Article Text

Abstract
Background Leber congenital amaurosis (LCA) and juvenile retinitis pigmentosa (RP) are inherited retinal diseases that cause early onset severe visual impairment. An accurate molecular diagnosis can refine the clinical diagnosis and allow gene specific treatments.
Methods We developed a capture panel that enriches the exonic DNA of 163 known retinal disease genes. Using this panel, we performed targeted next generation sequencing (NGS) for a large cohort of 179 unrelated and prescreened patients with the clinical diagnosis of LCA or juvenile RP. Systematic NGS data analysis, Sanger sequencing validation, and segregation analysis were utilised to identify the pathogenic mutations. Patients were revisited to examine the potential phenotypic ambiguity at the time of initial diagnosis.
Results Pathogenic mutations for 72 patients (40%) were identified, including 45 novel mutations. Of these 72 patients, 58 carried mutations in known LCA or juvenile RP genes and exhibited corresponding phenotypes, while 14 carried mutations in retinal disease genes that were not consistent with their initial clinical diagnosis. We revisited patients in the latter case and found that homozygous mutations in PRPH2 can cause LCA/juvenile RP. Guided by the molecular diagnosis, we reclassified the clinical diagnosis in two patients.
Conclusions We have identified a novel gene and a large number of novel mutations that are associated with LCA/juvenile RP. Our results highlight the importance of molecular diagnosis as an integral part of clinical diagnosis.
- Genetic screening/counselling
- Clinical genetics
- Ophthalmology
- Vision research
Statistics from Altmetric.com
Introduction
Leber congenital amaurosis (LCA) refers to a group of inherited retinal dystrophies that share the common feature of severe visual impairment within the first year of life. Clinically, LCA is defined by congenital blindness, congenital nystagmus, and lack of detectable signals on an electroretinogram (ERG).1 ,2 LCA affects 1 in every 50 000 individuals, but it accounts for 5% of all retinal dystrophies and 20% of blindness in school age children.3 ,4 To date, mutations in 19 genes are reported to cause LCA.5–12 Despite the breadth of current knowledge, genetic defects in about 30% of LCA cases remain unknown.11
The clinical phenotypes and genetic causes of LCA and juvenile retinitis pigmentosa (RP) largely overlap. Both diseases belong to a spectrum of retinal diseases termed early onset retinal dystrophies (EORD). In fact, LCA was initially considered to be a congenital form of RP.2 Compared with LCA, juvenile RP tends to have milder phenotypes and a later onset. Juvenile RP patients appear to have better visual function at birth than those with LCA, and later develop night blindness, narrowed visual fields, and eventually severe vision impairment. Mutations in several known LCA genes, such as CRB1 and RDH12, are reported to cause juvenile RP.13 Interestingly, mutations in other retinal disease genes, such as IQCB1 and KCNJ13, are also known to be associated with LCA or ‘LCA-like’ phenotypes.10 ,11 These observations may be explained by a combination of allelic differences, genetic background, and environmental modifications. Also, it has been demonstrated that the clinical phenotypes of many retinal diseases overlap with that of LCA.11 It is likely that in some cases visual impairment is the most obvious phenotype in the initial evaluation, and that other syndromic features appear at a later time. Therefore, given the limited evaluation possible in infants and in early childhood, some patients initially diagnosed with LCA may actually have a different retinal disorder, such as Alström syndrome or Joubert syndrome.11 Despite these observations, systematic screening for mutations in all known retinal disease genes on a large LCA patient cohort has not yet been reported.
Because of the genetic heterogeneity of LCA and other retinal diseases, an accurate molecular diagnosis can improve the clinical diagnosis, facilitate a more accurate description of prognosis, and allow gene specific treatment. One of the most common methods for molecular diagnosis of LCA is the Arrayed Primer Extension (APEX) chip (Asper Ophthalmics). It is a microarray based genotyping method that tests a subset of known mutations in known LCA genes, leading to molecular diagnosis in approximately 17–32% of LCA patients.14–16 With additional mutations added to the LCA APEX array, the estimated solving rate has been improved to about 50%.11 On the other hand, next generation sequencing (NGS) has been recently used for the molecular diagnosis of retinal diseases.17 ,18 Compared with the APEX chip, the NGS based approach is able to discover novel variants and genes. Recently, Coppieters and others described a workflow to screen the exons of known LCA genes, using amplicon PCR followed by NGS.19 However, this workflow was tested on a relatively small LCA patient cohort and did not cover other retinal disease genes.
The goal of this study was to develop a comprehensive molecular diagnostic method for LCA and potentially for other retinal diseases. For this purpose, we developed a targeted NGS method that allows us to systematically screen the exons of most known retinal disease genes at low cost (163 genes at the time of design, online supplementary files 1 and 2). We first evaluated this method on a standard control sample, and then applied it to the molecular diagnosis of a large cohort of unrelated and prescreened patients with the clinical diagnosis of either LCA or juvenile RP (n=179). Pathogenic mutations for 72 patients were identified by systematic NGS data analysis, Sanger sequencing validation, and segregation analysis. These 72 patients were classified into different confidence groups based on the clinical significance of their mutations. Among the 72 patients, 58 carried mutations in known LCA or juvenile RP genes and exhibited corresponding phenotypes, while 14 carried mutations in retinal disease genes that were not consistent with their initial clinical diagnosis. Guided by the molecular diagnosis, we revisited 12 out of the 14 patients. We found that homozygous mutations in PRPH2 can cause LCA/juvenile RP. We also reclassified or refined the initial clinical diagnosis for 10 patients.
Methods
Study subjects
We initially collected a cohort of 389 patients from around the world and with a variety of backgrounds. Using a combination of LCA APEX array, Sanger sequencing, homozygosity mapping, and phenotype directed genotyping methods (eg, preserved para-arteriolar retinal pigment epithelium in an LCA patient is associated with mutations in CRB1), we had previously identified the genetic causes for 210 patients (most of whom are LCA patients).13 ,20 The remaining 179 patients were included in this study. The available prescreening information for the 179 patients is listed in online supplementary table S5.
The 179 patients were seen at McGill University (RKK), University of Pennsylvania (SGJ), The Lighthouse of Chicago (GAF), University of Tennessee Health Science Center (AI), and University of Michigan (JRH), by ophthalmologists with expertise in retinal dystrophies. Informed consents and research protocols were approved by the respective institutional review boards or research ethics board and adhered to the tenets of the Declaration of Helsinki. Complete histories, pedigree analysis, and ophthalmic examinations were performed. Eye exams consisted of cycloplegic refractions, fixation testing, Snellen visual acuities (when possible), pupillary responses, slit lamp exams, dilated fundus exam by indirect ophthalmoscopy, retinal photography, and Goldmann visual field testing (when possible). In most cases, ERGs were done according to ISCEV (International Society for Clinical Electrophysiology of Vision) standards. LCA is defined by the phenotypes mentioned in the introduction and the absence of overt systemic features. Juvenile RP represents a milder disease with later onset of signs and symptoms. In juvenile RP patients, vision can appear normal at birth, and the first symptom is progressive night blindness, with progressive visual loss at around age 2 years, with or without nystagmus.
DNA was extracted from whole blood using the FlexiGene kit or the QIAamp DNA blood kit according to the manufacturer's protocol. The quantity and quality of DNA were verified by using NanoDrop.
Target DNA capture and NGS experiments
According to the manufacturer's protocol, Illumina paired-end libraries were generated. Briefly, ∼1 µg of genomic DNA was sheared into fragments of approximately 300–500 bp. The DNA fragments were end-repaired and an extra ‘adenine’ base was added to the 3′ end. Illumina Y-shape index adapters were ligated to the ends of the DNA fragments and eight cycles of PCR amplification were applied to each sample after ligation. The DNA libraries were quantified by the PicoGreen assay (Invitrogen). For each capture reaction, 24 to 48 libraries (3 µg of DNA in total) were pooled together. A design file (see online supplementary files 1 and 2) was submitted to Nimblegen for the design of the capture probe. NimbleGen SeqCap EZ Hybridisation and Wash Kits were used for the washing and recovery of captured DNA. Captured libraries were quantified and sequenced on the Illumina HiSeq 2000 as 100 bp paired-end reads, following the manufacturer's protocols. Illumina sequencing was performed at the BCM-FGI core.
Evaluation of our method's sensitivity to detect SNPs on the Hapmap sample
Single nucleotide polymorphisms (SNPs) genotyping data of HapMap sample NA11831 were downloaded from 1000 Genome omni database (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20110527_bi_omni_1525_v2_genotypes/). This sample had been genotyped using the Illumina OMNI2.5 SNP genotyping array. A total of 1190 genotyped single nucleotide polymorphisms (SNPs) in this sample are within our design region, including 919 homozygous reference SNPs, 107 homozygous alternative SNPs, and 164 heterozygous alternative SNPs. A total of 1184 SNPs were detected by our targeted NGS method. Among the detected SNPs, 1183 out of 1184 had the same genotype between the SNP array and NGS. The single disconcordant SNP rs3763073 was heterozygous C/T on the SNP array but homozygous C/C in targeted NGS. To resolve the conflict, we performed direct Sanger sequencing and confirmed that rs3763073 was indeed homozygous for the reference C, indicating that NGS detected the SNP correctly (data not shown).
Data analysis
Sequencing reads were aligned to the human genome reference version hg19 using Burrows-Wheeler Aligner (BWA).21 Base quality recalibration and local realignment were performed using the Genome Analysis Toolkit (GATK).22 AtlasSNP was used for SNP calling and AtlasIndel2 was used for indel calling.23 The 1000 genome database, dbSNP, ESP5400, NIEHS95 exomes, and our internal database were used to filter out common SNPs and indels, with allele frequency cutoffs at 0.5% for recessive variants and at 0.1% for dominant variants (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, Washington (http://evs.gs.washington.edu/EVS/), NIEHS Environmental Genome Project, Seattle, Washington (http://evs.gs.washington.edu/niehsExome/).24 ,25 Variant annotation was performed using ANNOVAR.26 The Refseq gene sequences below were used for the mutation coordinates: AIPL1:NM_014336, ALMS1:NM_015120, BBS1:NM_024649, BBS7:NM_018190, CEP290:NM_025114, CERKL:NM_001160277, CLN3:NM_001042432, CRB1:NM_201253, GUCY2D:NM_000180, INPP5E:NM_019892, IQCB1:NM_001023570, LCA5:NM_181714, LRAT:NM_004744, NR2E3: NM_016346, OTX2:NM_172337, PDE6A:NM_000440, PRPF31:NM_015629, RDH12:NM_152443, RPE65:NM_000329, RPGR: NM_000328, RPGRIP1:NM_020366, SAG:NM_000541, SNRNP200:NM_014014, SPATA7:NM_018418, TULP1:NM_003322. The pathogenicity of novel missense mutations was predicted by dbNSFP, whose prediction score is derived from five algorithms (SIFT, Polyphen2, LRT, MutationTaster, and PhyloP).27–32
PCR and direct Sanger sequencing
To validate the mutations detected by NGS, primers were designed (Primer3, http://biotools.umassmed.edu/bioapps/primer3_www.cgi) to PCR-amplify the 400–500 bp region flanking the mutation. To ensure the high quality of Sanger sequencing, the amplicon was designed to have a boundary at least 50 bp away from the mutation. The amplicon was then Sanger sequenced on Applied BioSystems (ABI) 3730×l capillary sequencer. The Sanger sequencing results were analysed with Sequencher software. The intronic mutation c.2991+1655A>G in CEP290 was not initially included in the original design of our exonic capture panel. Sanger sequencing of this mutation was performed and the results were combined with the NGS data.
Results
A cohort of 179 patients clinically diagnosed with LCA or juvenile RP
After prescreening for known mutations in LCA and juvenile RP genes using a combination of conventional genotyping methods, the genetic defects in 173 LCA and six juvenile RP patients remained unexplained (see online supplementary table S1 and Methods). We hypothesised that a portion of these cases were caused by mutations in known LCA and juvenile RP genes that were not included in the conventional screening methods, or caused by mutations in other retinal disease genes that had not been previously associated with LCA or juvenile RP. To Sanger-sequence all known retinal disease genes for such a large sample set would be prohibitively expensive and time consuming. Therefore, we utilised a targeted NGS based method for the comprehensive molecular diagnosis of these patients.
Targeted NGS of a standard control sample from HapMap project
A capture panel was designed to enrich the target DNA, which consisted of 649 804 bp covering 2560 exons in 163 known retinal disease genes that had been reported and recorded in the RetNet at the time of design (see online supplementary files 1 and 2, https://sph.uth.tmc.edu/retnet/). The enriched DNA was then sent for NGS.
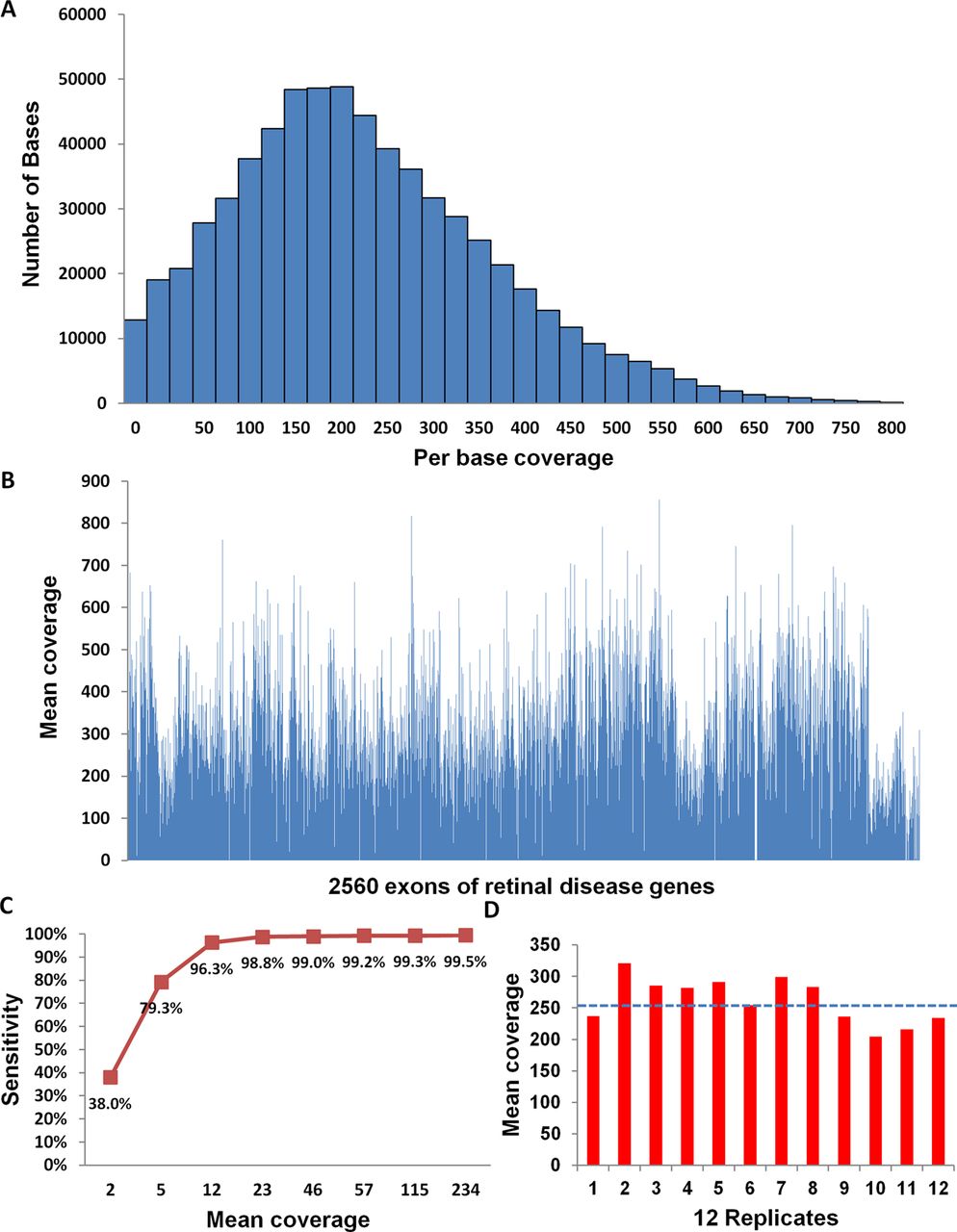
We first evaluated the coverage of our targeted NGS method on NA11831, a standard control sample from the original HapMap Centre d'Etude du Polymorphisme Humain (CEPH) cohort.33 DNA from NA11831 was captured and sequenced at high coverage. A total of 8 240 805 mappable reads were obtained, 39% of which mapped to the target region and resulted in a 234× mean per base coverage. As shown in figure 1A and B, the vast majority of the targeted regions were well covered. Indeed, 97% of the bases in target region had coverage >10× and 92% of the bases had coverage >50× (figure 1A). Also, 98% of the 2560 exons had mean coverage >5× (figure 1B). The low coverage exons were either within duplicate regions or those with a high GC content (see online supplementary tables S2 and S3).

Systematic evaluation of targeted next generation sequencing method on a Hapmap sample NA11831. (A) The number of bases within different coverage groups. (B) The mean coverage of 2560 exons in the design region. (C) The sensitivity to detect single nucleotide polymorphisms versus the according mean coverage. (D) The high and uniform mean coverage for 12 multiplexed replicates. The blue dot line represents the average coverage for 12 replicates.
To systematically evaluate the accuracy of our method, we compared the genotyping data obtained from NGS to that from the SNP array. As part of the 1000 Genome project, sample NA11831 had been genotyped using the Illumina OMNI2.5 genotyping array. A total of 1190 genotyped SNPs in this sample were within our design region and were used as standards to test the accuracy of our method. As a result, 99.5% of SNPs (1184/1190) were detected by NGS (minimum coverage=3). The six undetected SNPs were within low coverage exons (data not shown). The genotypes of all 1184 NGS SNP calls were validated by either SNP array or Sanger sequencing (see Methods). Therefore, high quality SNP genotyping results were obtained by targeted NGS with a sensitivity of 99.5% (1184/1190) and a genotype concordance of 100% (1184/1184).
To further explore the effect of coverage on the sensitivity of SNP detection, sequencing reads generated from NA11831 were randomly sampled in silico to achieve different levels of coverage from 3× to 234×. As shown in figure 1C, sensitivity increased sharply from 38% to 96% as the coverage increased from 2× to 12×, then gradually reached a sensitivity of 99% at around 23×. Based on this result, we chose to sequence patient samples at around 50× coverage to achieve nearly saturated sensitivity with a relatively low cost (cost is linear to the depth of coverage). At 50× coverage, up to 100 samples could be sequenced in one lane of Illumina HiSeq 2000. To develop a more cost effective method, we tested the robustness of sample multiplexing. We molecularly barcoded 12 replicates of NA11831 DNA and performed targeted NGS for these 12 replicates in one capture reaction. As shown in figure 1D, uniform and high coverage of these replicates was achieved.
Targeted NGS of 179 patients
Using the capture panel described above, we applied targeted NGS to DNA obtained from a large cohort of 179 unrelated patients with the diagnosis of LCA or juvenile RP. The sequencing reads were processed by our bioinformatics pipeline that performed reads alignment, recalibration, realignment, variants calling, filtering, annotation, and quality control (see Methods). An average of 62× coverage was achieved for the 179 patient samples. Within the design region, 84% of bases had coverage >10× and 70% of bases had coverage >20×, indicating that sufficient coverage was achieved for high sensitivity of variants detection (table 1, figure 2A). For each individual, about 407 SNPs and small insertions/deletions (indels) were identified. Since LCA and juvenile RP are rare Mendelian diseases, common variants with a frequency >0.5% (for recessive variants) or >0.1% (for dominant variants) in any of the following databases were filtered out: the 1000 genome database, dbSNP135, the ESP5400 database, the NIEHS 95 exomes database, and our internal database (see Methods). As a result, an average of eight rare variants in retinal disease genes that lead to protein coding change were identified per sample (table 1). Furthermore, mutations known to cause retinal diseases in the Human Gene Mutation Database (HGMD) or the primary literature were identified.34 Finally, dbNSFP, a program that compiles prediction score from five well established prediction algorithms (PhyloP, SIFT, Polyphen2, LRT, and MutationTaster), was used to predict the pathogenicity of novel missense changes.27–32 In this study, we only reported novel missense variants that are predicted to be pathogenic by at least three of the five algorithms (see online supplementary table S4). After all these stringent filtering steps, the remaining variants are likely to cause the disease in patients.
Target NGS statistics for 179 patients

The targeted next generation sequencing statistics for 179 patients. (A) The percentage of bases in design region in each coverage group for 179 patients. (B) The percentage of 179 patients in different categories. (C) The percentage of different types of pathogenic mutations identified in the 72 patients. LCA, Leber congenital amaurosis.
Identification of pathogenic mutations
To identify the potential pathogenic mutations among several rare variants in each patient, we looked for variants that matched the reported inheritance pattern of the respective genes:
-
Homozygous or compound heterozygous variants in recessive retinal disease genes, or
-
Reported heterozygous variants known to cause dominant retinal diseases, or
-
Novel heterozygous loss-of-function (LOF) variants in dominant retinal disease genes, if heterozygous LOF mutations in those genes are previously known to cause dominant retinal diseases.
All potential pathogenic variants identified above were validated by Sanger sequencing. Segregation analysis was performed if DNA from family members was available. Through this procedure, we identified pathogenic mutations for 72 out of 179 patients (40%). Among the 72 patients, 58 patients carried mutations in known LCA or juvenile RP genes and exhibited corresponding phenotypes, while 14 harboured mutations in retinal disease genes that were not consistent with their initial clinical diagnosis (figure 2B). A total of 83 distinct pathogenic mutations were identified in the 72 patients, including a large number of novel mutations (n=45) (table 2). Most of these mutations were missense (39%) and nonsense (35%) mutations (figure 2C).
Number of pathogenic mutations identified in this study
Molecular diagnosis of patients
Patients carrying mutations in known LCA or juvenile RP genes
In total, we identified 58 patients who carried mutations in known LCA or juvenile RP genes and exhibited corresponding phenotypes (tables 4⇓–6). According to the American College of Medical Genetics standards to report sequence variants, mutations identified in our study can be classified into three categories with different clinical significance: (1) reported mutations that are known to cause retinal diseases; (2) novel LOF mutations that are expected to cause retinal diseases; (3) novel missense mutations that are predicted to be pathogenic by in silico prediction algorithms and may be causative of retinal diseases (see online supplementary table S4).35 To demonstrate the different confidence levels for different patients, we classified these patients into three groups based on the clinical significance of their mutations: patients in group 1 and 2 carried reported or novel LOF mutations with higher confidence, while patients in group 3 harboured one or more novel missense mutations with lower confidence (table 3).
Classification of patients according to clinical significance of mutations
We identified 26 patients in group 1 who carried homozygous or compound heterozygous mutations that were known to cause recessive LCA or juvenile RP (tables 3 and 4, online supplementary table S1). For example, patient 3916 carried compound heterozygous reported nonsense mutations c.582C>G (p.Y194X) and frameshift deletion c.805_809del (p.A269GfsX2) in RDH12 (table 4). The patient exhibited LCA phenotypes and the two mutations were previously known to cause LCA (see online supplementary figure S1, table S1).36 ,37 In this group of patients, AIPL1 was the most frequently mutated gene, which appeared in five patients. The nonsense mutation c.834G>A (p.W278X) in AIPL1, the intronic mutation c.2991+1655A>G in CEP290, and the frameshift insertion c.805_809del (p.A269GfsX2) in RDH12 were the most frequent mutations, all carried by three patients.
Twenty-six patients carrying two reported mutations in LCA or juvenile RP genes
We identified 22 patients in group 2 who carried novel LOF mutations in known LCA or juvenile RP genes (tables 3 and 5). First, 13 patients carried homozygous or compound heterozygous novel LOF mutations. For example, a novel homozygous frameshift deletion c.613_614del (p.S205YfsX27) was identified in exon3 of LRAT in patient 4019. To our knowledge, this is the first reported disease allele outside LRAT exon2.48 ,51–53 The c.613_614del is predicted to change the 205–230 amino acids in the C terminus of LRAT protein, which is thought to be important for the LRAT protein enzymatic activity and its localisation to the endoplasmic reticulum membrane.54 ,55 Second, eight patients carried one reported mutation plus one novel LOF mutation. Third, patient 3561 carried a novel heterozygous frameshift insertion in OTX2. This insertion is likely to be pathogenic because a heterozygous protein truncating mutation in OTX2 was previously reported to cause ocular malformation and LCA.56 In this group of patients, CEP290 was the most frequently mutated gene, which appeared in seven patients.
Twenty-two patients carrying novel LOF mutations in LCA or juvenile RP genes
We identified 10 patients in group 3 who carried one or more novel missense mutations in known LCA or juvenile RP genes (table 6). Specifically, four patients carried homozygous or compound heterozygous novel missense mutations, three patients had a novel missense mutation plus a reported mutation, and three patients had a novel missense plus a novel LOF mutation (table 3). For example, patient 3319 carried a homozygous novel missense mutation (c.1439G>C, p.C480S) in CRB1 that changes a cysteine to a serine. The cysteine is conserved across mammals and this mutation is predicted to be damaging to protein function/structure by in silico prediction (see online supplementary table S4). Interestingly, similar missense mutations p.C480R and p.C480G at this residue were reported to cause LCA, further supporting the pathogenicity of p.C480S.43 In this group of patients, GUCY2D was the most frequently mutated gene, which appeared in four patients.
Ten patients carrying one or more novel missense mutations in LCA or juvenile RP genes
Patients carrying mutations in other retinal disease genes
We also identified 14 patients who carried mutations in retinal disease genes that were not consistent with their initial clinical diagnosis, representing 19% of the 72 diagnosed patients. Using the criteria mentioned above, we classified these 14 patients into three groups based on the clinical significance of their mutations.
We identified eight patients in group 1 who carried reported mutations known to cause retinal disease genes that were not consistent with their initial clinical diagnosis (tables 3 and 7). Within this group, seven patients carried homozygous mutations. In addition, juvenile RP patient 3311 carried a heterozygous reported mutation known to cause autosomal dominant RP (adRP).
Eight patients carrying mutations known to cause other retinal diseases
We identified two LCA patients in group 2 who carried homozygous or compound heterozygous novel LOF mutations in other retinal disease genes (tables 3 and 8). For example, patient 3688 carried a hemizygous novel splice site mutation c.248–1G>T in RPGR. Previously reported splice site mutations in RPGR were known to cause X-linked RP, supporting that this mutation may cause the retinal defects in patient 3688.
Two patients carrying novel LOF mutations in other retinal disease genes
We identified four patients in group 3 who carried homozygous novel missense mutations in retinal diseases genes that were not consistent with their initial clinical diagnosis (tables 3 and 9). For example, patient 1327 carried a homozygous novel missense mutation c.728G>A (p.C243Y) in Bardet–Biedl syndrome (BBS) gene BBS7. This mutation changes a cysteine residue that is conserved across vertebrates. It was predicted to be damaging by all of the five in silico prediction algorithms, supporting that this mutation is likely to be pathogenic (see online supplementary table S4).
four patients carrying homozygous novel missense mutations in other retinal disease genes
Revisiting patients carrying mutations in other retinal disease genes
In our study we observed that 14 patients carried mutations in genes that were not consistent with their initial clinical diagnosis. This observation may be explained by novel genotype–phenotype correlations, or by the difficulty assigning clinical diagnosis at the time of initial visit. In most cases, the first visit of a blind or low vision infant occurs shortly after birth. The initial clinical diagnosis may be difficult and influenced by the most obvious ophthalmic and visual findings at that time. To test these two possibilities, we managed to revisit 12 of these 14 patients.
Homozygous mutations in PRPH2 cause EORD with LCA/juvenile RP phenotypes
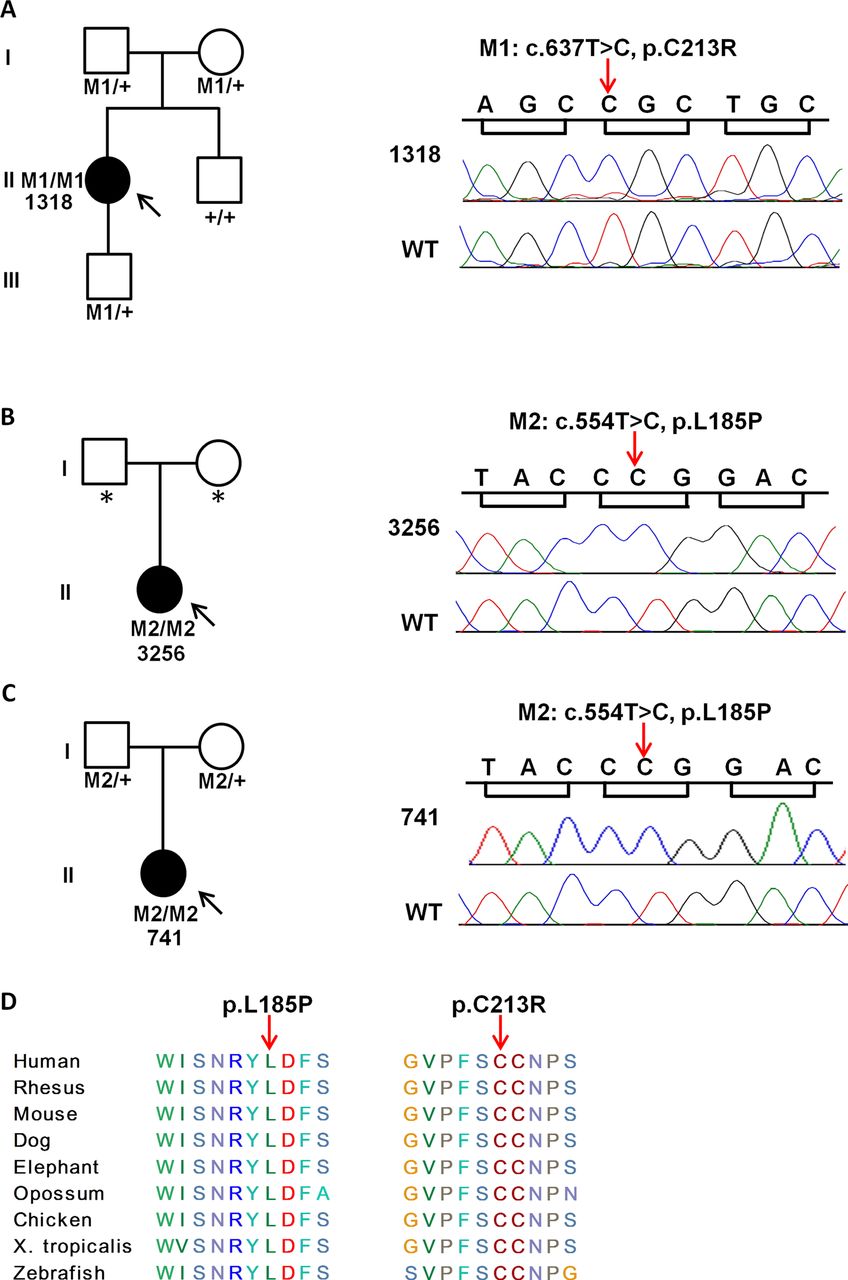
After revisiting, we confirmed the clinical diagnosis of LCA in patients 1318 and 3256 (figure 3, online supplementary table S1). Each patient carried a reported homozygous missense mutation in gene PRPH2: c.637T>C (p.C213R) and c.554T>C (p.L185P), respectively (table 7). PRPH2 encodes peripherin, a membrane glycoprotein that is important for the stabilisation and compaction of photoreceptor outer segment discs.68 The p.C213R mutation is associated with autosomal dominant pattern dystrophy, and the p.L185P mutation, together with a null mutation in ROM1, has been reported to cause digenic RP.65 ,66 However, it has not been reported that homozygous mutations in PRPH2 cause severe EORD. To further validate this finding, we sequenced PRPH2 in another 135 unsolved LCA or juvenile RP patients and found the same homozygous missense mutation p.L185P in PRPH2 in a third juvenile RP patient, 741. These mutations were confirmed by Sanger sequencing and their segregations with the disease in the families were examined (figure 4). All the patients with homozygous mutations in PRPH2 exhibited LCA or juvenile RP phenotypes, including visual impairment within the first year of life, nystagmus in the two LCA patients (1318 and 3256), non-detectable or reduced ERGs, and a very similar form of maculopathies in the fundus (figure 3, online supplementary table S1). By contrast, family members who carried heterozygous mutations in PRPH2 were asymptomatic but showed detectable maculopathy phenotypes. For example, the 56-year-old father of patient 741 had macular pattern dystrophy and clear-cut foveal changes, but his visual acuity was essentially normal in both eyes (see online supplementary figure S2A–C, table S1). Similarly, the mother and the son of patient 1318 were both carriers of the mutation c.637T>C (p.C213R) (figure 4). At 57 years of age, the mother was asymptomatic with 20/20 visual acuity but had a florid butterfly-shaped macular pattern dystrophy and a number of other retinal flecks upon examination (see online supplementary figure S2D). The 7-year-old son had a significant refractive defect whereby visual acuity was reduced due to partial amblyopia. His fundus showed a miniature form of foveal butterfly-shaped macular pattern dystrophy that was consistent with an early stage PRPH2 related phenotype (data not shown). The brother of patient 1318, who was homozygous wild-type for the mutation, had normal visual acuity (20/20) and no maculopathy (data not shown). To our knowledge, our study reported for the first time that homozygous mutations in PRPH2 cause EORD with LCA/juvenile RP phenotypes.
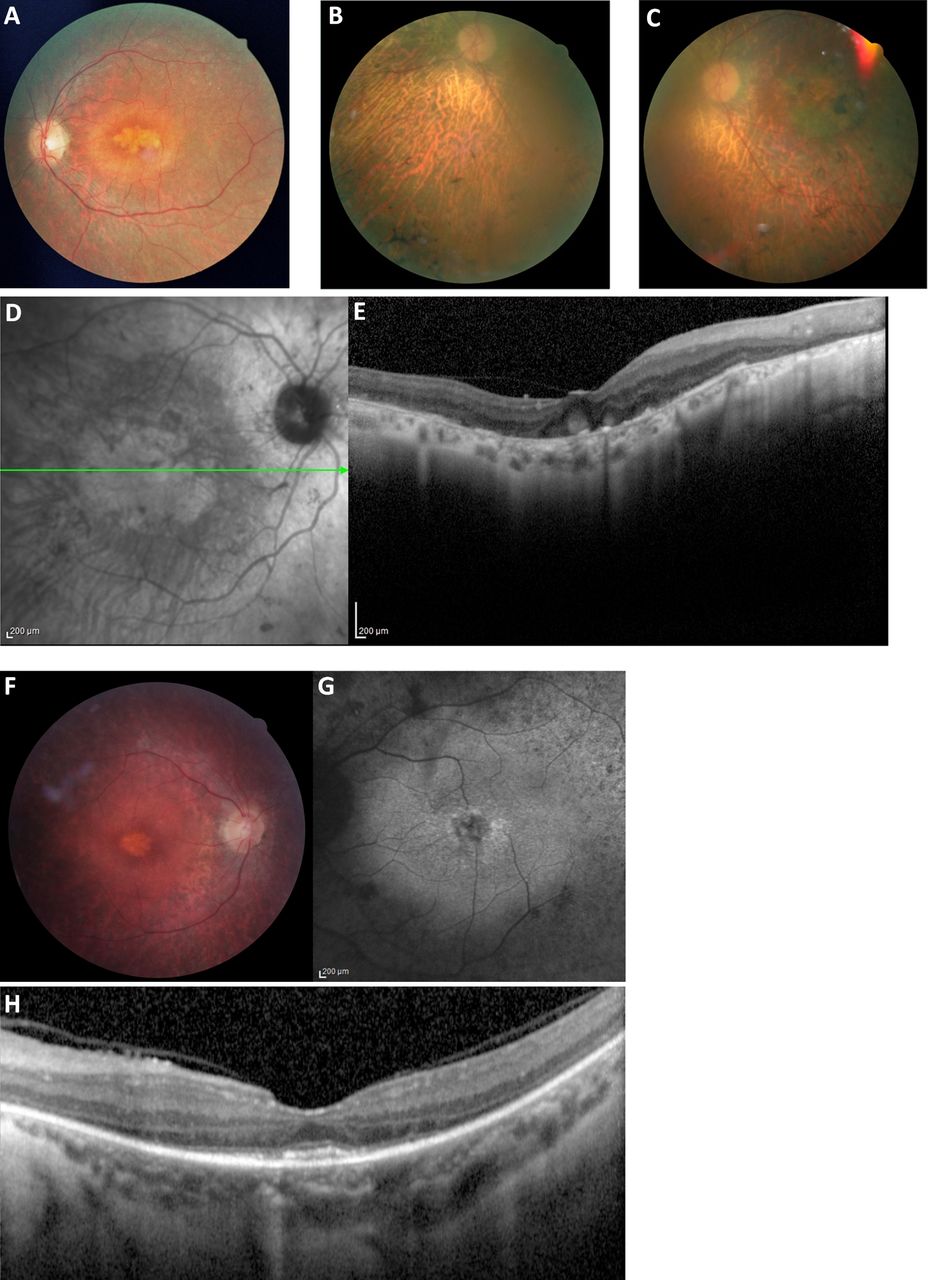
The phenotypes of patients 1318, 3256, and 741 who carry homozygous mutations in PRPH2. (A) Fundus photograph of patient 1318 shows a prominent multilobulated central atrophic maculopathy surrounded by concentric rings of yellow deposits, with vessel narrowing and fine diffuse peripheral retinal changes. (B–D) Fundus photographs of patient 3256 show pigment deposits both peripherally and in the macular region, extensive disease with choroidal sclerosis, vessel narrowing, and optic disc pallor; a central extensive atrophic maculopathy is seen in C and D. (E) Optical coherence tomography images of patient 3256 confirm the extensive maculopathy and unusual globular lesions in the foveal region. (F) Fundus photograph of patient 741 shows the obvious diffuse retinal dystrophy with retinal vessel narrowing, retinal pigment epithelium mottling and loss, and a multilobulated maculopathy. (G) Fundus autofluorescence of patient 741 shows diffuse retinal abnormalities, perifoveal hyper-autofluorescence and a multilobulated foveal abnormality. (H) Optical coherence tomography images of patient 741 shows inner segment/outer segment junction confined to the central macula, which explains well the fairly good visual acuity (20/40), and an unusual appearing deposit in the foveal regions. Note the similarity with the maculopathy shown in A, D, F, and G. The photographs of patients 1318, 3256, and 741 were taken at the age of 29, 66, and 30 years, respectively.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pathogenic mutations of PRPH2 identified in three patients. (A–C) Pedigree information of three patients and Sanger sequencing results for the two mutations in patients and controls. (A) The c.637T>C mutation is homozygous in patient 1318, heterozygous in both parents and the son, and homozygous wild-type in the brother. (B) The c.554T>C mutation is homozygous in patient 3256. (C) The c.554T>C mutation is homozygous in patient 741 and heterozygous in both parents. (D) Amino acid residues affected by the two missense mutations are conserved across different species. Solid symbols: affected; open symbols: unaffected; squares: male; circles: female; arrow: proband; asterisk: the DNA was not available for both parents of 3256; M1 and M2 represent the two mutations, respectively.
Revision of the initial clinical diagnosis in two patients
After revisiting, two patients were reclassified to retinal diseases that were consistent with their molecular diagnosis (tables 7 and 8, online supplementary table S1). The clinical diagnosis of the first patient 3425 who carried a reported homozygous nonsense mutation in the known Oguchi disease gene SAG was revised to Oguchi disease, which presents as congenital stationary night blindness, fundus discolouration, and slowed dark adaptation.67 The second patient 3494 carried novel compound heterozygous nonsense mutations in the Alström syndrome gene ALMS1 (Otable 8). Both mutations segregated with the disease in the family (see online supplementary table S1). Patient 3494 was initially diagnosed with LCA at the age of 8; however, revisiting this patient at the age of 11 revealed other syndromic features including obesity, diabetes mellitus, and learning difficulties (see online supplementary table S1). Furthermore, the fundus examination showed an atrophic bull's eye-like maculopathy, which was often seen in Alström syndrome patients (see online supplementary figure S3). These results indicate that molecular diagnosis can be a useful tool to revise or correct the initial clinical diagnosis.
The LCA-like or juvenile RP-like presentations in eight patients
Guided by the molecular diagnosis, revisiting the phenotypes of an additional eight patients revealed their ‘LCA-like’ or ‘juvenile RP-like’ phenotypes that may represent spectrums of corresponding retinal diseases (see online supplementary table S1). For example, patient 3688 carried a novel hemizygous splicing site mutation in X-linked RP gene RPGR (table 8). This patient exhibited ‘LCA-like’ phenotypes including nystagmus at birth, which is typically absent in X-linked RP (see online supplementary table S1). However, it is already known that X-linked RP patients may lose central and peripheral vision more rapidly than average RP patients.69 Similarly, two patients (647, 617) carried reported mutations in cone–rod dystrophy gene CERKL and enhanced S-cone syndrome gene NR2E3, respectively (table 7). They exhibited ‘LCA-like’ phenotypes, including congenital visual impairment and nystagmus at birth (see online supplementary table S1). However, based on the available clinical information, the phenotypes of the two patients may also represent severe spectrums of cone–rod dystrophy and S-cone syndrome, respectively. Patient 3311 carried a heterozygous mutation in PRPF31 that is known to cause RP with late onset and mild phenotypes.64 This patient exhibited early onset ‘juvenile RP-like’, possibly due to modifier effect from another gene (see online supplementary table S1, figure S4).
In addition, four patients (704, 1327, 3748, and 3773) carried mutations in BBS1, BBS7, CLN3, and INPP5E, respectively (tables 7 and 9). Mutations in these genes were known to cause syndromes that are characterised by visual impairment and other systemic features.70–72 It was also reported in some cases that these genes were associated with ‘LCA-like’ or ‘RP-like’ phenotypes without defects in other organs.60 ,62 ,73 In our study, revisiting these patients confirmed their severe retinal degenerations without other syndromic features (see online supplementary table S1). For example, patient 704 carried a reported homozygous missense mutation in BBS gene BBS1 (table 7). This mutation segregated with disease within the family (see online supplementary table S1). Revisiting this patient at the age of 53 confirmed the ‘juvenile RP-like’ phenotypes without other syndromic features (see online supplementary table S1). However, the retinal features of this patient were consistent with those observed in other BBS patients with BBS1 mutations (see online supplementary figure S5).72 Due to these molecular findings and retinal features, we should still follow up the potential development of syndromic phenotypes in these patients.
Collectively, these results suggest that the clinical manifestations of LCA/juvenile RP and related retinal diseases are overlapped, and that patients with ‘LCA-like’ or ‘juvenile RP-like’ phenotypes may actually carry mutations in non-canonical LCA/juvenile RP genes.74 Therefore, molecular diagnosis should be used to refine the clinical diagnosis and get a better understanding of the disease. To achieve a more accurate diagnosis for these patients, it is essential to screen for mutations in a larger set of retinal disease genes.
Discussion
In this study, we developed a targeted NGS based method for the molecular diagnosis of LCA and most other retinal diseases. We systematically evaluated this method on a HapMap sample and then applied it to 179 unrelated and prescreened LCA or juvenile RP patients. To our knowledge, our sample set represents the largest cohort of unrelated patients diagnosed with LCA or juvenile RP that is systematically screened for all known LCA genes and most other known retinal disease genes. In-depth analysis of this dataset led to several important findings.
A large number of novel mutations have been identified in our study, representing 54% (45/83) of the identified mutations in this patient cohort (table 2). Our observations are consistent with the 1000 genome project's finding that every individual's genome contains a large number of rare variants.25 Compared with common variants that arose earlier during the evolution, these recent rare variants may have greater impact on disease pathogenesis.75 Therefore, we expect that a significant number of novel mutations will continue to be discovered every time a new patient is sequenced. Since NGS based molecular diagnosis can capture novel mutations, it is likely to achieve a high diagnosis rate. Among the 45 novel mutations that we identified, 29 were LOF mutations and 16 were missense mutations. All these novel mutations are likely to be pathogenic. First, these mutations are rare in large control databases. Collectively the databases used in our study contain more than 7400 control individuals. Second, all of these mutations match the reported inheritance pattern of the respective genes. In particular, the pathogenicity of all novel missense mutations reported in our study was supported by five well-established algorithms (see online supplementary table S4). Our study adds a significant number of novel pathogenic mutations to our current knowledge of disease causing mutations. These mutations can serve as references and directly benefit the future molecular diagnosis of patients clinically diagnosed with LCA or juvenile RP.
We identified the genetic defects in 40% of our patient cohort. This lower ratio is primarily due to the fact that our patient cohort had been prescreened. Among the initial cohort of 389 patients, we had previously identified mutations in known LCA genes for 210 patients (see Methods). Among the remaining 179 patients included in this study, mutations in known LCA genes were identified in 56 patients. Therefore, about 68% ((210+56)/389) of our initial cohort can be explained by mutations in known LCA genes. This is concordant with the estimation that mutations in current known LCA genes explain about 70% of LCA cases.11 Among the 56 patients who carry mutations in known LCA genes, 24 patients have prescreening information available (see online supplementary table S5). We found that 16 patients had neither been screened by LCA APEX array nor been Sanger sequenced for the corresponding genes identified in this study. The remaining eight patients had been screened by LCA APEX array and/or Sanger sequencing for the corresponding genes identified in this study. Their mutations had not been identified in the prescreening because the mutations had not been covered by LCA APEX array and/or because Sanger sequencing only covered the frequently mutated exons of related genes.15
To our knowledge, our results demonstrate for the first time that homozygous mutations in PRPH2 cause EORD. The phenotypes of the three patients with homozygous mutations in PRPH2 were severe and quite consistent, especially with regard to the maculopathy phenotypes. By contrast, their family members who carried heterozygous mutations in PRPH2 had milder phenotypes. These results are consistent with the previous observations in PRPH2 mouse models. The rds/rds mouse that carried a homozygous null mutation in PRPH2 failed to develop photoreceptor outer segments and showed early onset and severe retinal degeneration, whereas the heterozygous rds/+mouse displayed milder retinal degeneration and visual loss, suggesting that dose dependent phenotypic expression is an essential feature in the working of the PRPH2 gene.76 ,77 Until the discovery of these three patients homozygous for PRPH2 mutations, the full severity of the retinal degeneration seen in the rds/rds mouse had not yet been observed in humans. In our study, individuals with heterozygous mutations in PRPH2 were asymptomatic but had detectable macular flecks upon subsequent examination, exhibiting the clinical presentation of a macular pattern dystrophy, which is fully consistent with previously reported PRPH2 mediated phenotypes.65 By contrast, the severe early onset retinal defects in the three patients with homozygous mutations in PRPH2 are novel and likely due to dose dependent effect.
It may be argued that the rds/rds mouse and our patients harboured different mutations and that individuals with the heterozygous p.L185P mutation in previously reported digenic RP families were originally reported as asymptomatic.78 However, the p.L185P mutation is now known to exert a measurable partial LOF effect. Work from Molday and co-workers established that this peripherin mutant is conditionally defective with respect to subunit assembly, and is capable of forming peripherin dimers but not tetramers.79 ,80 Furthermore, Kedzierski et al have shown that rds/+mice overexpressing L185P peripherin mutant indeed exhibited a mild phenotype. These mice had outer nuclear layer loss, partially disorganised outer segments, and reduced ERG responses. As observed in our patients homozygous for the p.L185P mutation, rds/rds mice overexpressing L185P peripherin mutant exhibited dramatically reduced levels of peripherin expression in their retinas, and a much more severe histological and electroretinographic retinal phenotype.81 Taken together, these findings suggest that, although asymptomatic, individuals harbouring the heterozygous p.L185P mutation should be expected to exhibit a subclinical phenotype and it is possible that, as in our cases, later in life they may all consistently develop asymptomatic macular flecks or other minor yet measurable phenotypic manifestations.
Interestingly, similar examples have been reported for many other genes.82 ,83 PITX3 is a gene that is usually mutated in dominant congenital cataracts and anterior segment dysgenesis. However, patients with two mutations in this gene exhibited microphthalmia and central nervous system (CNS) abnormalities.84 For another example, homozygous mutations in the low density lipoprotein (LDL) receptor gene were known to cause much more severe phenotypes of hypercholesterolaemia than heterozygous mutations.85
From a therapeutic standpoint, the implication for patients with retinal degenerations caused by homozygous mutations in PRPH2 is that their diseases can be modelled by the rds/rds mouse, which has been treated by gene augmentation therapy in proof-of-concept research.86 ,87 There is also a long history of investigation of the severe phenotype in this model, features of which can now be studied in the patients to determine how representative the model is in relation to the newly identified human condition.
There are two main explanations for the 60% of our patients for whom we were unable to find pathogenic mutations in this study. First, mutations that were not covered by our method, including intronic mutations, synonymous mutations, large structural variations, and copy number variations, may account for diseases in these patients. Second, these unsolved cases may due to novel disease-causing genes. Indeed, whole exome sequencing (WES) of some of these unsolved cases has led to the identification of a novel LCA gene NMNAT1.6 Therefore, we expect that additional novel disease-causing genes will be identified by performing WES on these unsolved cases.
Our results highlight the utility of molecular information in diagnosing clinically heterogeneous diseases. Assigning clinical diagnosis at the time of initial visit is difficult in some cases, and molecular diagnosis can guide the health care provider to reassess the phenotypes of their patients and achieve a more accurate diagnosis. Indeed, guided by their molecular diagnosis, two patients in our study were reclassified with other retinal diseases. Additionally, the clinical manifestations of different retinal diseases are sometimes overlapped, and molecular diagnosis can help us to better define the disease. In our report, eight patients exhibited ‘LCA-like’ or ‘juvenile RP-like’ presentations. Based on the available clinical information, the diagnosis of these patients may be either LCA/juvenile RP, or extreme spectrums of other related retinal diseases, due to the allelic differences or genetic background. Despite the phenotypic similarity between different clinical diagnoses, diseases can be well defined by the molecular diagnosis. Therefore, with the rapid drop of sequencing costs, comprehensive mutation screening that covers all known retinal disease genes should become an integral part of diagnosis in the near future.
In addition to aiding the diagnosis, molecular information can directly contribute to better patient management. Recently, studies on gene therapy for LCA have made significant progress.88–91 An accurate molecular diagnosis is the first step toward realising the promise of gene therapy. Additionally, it can clarify the prognosis and change the focus of the clinical follow-up. Patients with different molecularly defined diseases may receive a different prognosis and clinical interventions. For example, patients who exhibit LCA phenotypes but carry mutations in syndromic retinal disease genes should be followed for the development of syndromic features and be given corresponding clinical management. Finally, it can facilitate the genetic counselling and decision-making. Carrier tests or predictive tests for retinal diseases can inform prospective parents of their reproductive risk and possibly influence their decisions.
The low coverage regions in our design either had a higher GC content or were within duplicate regions (see online supplementary tables S2 and S3). Indeed, the GC content bias of coverage in Illumina sequencing data has been previously reported and the bias could be potentially introduced in many steps during sequencing.92–94 It was recently recognised that PCR amplification before sequencing may be the major source of GC content bias; protocols to minimise such bias were proposed accordingly.95 ,96 In addition, low coverage in duplicated regions is likely due to the inability to map reads to a single unique position. The relatively shorts reads (90∼300 bp) generated by most currently available NGS platforms lack enough sequence specificity to be mapped to a single location among multiple duplicated regions. To uncover the genomic information of duplicated regions, long range PCR or NGS sequencer producing longer reads may be utilised.
In summary, we were able to identify pathogenic mutations for 40% of this prescreened patient cohort. A total of 45 novel pathogenic mutations were found. Interestingly, we found that homozygous mutations in PRPH2 can cause LCA and juvenile RP. Our study highlighted the utility of comprehensive molecular information as an integral part of the diagnosis process to achieve more accurate diagnosis and potentially better disease treatment and management.
Acknowledgments
We sincerely thank all the patients and their families for their participation. We thank Ms Shirley Briand, Ms Alcira Vieiri and Ms Renee Pigeon for coordinating the blindness clinics at the McGill Ocular Genetics Laboratory. XW is supported by predoctoral fellowship: The Burroughs Wellcome Fund, The Houston Laboratory and Population Sciences Training Program in Gene Environment Interaction. HW is supported by NIH postdoctoral fellowship 5F32EY19430. JEZ is supported by NIH training grant T32 EY007102. RKK is supported by the Foundation Fighting Blindness Canada, the Canadian Institutes for Health Research, FRSQ, the Foundation for Retinal Research, and Reseau Vision. AI is supported by grant from Research to Prevent Blindness, Inc, New York, NY (unrestricted grant to UTHSC Department of Ophthalmology and a Physician Scientist Award to AI). This work is supported by grants from the Retinal Research Foundation and the National Eye Institute (R01EY018571 and R01EY020540) to RC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online figures
- Data supplement 4 - Online tables
Footnotes
-
XW and HW contributed equally to this study.
-
Contributors RC, XW and RKK designed the study. SGJ, AI, DB, JRH, GAF, EIT and DW performed the clinical study. XW, HW, VS, HFT, VK, KW, HR, IL, JEZ, SS, SB, AK, JS, FW and YL performed the molecular study. XW integrated the data and performed the analysis. XW, HFT, RC, RKK, JEZ, SGJ, AI wrote the manuscript.
-
Funding Retinal Research Foundation and the National Eye Institute.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.