Article Text

Abstract
Understanding the causal role of biomarkers in cardiovascular and other diseases is crucial in order to find effective approaches (including pharmacological therapies) for disease treatment and prevention. Classical observational studies provide naïve estimates of the likely role of biomarkers in disease development; however, such studies are prone to bias. This has direct relevance for drug development as if drug targets track to non-causal biomarkers, this can lead to expensive failure of these drugs in phase III randomised controlled trials. In an effort to provide a more reliable indication of the likely causal role of a biomarker in the development of disease, Mendelian randomisation studies are increasingly used, and this is facilitated by the availability of large-scale genetic data. We conducted a narrative review in order to provide a description of the utility of Mendelian randomisation for clinicians engaged in cardiovascular research. We describe the rationale and provide a basic description of the methods and potential limitations of Mendelian randomisation. We give examples from the literature where Mendelian randomisation has provided pivotal information for drug discovery including predicting efficacy, informing on target-mediated adverse effects and providing potential new evidence for drug repurposing. The variety of the examples presented illustrates the importance of Mendelian randomisation in order to prioritise drug targets for cardiovascular research.
- Epidemiology
- Genetics
- Study design
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Identification of efficacious therapies in large-scale randomised controlled clinical trials (RCTs) have contributed to the reduction of cardiovascular disease (CVD).1 2 However, despite these advances, CVDs (such as ischaemic heart disease and stroke) have persisted in being the leading causes of premature death3 and disability worldwide in 2015.4 Well-designed and conducted RCTs provide the most reliable evidence for efficacy of therapeutic interventions and by extension on the causal role of biomarkers targeted by the intervention in disease risk. However, RCTs are costly and high risk, thus it is informative to have preliminary evidence on causality prior to embarking on them, and in some situations it may not be practical or ethical to randomise patients to interventions.5 For example, despite prior classical observational evidence suggesting that blood pressure (BP) has a J-shaped relationship with CVD,6 it would be unethical to test the raising of BP in those at the low end of the systolic or diastolic BP distribution to test whether this conveys cardiovascular benefit, and for good reason as subsequent RCTs provided robust evidence that no such ‘causal’ J-shape between either systolic or diastolic BP and CVD exists.7 When such trial data are unavailable, evidence may be obtained from classical observational epidemiological studies, but these are subject to several potential biases8 arising from confounding (where the association between an exposure of interest and outcome is driven through another characteristic—eg, an association between yellow teeth and lung cancer is entirely due to, and confounded by, smoking), reverse causality (where the disease itself leads to alterations in a biomarker, such as C-reactive protein and coronary heart disease (CHD); see figure 1 for a pictorial example), information bias (an example being where an exposure may be imprecisely measured and thus leads to underestimation of the exposure–disease association known as regression dilution bias if the measurement error is random) and selection bias (where individuals included in studies are not representative of the general population) (see box 1 for definitions; figure 2 for pictorial example).
Brief overview of the main biases in classical epidemiological studies8
Bias may be defined as any systematic error in an epidemiological study that results in an incorrect estimate of the association between the exposure and the outcome. The main biases are:
Confounding: Occurs when an observed association between exposure and outcome can be totally or partially explained by another factor.
Information bias: This is a result of mismeasurement of the exposure or outcome obtained from the individuals included in the study. This may mean that individuals are assigned to the wrong outcome category, leading to an incorrect estimate of the association between exposure and outcome. For a continuous exposure, mismeasurement can lead to underestimation of the association between the exposure and outcome, which can lead to a special form of information bias known as regression dilution bias.
Regression dilution bias: Random measurement error occurs when the measured values of an exposure (such as systolic blood pressure) fluctuate randomly around the true values, such that some measured values will be higher than the true values and other measured values will be lower. Regression dilution bias occurs when random measurement error in the values of the exposure (such as systolic blood pressure) causes an attenuation or ‘flattening’ of the slope of the line describing the association between the exposure and the outcome (eg, CHD).
Selection bias: This can occur when individuals included in the study are different from the target population that the investigators are trying to study.

Examples of (A) confounding and (B) reverse causality in observational epidemiology. (A) The arrows denote the direction of proposed causality and the cross denotes that the postulated direct link between yellow teeth and lung cancer is false. (B) The arrows denote the direction of proposed causality and the cross denotes that the postulated direct link between C-reactive protein (CRP) and CHD is false and in fact the current evidence suggests that CHD raises levels of CRP (ie, the arrow goes in the opposite direction).

Example of regression dilution bias in observational epidemiology. The sizes of the boxes are inversely proportional to the amount of statistical information. The HRs are plotted on a natural logarithmic (doubling scale). The black boxes (and the black dotted line) show the association between mismeasured systolic blood pressure and CHD; the red boxes (and the red dotted line) shows the association between systolic blood pressure and CHD if systolic blood pressure was measured without error. This illustrates that the slope of the association is underestimated when an exposure that is subject to random measurement error is related to a disease outcome. SBP, systolic blood pressure.
Although RCTs are the ‘gold standard’ for inferring the causal role of a biomarker in the development of disease, alternative non-interventional approaches have been increasingly used in recent times in clinical research9 10 in the absence of an RCT evidence base. A particular type of analysis is one where a genetic variant (either in isolation or combination with multiple genetic variants) is used to conduct ‘Mendelian randomisation’, and these studies have become increasingly common in health-related research over the last decade. This has been facilitated by improved genotyping platforms analysing millions of single-nucleotide polymorphisms (SNPs) and availability of data from global genetic consortia that have investigated tens to hundreds of thousands of participants, which collectively have contributed to better understanding of the genetic architecture of heart disease and cardiovascular risk factors including blood lipids, blood pressure, body mass index (BMI) and diabetes.11–14 Such discoveries have facilitated reliable identification of SNPs that associate with biomarkers that can then be used in Mendelian randomisation analyses to test the causal relevance of these biomarkers in disease risk.
The aim of this review is to explain the rationale for Mendelian randomisation, describe the advantages and potential limitations of this type of study design, and provide examples of how Mendelian randomisation has been used in and benefited cardiovascular research.
What is a Mendelian randomisation study?
In simple terms, a Mendelian randomisation study is one in which genetic variants are used to investigate the causal relationship of a biomarker on risk of disease.15 16 The concept was first described by Katan in 1986 in relation to cancer, at the time there was concern, from observational evidence, that blood cholesterol lowering may lead to altered risk of cancer, and Katan suggested using genetic variants in the APOE locus (that associate with plasma cholesterol concentrations) to assess the causal role of cholesterol with cancer.17
The fundamental principle of Mendelian randomisation is that if genetic variants that either alter the level of, or imitate the biological effects of, a modifiable biomarker that is causal in disease, then these genetic variants should also be associated with disease risk to the extent predicted by the effect of the genetic variant with the biomarker.16 This can be thought of as an analogy to a RCT: when stronger doses of drugs are used that have a greater effect on a causal biomarker (eg, use of more potent statins that have a greater reduction on low density lipoprotein cholesterol (LDL-C) levels), the resultant effect on risk reduction for CHD is greater.18 Thus, use of genetic variants that have stronger effects on LDL-C should have stronger relationships with risk of CHD, and this is exactly what is seen.19
The name ‘Mendelian randomisation’ refers to the random assortment of alleles during meiosis where DNA is transferred from parent to offspring at the time of gamete formation, a process named Mendel’s second law.20 This means that the inheritance of any particular genetic variant in an individual’s DNA should be independent of other characteristics, thus, when individuals in the population are grouped by a particular genotype that associates with difference in a biomarker, they should be similar in all respects other than one group has a genetically higher biomarker (such as LDL-C) and the other group has a genetically lower biomarker. Perhaps the easiest way to understand a Mendelian randomisation study design is by way of an analogy with an RCT design (see figure 3), and indeed Mendelian randomisation has been described as ‘nature’s randomised trials’.21

Comparison of a conventional trial with a Mendelian randomisation study. This illustrates the analogy between a conventional randomised controlled trial and a Mendelian randomisation study. CV, cardiovascular.
What are the key rules of Mendelian randomisation and why are they important?
There are three key rules for the conduct of a valid Mendelian randomisation study: (1) that the genetic variant associates with the biomarker, (2) the genetic variant is not associated with confounders of the biomarker to outcome association and (3) that the genetic variant only influences risk of disease through the biomarker of interest. Violation of any of these three rules can lead to a biased estimate, meaning that the causal estimate may not be reliable.
What are the advantages of a Mendelian randomisation study?
The principal advantages of a Mendelian randomisation study are that genetic variants are (1) non-modifiable, and therefore not susceptible to ‘reverse causality’; (2) should not be influenced by confounding, due to Mendel’s second law; and (3) measured with precision, thereby reducing regression dilution bias due to random measurement error.22 23 This means that the use of Mendelian randomisation can overcome the main sources of bias from classical observational epidemiology (figures 1 and 2) and provide more reliable estimates of the likely underlying causal relationship of a biomarker with risk of disease. Additional benefits include the fact that genetic differences in the biomarker, if untreated, remain constant, are not influenced by selection bias and reflect prolonged or lifelong differences.
How to estimate the causal effect of a biomarker on disease using Mendelian randomisation
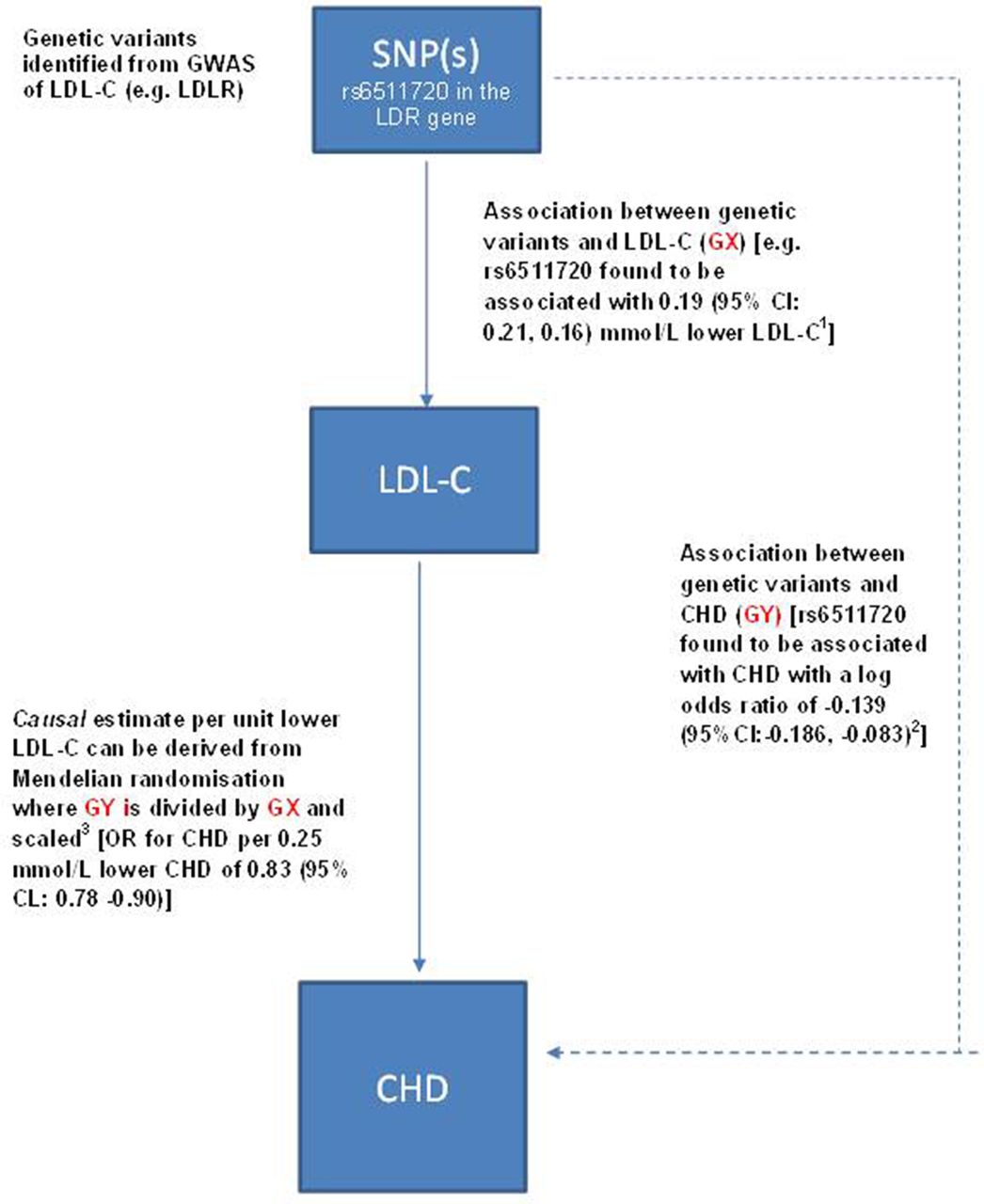
Figure 4 illustrates the generation of a Mendelian randomisation estimate: using a conventional approach, this is simply derived by scaling the SNP-to-disease estimate (GY) by the SNP-to-biomarker estimate (GX) to derive a causal estimate corresponding to a unit increase in the biomarker. This is termed the ratio method and can be used for an individual SNP or multiple SNPs in combination.24

Mendelian randomisation to test causality of a biomarker in disease: applied to LDL-cholesterol and risk of CHD. This example uses a genetic variant to estimate the causal relevance of LDL-C in CHD. Although for simplicity we use a single genetic variant, for a non-protein trait such as LDL-C, Mendelian randomisation should ideally employ multiple genetic variants in combination identified from genome-wide association studies of LDL-C as this more accurately reflects the underlying genetic architecture of the trait and thus gives a more reliable estimate for causality.(1) Association of LDLR SNP rs6511720 with LDL-C based on a meta-analysis of 137 818 participants reported by Ference et al JACC (2012); 6025 2631–2639.(2) Association of rs651170 with CHD based on a meta-analysis of 77 041 CHD cases reported by Ference et al JACC (2012); 6025 2631–2639(3) The causal estimate of LDL-C with CHD is found by taking the exponential of scaled value based on GX and GY to obtain the OR and its associated 95% CI. For this example a 0.19 mmol/L lower LDL-C (GX) was associated with a log OR (GY) of −0.1393 (that corresponds to an OR of 0.87=exp[−0.1393]). The causal estimate is required for a 0.25 mmol/L lower LDL-C so this can be obtained by 0.25 × [−0.1393/0.19]=−0.1833 exp(−0.1833)=0.83. The SE and CI are more challenging to calculate and the details are contained in Burgess et al’s Statistical Methods in Medical Research.24 SNP, single-nucleotide polymorphism.
What are the limitations of Mendelian randomisation?
Although Mendelian randomisation studies have advantages over classical observational epidemiological studies, the ability to reliably determine causality can be hindered by three main potential limitations that are now described and a fuller description is available elsewhere.25
Main limitations of Mendelian randomisation studies
Inadequate statistical power: Statistical power is the probability that the null hypothesis (typically of no effect) can be rejected if there is a true association of the biomarker with disease risk. As genetic variants typically explain a small proportion of the variance in biomarkers, the statistical power to detect an association between the variant and outcome in an applied Mendelian randomisation context can be low unless sample sizes are large. A large sample size is particularly important as it can inform whether a null finding is representative of a true null causal association, or simply a lack of power to detect an effect size of clinical interest. Often to increase the sample size, researchers will conduct meta-analyses of appropriately selected Mendelian randomisation studies in order to detect effect sizes of potential clinical interest.24
Weak instrument bias: A genetic variant is considered to be a ‘weak instrument’ if the statistical evidence for the association of the genetic variant with the biomarker is not strong. The F-statistic based on the genetic variant–biomarker association (GX in figure 3) is usually quoted as a measure of the strength of an instrument with F-statistics >10 deemed to be adequate.24
Pleiotropy: This is the phenomenon by which a single gene or multiple genetic variants combined into a gene score can associate with multiple biomarkers. In the case that the genetic instrument associates with biomarkers on distinct pathways to the exposure of interest, this is termed ‘horizontal pleiotropy’ (see figure 4A for illustration). Conversely, when the association with biomarkers is simply representing the downstream effects of the genetic variant on the exposure of interest, this is termed ‘vertical pleiotropy’ (see figure 4B for illustration).28 Whereas the presence of vertical pleiotropy is informative of pathways from exposure through to disease, the presence of horizontal pleiotropy can lead to severe bias in the Mendelian randomisation estimate.
Inadequate statistical power
Most genetic variants only have a modest effect on a given biomarker (ie, they only explain a small amount of the variance). A small amount variance explained by the genetic variant(s) does not hamper the conclusions that can be drawn from a Mendelian randomisation study, but it does have implications, however, for obtaining adequate statistical power (see box 2 for definition). As genetic variants typically have only small effects on the exposure of interest, this means that very large numbers of cases are typically required to detect a causal relationship for the outcome of interest. Statistical power can be increased by combining multiple genetic variants together, into a gene score, which increases the proportion of variance of the biomarker explained. Furthermore, weighting the SNPs by their association with the biomarker of interest from prior genome-wide association studies provides additional statistical power.
Weak instrument bias
Even though a genetic variant typically explains only a small amount of the variation in a biomarker, it is important to ensure that there is a strong relationship between the genetic variant and the biomarker. Otherwise, so-called ‘weak instrument bias’ can arise, which refers to a genetic variant that does not have a sufficiently strong association with the biomarker (see box 2 for definition). Methods to quantify the strength of the relationship between the genetic variant(s) and biomarkers exist, including the F-statistic.26 27
Associations of the genetic variants with other traits: confounding and pleiotropy
As in a standard RCT, it is possible to check that the baseline characteristics are balanced between the ‘randomised groups’ in a Mendelian randomisation study by comparing individuals with and without the genetic variant. This in essence investigates whether the genetic variant shows associations with biomarkers other than the one under investigation. The genetic variant(s) should only be associated with the biomarker (and its pathway) under investigation; otherwise, it may not be valid to use this genetic variant in a Mendelian randomisation study.23
A genetic variant or multiple genetic variants (when used in combination) may associate with other biomarkers, a phenomenon known as ‘pleiotropy’ (see box 2 for definition). When those biomarkers are on discrete pathways to the biomarker of interest, this is termed horizontal pleiotropy,28 and the use of the genetic variant in this circumstance may result in a confounded estimate from Mendelian randomisation. For example, use of genetic variants in loci that associate with telomere length29 but that also associates with cancers (and therefore, by extension, cancer therapy, some of which are deleterious to vascular health) could give a biassed estimate for the association of telomere length in the development of CHD as it is not clear whether telomere length itself is causing CHD or whether it is due to a pathway separate to it (red or blue arrows in figure 5A). Recent advances in methodology include Mendelian randomisation-Egger regression (based on the method used to assess small study bias in meta-analysis30), which can quantify the amount of bias due to horizontal pleiotropy and can provide a valid causal estimate even in the presence of horizontal pleiotropy.31
An alternative form of pleiotropy is ‘vertical pleiotropy’, which exists when the genetic variant(s) associates with other biomarkers downstream of the main biomarker of interest. For example, an association of SNPs used to measure the causal role of BMI in CHD with systolic blood pressure32 would represent vertical pleiotropy, and this form of pleiotropy does not invalidate the Mendelian randomisation analysis (see box 2; figure 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Example of (A) horizontal pleiotropy and (B) vertical pleiotropy in a Mendelian randomisation study. The arrows denote the direction of proposed causality. In scenario (A), whether CHD is a consequence of telomere length or whether the association is confounded by an association of the genetic variants with cancer chemotherapy (which itself has deleterious effects to the cardiovascular system) is not known. Thus, the potential independent association of genetic variants with cancer therapy could represent a horizontally pleiotropic pathway and thus give an invalid causal estimate for Mendelian randomisation. In scenario (B), the SNPs associated with BMI are also associated with systolic blood pressure; however, this simply reflects a downstream effect of BMI (as BMI is recognised to causally affect blood pressure) and is likely on the pathway between BMI and risk of CHD. Thus, while the potential presence of horizontal pleiotropy in scenario (A) makes it unclear whether telomere length plays a causal role in CHD, in scenario (B) the vertical pleiotropy is informative of potential mechanisms from exposure through to disease. BMI, body mass index; SBP, systolic blood pressure; SNP, single-nucleotide polymorphism.
Recent applications of Mendelian randomisation studies
Mendelian randomisation studies have become more prevalent in the literature as they are quicker and cheaper to conduct than RCTs and can utilise existing data from large genetic consortia in order to provide preliminary information on important research questions.
Predicting efficacy
There is considerable interest in predicting the efficacy of potential therapeutic targets as early as possible in the drug development process, as genetic support for a drug target can substantially enhance the probability that a RCT of a therapy targeting such a drug target succeeds.33 A recent example involved lipoprotein-associated phospholipase A2 (Lp-PLA2). Lp-PLA2 is an inflammatory biomarker that accumulates in unstable and unruptured plaques, and observational studies have suggested that higher circulating levels of Lp-PLA2 mass and activity are associated with higher risk of major cardiovascular outcomes.34 A loss of function variant in PLA2G7 gene that encodes Lp-PLA2 was used to assess the causal role of Lp-PLA2. PLA2G7 V279F (rs76863441) was genotyped in 91 428 individuals from the China Kadoorie Biobank (CKB),35 with follow-up through health record linkage for median 7 years. The primary outcome was incidence of major vascular events (vascular death, myocardial infarction, stroke), and the study had over 90% power to detect a 20% lower risk per F variant at p<0.01. These analyses found that V279F was not associated with major vascular events or other vascular outcomes.36 Results from the CKB study were broadly consistent with findings for cardiovascular outcomes from phase III randomised trials of the Lp-PLA2 inhibitor darapladib.37 38 The broader implication is that billions of dollars were spent on what turned out to be failed clinical trials for darapladib. While the negative outcomes could not have been fully anticipated when the trials were set up, had the genetic investigations been pursued prior to embarking on the phase III trials, darapladib drug development could have been sidelined and more promising targets prioritised in its place.
Predicting target-mediated adverse effects
Statins are one of the most widely prescribed medications for lowering LDL-C for primary and secondary prevention of CVD, and several large-scale trials1 39 and individual patient data meta-analyses of large-scale RCTs have clearly demonstrated that they are effective, compared with placebo, at lowering risk of CVD.40 Even though statins are very safe, they are not without side effects, and there is controversy about the frequency of these adverse effects and how they should be reliably investigated, such as long-term follow-up of trials or using observational data.41 One of the adverse effects of statins is type 2 diabetes (T2D), and meta-analysis of RCTs and Mendelian randomisation studies have shown this to be an on-target effect of hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase.42 The question naturally arises whether other LDL-C lowering drugs (such as ezetimibe or PCSK9 inhibitors) will have the same diabetogenic effects. Lotta et al 43 used data from a meta-analysis of 50 000 T2D cases and 270 000 controls and found that the LDL-C SNPs in the NPC1L1 gene (a genetic proxy for ezetimibe) and HMGCR gene (a genetic proxy for statins) were associated with an OR of T2D of 2.42 (95% CI: 1.70 to −3.43) and 1.39 (1.12 to −1.73) per genetically predicted 1 mmol/L lower LDL-C, respectively. Two recent Mendelian randomisation studies44 45 have provided orthogonal evidence that LDL-C lowering through PCSK9 inhibition is, as with statins, likely to lead to increased risk of T2D, and more generally, a recent study using SNPs across the genome provides evidence that an increased risk of T2D may arise as a general consequence of LDL-C lowering.46
Repurposing drugs
Mendelian randomisation can be used to inform on potential repurposing of drugs, for example through use of a ‘phenome-wide scan’, which is facilitated by the availability of large-scale prospective biobanks with incident diagnoses procured through electronic health records, such as the China Kadoorie35 and UK biobanks.47 Taking the example above of Lp-PLA2 in the CKB, there was no evidence of an association of genetic variants in V279F with risks of other diseases.48 While this supports safety data from clinical trials of darapladib,37 38 it also helps clarify that any repurposing of darapladib for other outcomes (particularly the 41 different outcomes investigated in the CKB study) is unlikely to be fruitful. Mendelian randomisation can also be used to investigate for potential pharmacogenetic associations (whereby one or more genetic variants are used to identify patients more likely to respond to a drug and/or patients less likely to suffer an adverse drug reaction): by stratifying a Mendelian randomisation analysis by these genetic variants, clarity can be provided as to whether subpopulations are likely to derive greater benefit from a drug. This may be helpful prior to embarking on de novo RCTs targeting such subgroups.49
Summary
Mendelian randomisation studies have been particularly successful in cardiovascular epidemiology demonstrating strong evidence of causality for established and novel biomarkers (such as lipoprotein(a)) and drug targets (such as PCSK9), and providing reliable evidence for drug targets that have not shown to be causal in subsequent trials.50 Mendelian randomisation can also be applied when the exposure is a modifiable behaviour rather than a biomarker (eg, alcohol intake or smoking).51 52 Developments in genotyping and availability of large-scale genetic consortia with hypothesis-free reliable discovery of new genetic variants for biomarkers that may play causal roles in disease development and application of such genetic variants in Mendelian randomisation analyses presents tantalising opportunities to identify potentially modifiable exposures that, if shown to be causal, may be tested in future intervention studies. Thus, well-conducted Mendelian randomisation studies can be extremely valuable as they use existing genetic data to provide qualitative information of treatment efficacy, target-mediated adverse effects and opportunities for drug repurposing, which can be very informative to prioritise biomarkers to take forward into phase II/III clinical trials in humans.
References
Footnotes
Contributors DAB drafted the original report, critically reviewed and updated the manuscript. MVH helped to revise the manuscript and critically reviewed the scientific content.
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.
Correction notice This paper has been amended since it was published Online First. Owing to a scripting error, some of the publisher names in the references were replaced with ‘BMJ Publishing Group’. This only affected the full text version, not the PDF. We have since corrected these errors and the correct publishers have been inserted into the references.