulated 200-fold in EBV-infected cells20 and is upregulated in subtypes 2, 3, 5, 6 and 7, but not in the normal controls.6 One subject with EBI2 upregulation was a 26-year-old female whose CFS had been triggered by laboratory-documented EBV infection, and who had a chronic course with detectable EBV replication in blood for several years after the acute phase. This suggests the possibility that EBI2 may be a surrogate marker for ongoing EBV replication in patients with CFS, although this remains to be clarified. If this is true, then this would be very useful to inform the decision as to which patients with CFS should be treated with valganciclovir, which has been shown to be beneficial in CFS.21
Three patients had markedly raised levels of NTE, while all normal controls had uniformly low levels; CFS subtypes with significantly raised NTE levels were 1, 2, 5 and 7, of which subtypes 1, 2 and 7 were the most severely affected subtypes. We have previously documented upregulation of NTE in CFS.22 NTE is the primary site of action of organophosphate (OP) compounds such as sarin, which causes axonal degeneration and paralysis resulting from inactivation of its serine esterase activity,23 and in the adult chicken nervous system, OP-modified NTE initiates neurodegeneration. Exposure to OP compounds may trigger CFS/ME24 and Gulf War illness.25
IL10RA is a gene that is critical for T cell activation and immune system homeostasis, as polymorphisms in it have been shown to be associated with development of lymphoma, chronic obstructive pulmonary disease, autoimmunity, severity of hepatitis C infection, and multiple sclerosis.26–30 In the present study IL10RA was upregulated,6 although we have previously found it to be downregulated in patients with CFS.22 There were more subjects in our pilot study19 who were bed bound than in the present study,6 and IL10RA levels appear to be a marker of severity in CFS (as they are closely correlated with SF-36 general health score), with lower levels reflecting increasing severity (data not shown).
It is interesting that disease associations identified in the various subtypes are mostly those that are already recognised in CFS. However, for any one disease association, there are important variations between the subtypes. For example, for “neurological disease”, which applies to all subtypes, the number of genes in this category varies from subtype to subtype (table 2). Assuming that differential expression of these genes reflects, at least in part, the pathogenesis of CFS, the gene contribution to each disease association presumably affects the final phenotype and risk of complications (eg, lymphoma).31 32 It is also interesting that these genomically derived subtypes represent distinct clinical syndromes, and that those that were most severe were also those with anxiety/depression, as would be expected in a disease with a biological basis.
Oestrogen receptor signalling is implicated in CFS subtype 7. Interestingly, it has previously been reported that patients with CFS exhibit a downregulation of oestrogen receptor β.33 34 Oestrogen is an immunomodulator that has multiple effects on the immune system and on other hormones, which can themselves affect the immune response.35
Following repeat testing, and confirmation of these findings, it will be important to find a means by which we can determine the subtype of individual patients with CFS. For the purpose of subtype diagnosis, use of an 88-gene qPCR-derived signature is cumbersome and so it will be important to determine the most predictive genes within this signature, whose up- or downregulation reliably predicts subtype status. Using this approach, and depending on further research, we may then be able to use a shortlist of 10–20 CFS-associated genes to subtype individual patients in clinical settings.
We believe these 88 genes to reflect real biological features of these patients with CFS, and this is supported by the fact that differential expression of 16 of these genes has been reported previously by our group.22 If these findings are confirmed, there are various options for clinical trials using existing therapies that have been shown to be safe, based on targeting of key genes in patients of different CFS subtypes, namely IL6ST, TNF, CXCR4, APP and FNTA. Interestingly, one anti-tumour necrosis factor drug (etanercept) has already been trialled using an 8-week regimen in six patients with CFS, with reported clinical benefit in fatigue and pain in all subjects. Although this has not been published as a paper, the results were presented by Kristin Lamprecht and colleagues from Minnesota at the International Association for CFS meeting in Seattle in 2001.36 Unfortunately, this was not followed up because the Peterson group moved out of CFS research around this time (P Peterson, personal communication).
In conclusion, we report in detail the genomic and phenotypic differences in seven genomically defined subtypes of CFS. Further work is required to validate these findings, and this work is underway in our laboratory.
REFERENCES
Footnotes
Competing interests: None.
Funding: We thank Sir Joseph Hotung for funding of the salary of JK, and the CFS Research Foundation, Hertfordshire, UK, for generous funding of this project.