

Abstract
The DNA damage response (DDR) orchestrates a network of cellular processes that integrates cell-cycle control and DNA repair or apoptosis, which serves to maintain genome stability. DNA-PKcs (the catalytic subunit of the DNA-dependent kinase, encoded by PRKDC), ATM (ataxia telangiectasia, mutated), and ATR (ATM and Rad3-related) are related PI3K-like protein kinases and central regulators of the DDR. Defects in these kinases have been linked to neurodegenerative or neurodevelopmental syndromes. In all cases, the key neuroprotective function of these kinases is uncertain. It also remains unclear how interactions between the three DNA damage-responsive kinases coordinate genome stability, particularly in a physiological context. Here, we used a genetic approach to identify the neural function of DNA-PKcs and the interplay between ATM and ATR during neurogenesis. We found that DNA-PKcs loss in the mouse sensitized neuronal progenitors to apoptosis after ionizing radiation because of excessive DNA damage. DNA-PKcs was also required to prevent endogenous DNA damage accumulation throughout the adult brain. In contrast, ATR coordinated the DDR during neurogenesis to direct apoptosis in cycling neural progenitors, whereas ATM regulated apoptosis in both proliferative and noncycling cells. We also found that ATR controls a DNA damage-induced G2/M checkpoint in cortical progenitors, independent of ATM and DNA-PKcs. These nonoverlapping roles were further confirmed via sustained murine embryonic or cortical development after all three kinases were simultaneously inactivated. Thus, our results illustrate how DNA-PKcs, ATM, and ATR have unique and essential roles during the DDR, collectively ensuring comprehensive genome maintenance in the nervous system.
SIGNIFICANCE STATEMENT The DNA damage response (DDR) is essential for prevention of a broad spectrum of different human neurologic diseases. However, a detailed understanding of the DDR at a physiological level is lacking. In contrast to many in vitro cellular studies, here we demonstrate independent biological roles for the DDR kinases DNA-PKcs, ATM, and ATR during neurogenesis. We show that DNA-PKcs is central to DNA repair in nonproliferating cells, and restricts DNA damage accumulation, whereas ATR controls damage-induced G2 checkpoint control and apoptosis in proliferating cells. Conversely, ATM is critical for controlling apoptosis in immature noncycling neural cells after DNA damage. These data demonstrate functionally distinct, but cooperative, roles for each kinase in preserving genome stability in the nervous system.
Introduction
Maintaining genomic integrity in the developing and mature nervous system is essential to prevent neurologic disease (Barzilai, 2013; McKinnon, 2013; Madabhushi et al., 2014). There are many sources of endogenous DNA damage. For instance, replication-associated DNA damage during neurogenesis is frequent, and reactive byproducts of metabolism or transcription-associated DNA damage are also a constant threat to genome integrity (Aguilera and García-Muse, 2012; McKinnon, 2013). To ensure normal neural development, there is a broad armamentarium of DNA damage response factors that can correct multiple types of common endogenous DNA lesions.
Several key protein kinases respond to specific types of DNA lesions to activate signaling cascades that coordinate the DNA damage response (DDR). DNA-PKcs (encoded by PRKDC), ATM, and ATR are related phosphatidylinositol-3-kinase-like serine/threonine kinases that can initiate cell cycle arrest, DNA repair, or apoptosis (Lavin, 2008; McKinnon, 2012; Shiloh and Ziv, 2013). The activation of individual kinases is linked to the type of DNA lesion generated. For example, DNA-PKcs is recruited to double-strand breaks (DSBs) by the heterodimer complex Ku70/80 to form the holoenzyme DNA-PK and facilitate DNA repair via nonhomologous end-joining (NHEJ), a main DNA repair pathway for DSBs in mammalian cells (Lieber, 2010; Chapman et al., 2012; Davis and Chen, 2013; Jette and Lees-Miller, 2015). Compared with core NHEJ components, such as Ku70/80, XRCC4, or DNA Ligase IV (LIG4), which lead to widespread neural apoptosis and embryonic lethality, or compromised development when inactivated, mice lacking DNA-PKcs are viable (Gu et al., 2000; Douglas et al., 2005). Consequently, DNA-PKcs function during neurogenesis in the context of DNA damage and NHEJ remains uncertain. Additionally, DNA-PKcs has also been linked to processes outside of NHEJ, such as transcription regulation, including the response to insulin (Wong et al., 2009; Goodwin and Knudsen, 2014).
ATM can be activated by DNA DSBs and is recruited to this damage by the MRN (Mre11/Rad50/Nbs1) complex to signal cell cycle arrest, facilitate DNA repair, or activate apoptosis in immature neural cells (Shull et al., 2009; Stracker and Petrini, 2011; Marechal and Zou, 2013; Paull, 2015). In contrast, ATR is activated by replication protein-A bound single-stranded DNA generated by MRN-mediated resection of breaks (Nam and Cortez, 2011; Marechal and Zou, 2013). Both ATM and ATR have also been ascribed roles not reflective of DNA damage signaling, including synaptic vesicular function and autophagy (Li et al., 2009; Tripathi et al., 2016). Loss of ATM can result in the neurodegenerative syndrome ataxia telangiectasia, and hypomorphic ATR mutation the neurodevelopmental disorder ATR-Seckel syndrome (O'Driscoll et al., 2004; McKinnon, 2012). PRKDC mutations have been identified in two immune-compromised individuals associated with microcephaly (van der Burg et al., 2009; Woodbine et al., 2013).
How these three kinases collectively coordinate the responses to DNA damage within a physiological context remains unclear. It has been suggested that redundancy exists between these kinases, as they can phosphorylate similar substrates (e.g., p53) and dual loss of ATM and DNA-PKcs results in synthetic lethality (Gurley and Kemp, 2001; Matsuoka et al., 2007; Lavin, 2008; Bensimon et al., 2010). Here, we show that during neurogenesis DNA-PKcs functions to maintain genome integrity in the nervous system in a complementary manner to ATM and ATR, with each kinase fulfilling distinct and separate roles. Collectively, our data reveal both new insights into DNA-PKcs function and how these important DNA damage-responsive kinases cooperatively maintain genomic integrity in the nervous system.
Materials and Methods
Animals.
Atm, p53, and Atr mice and genotyping have been described previously (Lee et al., 2012). Prkdc mice (Gao et al., 1998a) were genotyped by PCR using the following primers: wild-type alleles were amplified with forward primer 5′-TGACAGCAAGTGCCTGTAAAGTGC and reverse primer 5′-ATAGTCCCTTCAGACAGCCAGC; and the Prkdc null allele was identified using reverse primer 5′-GAAGCGGGAAGGGACTGGCTGCTA (in lines with no cre) or 5′-CGCAGCGCATCGCCTTCTATCGCC (in lines with cre) and the same forward primer as the wild-type allele. Nestin-cre and Emx1-cre mice were obtained from The Jackson Laboratory (JAX mouse identifier #003771, RRID:IMSR_JAX:003771 and #005628, RRID:IMSR_JAX:005628). Mice of either sex were used for experiments, although when possible littermates were sex-matched. All animals were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility, and the Institutional Institutional Animal Care and Use Committee approved all experiments.
Histology.
Mice were perfused transcardially with 4% PFA while embryos were drop-fixed in 4% PFA, and tissues were cryoprotected in 25% PBS-buffered sucrose solution, embedded in O.C.T., and sectioned sagittally at 10 μm using an HM500M cryostat (Microm). Immunohistochemistry was performed after antigen retrieval as described previously (Lee et al., 2012). The following antibodies were used: anti-Tbr1 (1:100; Abcam, catalog #ab31940 RRID:AB_2200219), anti-Tbr1 (1:250; Synaptic Systems, catalog #328 005 RRID:AB_2620072, when used for dual IHC with Tbr2, Brn2, pKAP1, and γH2AX), anti-Tbr2 (1;200; Abcam, catalog #ab23345 RRID:AB_778267), anti-Cux1 (1:100; Santa Cruz Biotechnology, catalog #13024 RRID:AB_2261231), anti-Ctip2 (1:100, Abcam, catalog #ab18465 RRID:AB_2064130), anti-Brn2 (1:200; GeneTex, catalog #GTX114650 RRID:AB_10619683), anti-KAP1-phospho-Ser-824 (1:500; Bethyl Labs, catalog #A300-767A), anti-active caspase-3 (1:500; BD Biosciences, catalog #559565 RRID:AB_397274), anti-phospho-H2AX-Ser-139 (1:200; Cell Signaling Technology, catalog #2577L RRID:AB_2118010), anti-phospho-H3-Ser-10 (1:500; Cell Signaling Technology, catalog #9701L RRID:AB_331535), and anti-Tuj1 (1:500; Covance Research Products, catalog #MMS-435P RRID:AB_2313773). Immunostaining of active caspase-3 and pKAP-1(S824) was visualized with a VIP substrate kit (Vector Laboratories, catalog #SK-4600 RRID:AB_2336848) and biotinylated secondary antibody and avidin-biotin complex (Vectastain Elite kit; Vector Laboratories, catalog #PK-6100 RRID:AB_2336819). Sections were counterstained with 0.1% methyl green (Vector Laboratories), dehydrated, and mounted with DPX (Fluka). For fluorescence, FITC- or Cy3-conjugated secondary antibody (Jackson Immunologicals) were used and counterstained with DAPI (Vector Laboratories, catalog #H-1200 RRID:AB_2336790) or propidium iodide (Vector Laboratories, catalog #H-1300 RRID:AB_2336791). TUNEL staining was done using the ApopTag system (Millipore Bioscience Research Reagents).
In situ cell counts.
P5 and E15.5 brains were subjected to quantitative analysis to determine the indices of apoptosis and mitosis, respectively, during neurogenesis. Three brains per genotype were analyzed for each time point, and three representative sections per brain (nine sections per genotype) were used for quantification. Immunopositive cells for active caspase-3 were measured within 10× images of the P5 CA1 region. The neocortex was divided into four areas: ventricular zone (VZ), subventricular zone (SVZ), intermediate zone (IZ), and cortical plate (CP). pH3(ser10) cells in the apical region of the VZ were measured within 40× images of E15.5 embryonic cortices. Active caspase-3 in the E15.5 neocortex was quantified by counting in 110 μm2 areas taken from 10× images and the percentages reported. For the P5 cerebellum, cells were measured within a 68 μm2 area for active caspase-3. The developing P5 cerebellum was divided into two areas: white matter (WM) and the external granule layer (EGL). Postnatal day 11 brains were stained and quantified by percentage of cells that were positive for a particular marker. Graphs represent mean values of replicates, error bars the SEM, and p < 0.05 was considered significant. When comparing two genotypes, p values were calculated using unpaired Student's t test (GraphPad, Prism version 7.0 RRID:SCR_002798). For comparisons involving multiple genotypes, a one-way ANOVA was conducted and, when significant, followed post hoc with Tukey's multiple comparisons (GraphPad, Prism version 7.0, RRID:SCR_002798).
Western blots.
Protein extracts were prepared as described previously (Lee et al., 2012). After separation using NuPAGE 4%–12% Bis-Tris protein gels, proteins were identified using anti-DNA-PKcs Ab-4 mixture (1/100; Lab Vision, catalog #MS-423-P1 RRID:AB_61152), anti-phospho-ATM-ser1981 (1:1000; R&D Systems, catalog #AF1655 RRID:AB_2062935), anti-ATR (1:500; Santa Cruz Biotechnology, catalog #sc-1887 RRID:AB_630893), anti-Chk2 (1:1000; Millipore, catalog #05-649 RRID:AB_2244941), anti-p53-ser15 (1:1000; Cell Signaling Technology, catalog #9284 RRID:AB_331464), anti-ATM (1:1000; Cell Signaling Technology, catalog #2873S RRID:AB_2062659), anti-phospho-KAP1-ser824 (1:1000; Bethyl, catalog #A300-767A RRID:AB_669740), and anti-KAP1 (1:2000; Abcam, catalog #ab10484 RRID:AB_297223). HRP-conjugated secondary antibodies were used to visualize proteins using chemiluminescence. Equal protein transfer was confirmed using Ponçeau staining and/or anti-actin immunoblotting (1:500; Santa Cruz Biotechnology, catalog #sc-1616 RRID:AB_630836) was used as a loading control.
Results
DNA-PKcs loss hypersensitizes to apoptosis in the neocortex and postnatal brain
Although many NHEJ core components are essential for neural development (Barnes et al., 1998; Gao et al., 1998b; Gu et al., 2000), DNA-PKcs is dispensable, with no overt neural phenotype reported in Prkdc−/− mice (Gu et al., 2000). However, Scid mice (resulting from DNA-PKcs mutation) have been reported to show increased cell death in the developing nervous system, and cultured neurons from these mice showed increased sensitivity to staurosporine and kainic acid (Chechlacz et al., 2001; Vemuri et al., 2001; Liu et al., 2009). We therefore sought to determine how DNA-PKcs functions during the neural DDR, and additionally in the context of the related kinases ATM and ATR.
As neurogenesis is particularly susceptible to DNA damage (McKinnon, 2013), we used the neocortex to delineate DNA-PKcs function, a tissue where the spatiotemporal effects of genotoxic stress can be readily assessed. In the developing murine cortex, the VZ contains neural progenitors and the SVZ contains a mixed population of intermediate progenitors and immature noncycling neural cells, whereas the IZ contains mostly nonproliferating cells undergoing migration and differentiation. More mature neurons and glial cells reside in the CP, before establishment of the final six-layered tissue (Molyneaux et al., 2007).
Following exposure of the E15.5 embryo to ionizing radiation (IR), DNA damage signaling is activated that ultimately results in apoptosis (Orii et al., 2006; Gatz et al., 2011; Barazzuol et al., 2015). We initially determined the regional impact of DNA-PKcs loss throughout the E15.5 neocortex using immunostaining of activated caspase-3 as a surrogate marker for apoptosis. Compared with control tissue, which showed activation of caspase-3, the loss of DNA-PKcs substantially enhanced this response, and the greatest difference was seen in the nonreplicating differentiating IZ and CP (Fig. 1A,B). Quantitation of active caspase-3 immunostaining showed a marked increase in apoptotic cells in the Prkdc−/− CP, indicating that DNA-PKcs is important for suppressing cell death in differentiating neuronal regions (Fig. 1B). Notably, and similar to the developing cortex, we also observed a dose-dependent hyperactivation of apoptosis in the postnatal P5 hippocampus (Fig. 1C). Both the Prkdc−/− cornu ammonis (CA1–3) and dentate gyrus (DG) showed a marked increase in activated caspase-3, indicating a hypersensitivity to DNA damage. Quantitation of the CA1 region (as an example) after increasing doses of IR showed a significant increase in sensitivity in the Prkdc−/− tissue (Fig. 1D). This suggests that the neuroprotective role DNA-PKcs after DNA damage features broadly in the nervous system.
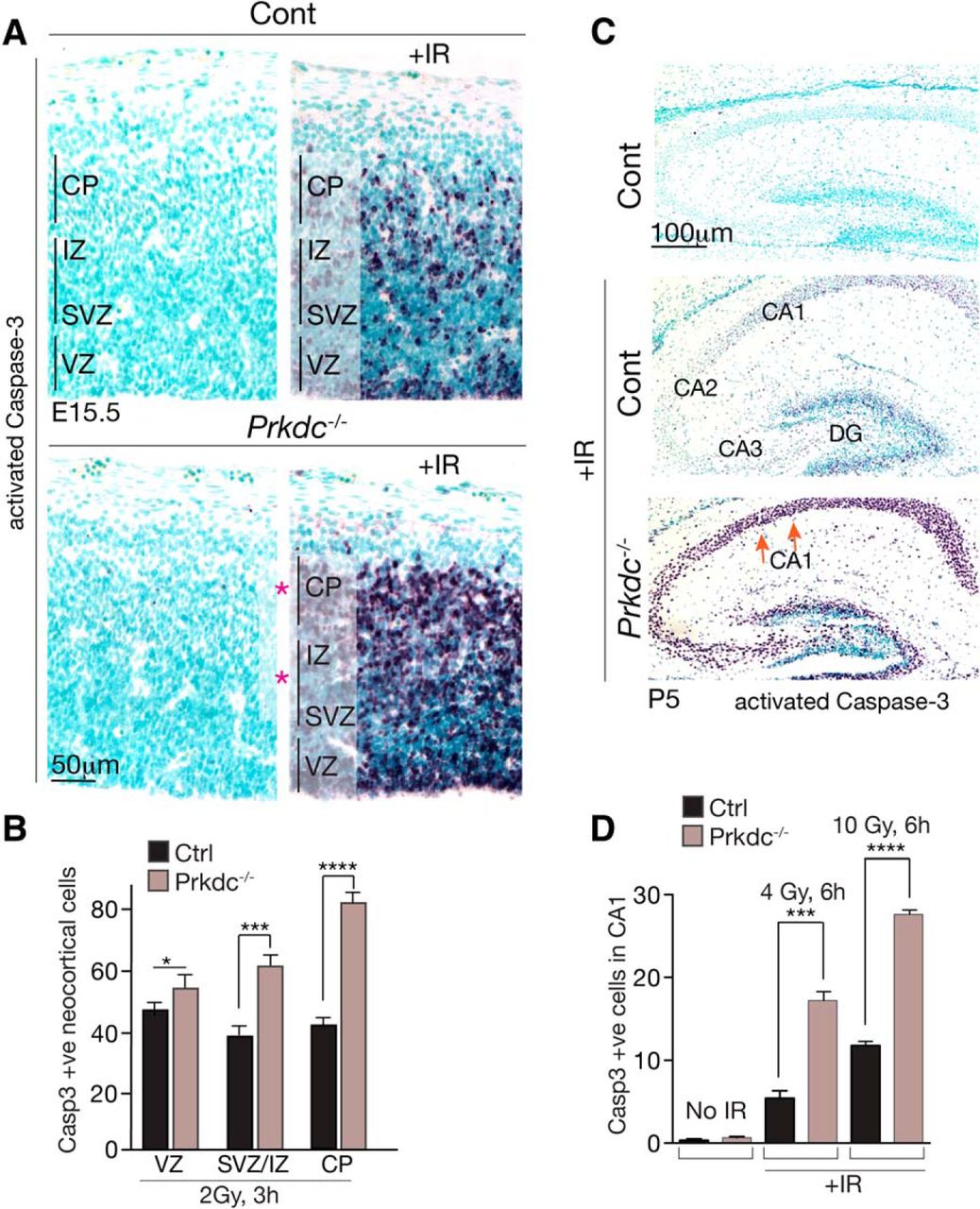
DNA-PKcs is neuroprotective in the developing cortex. A, Immunohistochemical identification of activated caspase-3 in E15.5 sagittal sections of murine neocortex shows increased apoptosis in the Prdkc−/− tissue 3 h after 2 Gy of IR. Red asterisks indicate regions of the Prdkc−/− neocortex associated with increased apoptosis. Sections were counterstained with methyl green. Vertical lines indicate regions that were quantified. B, Increased apoptosis in the Prkdc−/− neocortex occurs in all areas, but there are quantitative differences related to the specific stage of neuronal differentiation. The VZ shows the smallest difference (*p < 0.05), with a more significant increase seen in the combined SVZ/IZ (***p < 0.001); the most significant increase (****p < 0.0001) in activated caspase-3 staining was found in the immature noncycling cells of the developing CP. C, Sagittal sections of the P5 postnatal Prdkc−/− hippocampus showed significantly enhanced levels of apoptosis after IR in the CA and the DG regions compared with WT controls. D, Quantification of IR-induced apoptosis in the CA1 region of Prkdc−/− after radiation (4 and 10 Gy) is associated with significant dose-dependent increases in apoptosis compared with WT tissue. Graphs represent mean values of replicates (n = 3 animals per group). Error bars indicate SEM.
To identify the cells hypersensitive to DNA damage in the Prkdc−/− neocortex, we used various markers that identify cortical neuronal cell types. We used Tbr1, which is a transcription factor that regulates the laminar identity of postmitotic cortical neurons (Hevner et al., 2001; Bedogni et al., 2010), and Tbr2, a transcription factor required for generation of intermediate progenitor cells during corticogenesis, which is expressed in the SVZ (Englund et al., 2005; Sessa et al., 2008; Mihalas et al., 2016). Unirradiated WT and Prkdc−/− E15.5 tissue showed equivalent immunostaining patterns for Tbr1, indicating similar developmental profiles (Fig. 2A). However, after 4 Gy of IR, there was a substantial loss of ∼70% of the Tbr1+ neurons in the Prkdc−/− CP compared with only ∼10% in WT (Fig. 2A). For other cortical regions, we used Tbr2, which at E15.5 defines immature cells in the SVZ (Bulfone et al., 1999). Similar to the Tbr1+ CP cells, the Tbr2+ SVZ population in the Prkdc−/− tissue was also substantially affected, with most (>95%) of these cells being lost after IR in the Prkdc−/− neocortex, compared with ∼15% in WT (Fig. 2B). These data also indicate that the activated caspase-3 cells shown in Figure 1A subsequently undergo apoptosis.

DNA-PKcs loss selectively sensitizes cortical regions to DNA damage during development. A, Immunohistochemistry of sagittal E15.5 embryonic cortex using antibodies that recognize Tbr1 and Tbr2 distinguish different cortical regions, as shown in the illustration. Numbers of Tbr1+ neural progenitors are similar between E15.5 WT and Prdkc−/− unirradiated neocortex. After radiation, there is a marked reduction in Tbr1+ cells in the Prdkc−/− tissue compared with WT, revealing hypersensitivity in the CP to DNA-PKcs loss. B, Tbr2+ cells are also hypersensitive in the Prdkc−/− neocortex, with a loss of this population 6 h after radiation. Quantification of IR-induced cell loss in the Tbr1+ and Tbr2+ neocortical cell populations shows a significant loss of both Tbr1+ and Tbr2+ cells in the Prdkc−/− CP. ***p < 0.0001.
To further determine the relative DNA damage susceptibility of neural populations, we used additional markers that define subpopulations within the Tbr1+ neuronal cells of the developing neocortex. Ctip2 is a transcription factor localized to subcerebral projection neurons of the CP (Arlotta et al., 2005), whereas Brn2 is involved in regulating upper layer cells of the CP (Dominguez et al., 2013; Oishi et al., 2016). Consistent with a heightened susceptibility of Prkdc−/− Tbr1+ progenitors to DNA damage (Fig. 2A), interrogation of these cells using Ctip2 revealed that a large fraction of Tbr1+ cells (∼80%) in this neocortical region (i.e., Tbr1+Ctip2+ cells) were hypersensitive and eliminated after IR (Fig. 3A). In stark contrast, the same Tbr1+Ctip2+ populations in the WT tissue were mostly resistant and showed only minor cell loss (Fig. 3A). Additionally, within this region, Brn2+ cells were even more sensitive in the Prkdc−/− tissue, as this population was completely lost after IR, compared with an ∼10% reduction observed in WT (Fig. 3B). Thus, DNA-PKcs protects the developing neocortex in the face of genome damage; and within this tissue, certain neurons (e.g., Tbr1+Brn2+) are particularly hypersensitive to DNA damage when DNA-PKcs is absent. This differential sensitivity is consistent with differentiation status of the neurons being a key determinant in susceptibility to IR (McKinnon, 2013), as the Brn2+ cells are a less differentiated cell type than the Ctip2+cells at E15.5 (Molyneaux et al., 2007).
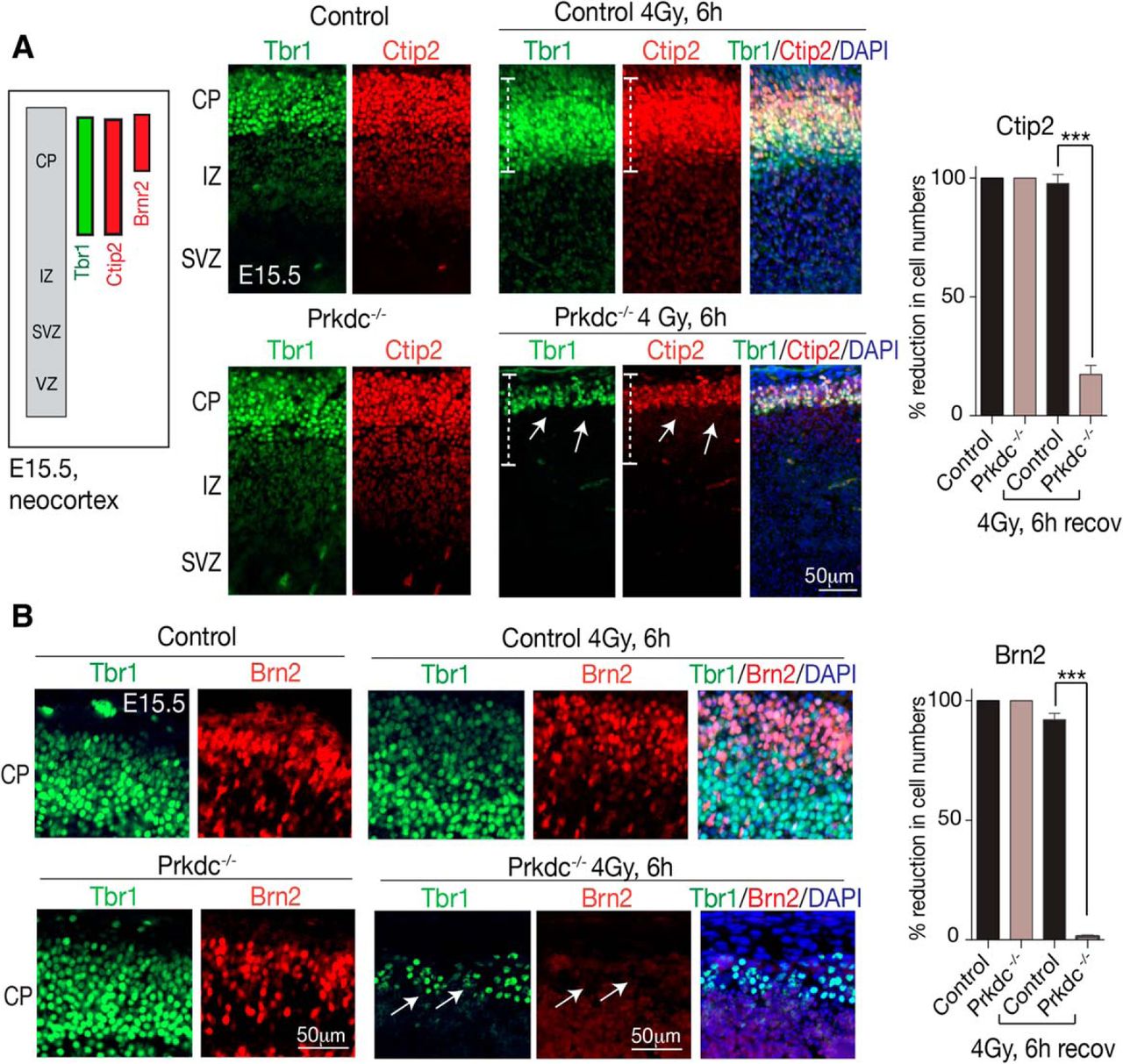
Differential sensitivity to DNA damage is observed within the developing CP. A, Sagittal sections of E15.5 neocortex show that within the Tbr1+ upper CP layer are neuronal subpopulations at various stages of differentiation and maturity that can be identified using Ctip2 or Brn2. After IR, Tbr1+/Ctip2+ populations (white dashed lines) are markedly reduced in the Prdkc−/− neocortex (white arrows), whereas in the WT tissue these are unaffected. B, Tbr1+/Brn2+ progenitors are almost entirely eliminated in Prdrkc−/− (white arrows), whereas in WT tissue the numbers of Brn2+ cells are reduced slightly compared with unirradiated controls (as quantitated in the adjacent graphs for Ctip2 and Brn2). ***p < 0.0001.
The neuroprotective role of DNA-PKcs is enhancement of NHEJ
IR hypersensitivity in the neocortex after DNA-PKcs loss could occur because of defective DNA repair (NHEJ) or attenuated DNA damage signaling due to absent kinase activity, both of which could result in increased DNA damage leading to apoptosis. Of the two possible DSB repair pathways, NHEJ is the pathway used by nonproliferating neural cells (Shull et al., 2009), as HR requires cells in S/G2 phase, where they contain sister chromatids as the repair template (Orii et al., 2006; Jasin and Rothstein, 2013). This implies that, if DNA-PKcs is primarily functioning as a DNA repair factor, its loss would likely impact DNA damage resolution of nonproliferative cortical cells.
To distinguish between these possible roles for DNA-PKcs, we examined DNA damage markers that identify activation of the DNA damage response. We initially assessed phosphorylation of the chromatin-associated protein KAP1 (KRAB-associated protein 1; TRIM28). Phosphorylation of KAP1 (pS824) occurs after DNA DSBs and is an ATM-dependent phosphorylation event, which is important for accessing DSBs in heterochromatin for repair (Ziv et al., 2006; Goodarzi et al., 2011). Compared with WT tissue, phosphorylated KAP1 in Prkdc−/− was abundant in the differentiating CP 1 h after radiation. By 6 h, pKAP1 remained in the CP in the mutant, consistent with slower DNA DSB repair in the Prkdc−/− noncycling differentiating cells (Fig. 4A). Notably, these DDR signaling events are ATM-dependent as they were absent from both AtmNes-cre and Prkdc−/−;AtmNes-cre tissue. These data imply that DNA-PKcs loss compromises efficient DNA repair and increases DNA DSBs, which activates ATM causing KAP1 phosphorylation. To identify cell types with persistent pKAP1 in the CP, we used Tbr1 costaining (Fig. 4B). In the WT tissue, infrequent pKAP1+ cells were present throughout the intact Tbr1+ CP layer at 6 h after IR, implying DNA damage resolution. In contrast, widespread Tbr1+ cell loss after IR was evident in Prkdc−/− tissue (Fig. 4B). The remaining Tbr1+ cells in the CP showed persistent pKAP1, implying that DSBs remained, likely due to a repair defect after DNA-PKcs loss. Thus, these data suggest that DNA-PKcs reduces the burden of DNA damage in Tbr1+ neuronal cells via resolution of DSBs.

Differential sensitivity of the neocortex to IR reflects the activation of the DNA damage response involving phosphorylation of KAP1. A, Immunostaining for phosphorylation of KAP1(Ser824) at 1 h after IR in E15.5 sagittal sections. pKAP1 is markedly increased in Prkdc−/− and persists at 6 h compared with controls; KAP1 phosphorylation is ATM-dependent and absent from AtmNes-cre tissue. PCNA immunostaining identifies proliferating progenitors. Sections were counterstained with DAPI (blue). B, In unirradiated E15.5 WT or Prkdc−/− tissue, no pKAP1 is observed. After irradiation, phosphorylated KAP1 persists throughout the remaining Tbr1+ cells of the Prkdc−/− CP, consistent with unrepaired DNA resulting in activation of the DNA damage response. Quantitation of the levels of Tbr1 and pKAP1 in WT and Prkdc−/− cortex. ***p < 0.0001.
We next examined DNA damage signaling in the P11 cortex. We chose this developmental stage because these more mature neural populations do not engage apoptosis, and so assessment of DNA strand breaks via γH2AX in the absence of those formed during apoptosis is possible (McKinnon, 2013). Similar to the neocortex, we found no differences in histology between WT and Prkdc−/− at P11 using markers to distinguish all six cortical layers (Fig. 5A; as an example, Cux1 that delineates layers II-IV is shown). However, compared with the WT cortex, radiation of Prkdc−/− resulted in markedly elevated and persistent DNA DSBs in this tissue as indicated by γH2AX formation (Fig. 5B); the expanded inset image shows clear puncta of γH2AX, typical of DNA strand breaks, rather than pan-nuclear staining typical of apoptosis (Shimada et al., 2015). γH2AX colocalizes with Tbr1+ cells, indicating that DNA-PKcs functions in neurons to reduce DNA damage accumulation. Similarly, pKAP1 levels are also elevated in Prkdc−/− tissue, colocalizing with Tbr1+ cells (Fig. 5C). Comparative quantitation of γH2AX and pKAP1 present in irradiated P11 WT and Prkdc−/− tissue is shown in Figure 5D.

KAP1 and H2AX phosphorylation is enhanced in the Prkdc−/− P11 cortex. A, At P11, the six layer structure of the cortex is present (illustration), and both WT and Prkdc−/− are histologically similar, with Cux1 immunostaining of sagittal E15.5 sections used as an example. B, Phosphorylated H2AX(ser134), which identifies DNA DSBs, is largely absent from both WT and Prkdc−/− embryonic tissue, but after radiation γH2AX is abundant in the Prkdc−/− cortex compared with WT and associated with Tbr1+ neurons. The expanded view indicates the typical punctate staining pattern for γH2AX after DNA damage. C, DNA damage after IR also causes widespread activation of KAP1 in neurons in the Prkdc−/− cortex as seen by immunolocalization of phosphorylated KAP1 and Tbr1. D, Quantitation of the levels of γH2AX and pKAP1 in WT and Prkdc−/− cortex. ***p < 0.0001.
Finally, to compare DNA-PKcs function in adult brain to earlier developmental stages, we examined radiation-induced DNA damage signaling in the adult cortex and DG. As shown in Figure 6A, compared with WT, elevated DNA damage signaling determined using pKAP1 immunohistochemistry was found in Prkdc−/− DG and cortex after IR treatment. Additionally, we also found widespread endogenous DNA breaks as shown by γH2AX in Prkdc−/− cortex compared with WT (Fig. 6B), indicating that DNA-PKcs protects the adult brain from endogenous DNA damage accumulation.
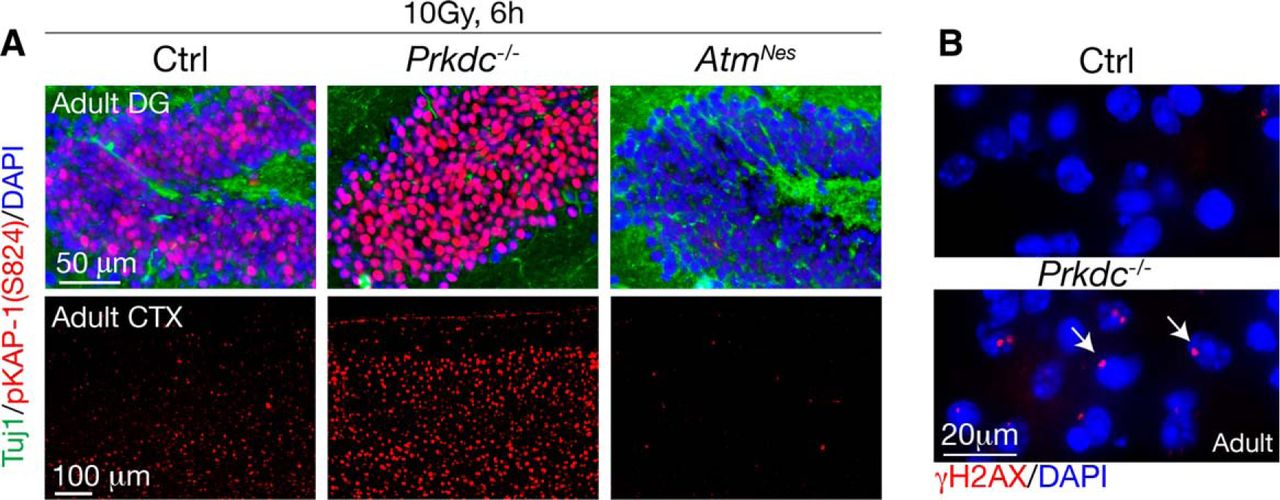
DNA-PKcs restrains DNA damage in adult neural tissue. A, In irradiated adult dentate gyrus or cortex, DNA damage signaling is elevated in Prkdc−/− tissue. Consistent with phosphorylation of KAP1 being ATM-dependent, AtmNes-cre tissue shows no IR-induced pKAP1. B, In 6-month-old Prkdc−/− cortex, DNA damage accumulates as shown by the presence of endogenous DNA strand breaks, as indicated using γH2AX immunostaining. The region of the cortex shown is an area directly superficial to the hippocampus.
Hypersensitivity and enhanced DNA damage signaling occur in Prkdc−/− cerebellar progenitors
We expanded our analysis from cortical structures to include the cerebellum to further corroborate the impact of DNA-PKcs loss toward neural genome integrity. The cerebellum continues development after birth (for ∼3 weeks in the mouse) as the EGL generates granule neurons that migrate inwards to form the inner granule neuron layer as the cerebellum matures (Butts et al., 2014).
After IR treatment, increased activated caspase-3 levels were observed in the Prkdc−/− P5 cerebellum compared with controls, with significantly more death in both the EGL and WM of the mutant tissue (Fig. 7A). Thus, loss of DNA-PKcs in the cerebellum also hypersensitizes to apoptosis after DNA damage, further indicating that DNA-PKcs broadly restrains the detrimental effect of DNA damage throughout the nervous system.

DNA-PKcs protects the cerebellum from excessive DNA damage. A, Quantification of active caspase-3 in the irradiated P5 cerebellum shows increased apoptosis in both the Prkdc−/− EGL and WM. Graphs represent mean values of replicates (n = 3 animals per group). Error bars indicate SEM. **p < 0.01. ***p < 0.001. B, Phosphorylation of p53(ser15), KAP(ser824), and ATM(1981) was increased in irradiated P5 Prkdc−/− cerebellar tissues compared with controls; CHK2 activation is shown as a band-shift (top arrow). These events are ATM-dependent and absent in AtmNes-cre tissue. Ponçeau staining shows equal protein transfer. Ratios indicate levels of radiation-induced phosphorylation of the respective DNA damage signaling components.
We then used Western blot analyses of WT and Prkdc−/− cerebellar tissue to identify key protein modifications characteristics of the DDR, to assess the impact of DNA-PKcs loss toward DNA damage signaling. We used whole P5 cerebella at 2 h after IR (a time before apoptotic morphology is found in the immature EGL), enabling unambiguous identification of DNA damage-induced signaling events. We found radiation-activated CHK2, as indicated by retarded gel mobility, and enhanced phosphorylation of p53 (pSer15) and KAP1 (pSer824) in Prkdc−/− tissue compared with controls (Fig. 7B). ATM phosphorylation was also increased in Prkdc−/− tissue after IR, consistent with elevated levels of unrepaired DNA breaks (Fig. 7B). Tissues from AtmNes-cre and Prkdc−/−;AtmNes-cre serve as controls for Chk2, p53, and KAP1 phosphorylation after IR as these are ATM-dependent events. These data suggest that increased DNA damage in the Prkdc−/− tissue after IR leads to enhanced ATM-dependent DNA damage signaling, which ultimately results in elevated apoptosis. The increased phosphorylation levels of p53 and KAP1 further indicate that loss of DNA-PKcs exacerbates DNA damage signaling because of unrepaired DNA.
DNA-PKcs, ATM, and ATR integrate DNA damage signaling during neurogenesis
The physiological interrelationship between the DNA damage-responsive PI3K kinases remains unclear. In the immune system, ATM and DNA-PKcs coordinate DSB repair and apoptosis during class switch recombination (Callén et al., 2009; Lee et al., 2013), and ATM and DNA-PKcs can both phosphorylate H2AX after DNA damage (Stiff et al., 2004). To understand how DNA-PKcs function is integrated in the DDR during neural development, we generated mice with a combinatorial loss of DNA-PKcs with ATM and ATR. We generated Prkdc−/−;AtmNes-cre and Prkdc−/−;AtrNes-cre double knock-outs (DKO) and Prkdc−/−;(Atm;Atr)Nes-cre triple knock-out animals (TKO). Despite synthetic lethality after germline inactivation of ATM and DNA-Pkcs (Gurley and Kemp, 2001), Prkdc−/−;AtmNes-cre mice showed no overt phenotype. Prkdc−/−;(Atm;Atr)Nes-cre were also born alive, although they only survived for a day, an accelerated phenotype compared with AtrNes-cre single-mutant mice (Lee et al., 2012).
Because of the reports linking overlapping functions of these kinases (Matsuoka et al., 2007) and our observation that double- or triple-mutant embryos showed no obvious defects at E15.5, we used Western blots to confirm loss of the respective proteins. Western blot analysis of WT tissue compared with that from DKO (Prkdc−/−;AtmNes-cre) and TKO confirms these proteins were absent from E15.5 embryos (Fig. 8A). Moreover, analysis of DNA damage signaling by examining p53 and KAP1 phosphorylation also confirmed this process was defective after ATM loss (Fig. 8A). We then determined apoptosis after exposure to IR by active caspase-3 immunostaining. We found that, whereas active caspase-3 levels were increased in Prkdc−/−, apoptosis was greatly reduced in AtmNes-cre tissues (Fig. 8B,C). In contrast, ATR loss led to a sparing of replicating cells in the VZ but had little effect on DNA damage-induced apoptosis in noncycling cells in the SVZ and CP (Fig. 8B,C), which is consistent with its requirement for sensing replication stress (Nam and Cortez, 2011).

Integration of DNA damage signaling by DNA-PKcs and ATM and ATR. A, Western blot analysis confirms protein deletion of DNA-PKcs, ATM, and ATR in the various compound genotypes. Minor residual ATM or ATR may be contaminating wild-type cells or incomplete gene deletion. However, defective phosphorylation of p53 and KAP1 in the P5 cerebellum after radiation confirms compromised DDR signaling in mutant tissue. B, Sagittal section of E15.5 embryos shows that radiation induced apoptosis in the neocortex using activated caspase-3 immunostaining. ATM deletion (AtmNes-Cre) substantially reduces apoptotic signaling throughout the cortex. The combined loss of ATM and ATR eliminates IR-induced apoptotic signaling, whereas DNA-PKcs loss sensitizes immature, noncycling cortical neurons to IR-induced apoptosis, even in the absence of ATM and ATR. Boxes represent CP. C, Quantitation of activated caspase-3 levels in different cortical regions shows the relative contribution of the various kinases to IR-induced apoptosis in specified cortical areas. ****p < 0.0001. D, Coincident loss of DNA-PKcs with AtmNes-cre or (Atr;Atm)Nes-cre still sensitizes the CP to DNA damage. ****p < 0.0001. **p < 0.01. Dual inactivation of ATM and ATR resulted in an absence of active caspase-3-positive cells in the VZ. Genotype legends shown are in addition to those already presented in C. E, Active caspase-3 immunostaining of irradiated E17.5 (Atm;Atr;p53)Emx1-cre;Prkdc−/− brain compared with WT control shows that apoptosis in the TKO is p53-dependent.
The combined loss of Atm and Atr eliminated IR-induced apoptotic signaling as demonstrated by a lack of active caspase-3 immunostaining throughout all regions of the neocortex (Fig. 8B). In comparison, Prkdc−/−;AtmNes-cre embryos showed reduced active caspase-3 signal compared with Prkdc−/− (Fig. 8D) consistent with ATM being a key regulator of apoptosis after DNA damage (Herzog et al., 1998). In contrast, dual inactivation of DNA-PKcs and ATR resulted in radiation-induced apoptosis similar to DNA-PKcs (Fig. 8B,D), reflecting the primary role of ATR in VZ progenitors (Nam and Cortez, 2011; Lee et al., 2012; Zhou et al., 2012). ATM can also activate DNA damage-induced apoptosis in the VZ (Shull et al., 2009; Gatz et al., 2011). However, after radiation apoptosis still occurred in the TKO embryo, albeit this was mostly restricted to the differentiated cells (Fig. 8B,D). The specific activation event (possibly still a protein kinase) leading to IR-induced apoptosis in the absence of all three DNA damage-response kinases is unknown. Moreover, similar to IR-induced apoptosis during neurogenesis being p53-dependent (Lee et al., 2001; Orii et al., 2006), this was also the case in the TKO (Fig. 8E). Collectively, these data illustrate distinct spatiotemporal functions for each kinase as DNA-PKcs prevents damage accumulation in postmitotic cells, where ATM is the kinase that transduces DNA damage signaling. In contrast, in proliferative progenitors, ATR monitors replication stress and modulates DNA damage-induced apoptosis.
ATR uniquely controls the IR-induced G2 checkpoint
The data above indicate distinct roles for DNA-PKcs, ATM, and ATR in signaling DNA damage during neural development. The response to DNA damage also involves cell cycle checkpoint activation, and ATM and ATR in particular have been directly linked to this process. Whereas ATM modulates the DNA damage-induced G1 and S phase checkpoint, ATR is considered important for establishing the G2 and S phase checkpoints in vitro (Bartek et al., 2004; Shiloh and Ziv, 2013). To establish whether these kinases have unique roles in modulating cell cycle checkpoints in vivo in the setting of the nervous system, we analyzed IR-induced cell cycle dynamics in cortical progenitors after DNA damage. Proliferating neural cells in the VZ undergo dynamic nuclear positioning reflective of cell cycle phases, where mitotic cells reside at the apical surface of the VZ and cells in S phase at the basal surface (Spear and Erickson, 2012). Progenitors in M phase can be identified using antibodies for histone H3 phospho-ser10 (pH3), which normally distribute along the apical surface of the VZ (Fig. 9A). Therefore, we compared the various kinase mutants after radiation and used pH3 to identify M phase cells. We found much reduced pH3 immunostaining in Prkdc−/− and AtmNes-cre tissue after radiation, whereas abundant pH3-immunopositive M phase cells were present in AtrNes-cre tissue (Fig. 9B). Importantly, in unirradiated tissue, no differences were found in the distribution of pH3 between genotypes (Fig. 9C). This implies that ATR activates cell cycle arrest to prevent irradiated cells continuing through the cell cycle, likely to facilitate DNA repair. This may account for sparing of the VZ cells in AtrNes-cre as arrested cells will resist apoptosis.

ATR controls the DNA damage-induced G2 checkpoint. A, Apical mitotic cortical progenitors in G2/M in unirradiated WT sagittal E15.5 cortex sections were identified using histone pH3 (Ser10) (p-H3; red). Adjacent image, Expanded view of the boxed region. A neuronal differentiation marker (Tuj1; green) is included as a comparison. B, Irradiated WT and various mutant neocortices show pH3 accumulation after DNA damage in AtrNes-cre tissue, indicating that ATR controls the G2/M checkpoint. Coincident loss of ATR or ATM with PRKDC still results in accumulation of pH3 after DNA damage. Inset, Expanded from white boxed regions. Sections are from embryos recovered 6 h after 4 Gy of radiation. C, Quantitation of pH3-positive cells after radiation in the various genotypes shows that ATR regulates the neocortical G2/M checkpoint. Graphs represent mean values of replicates (n = 3 animals/group). Error bars indicate SEM. ****p < 0.0001.
To determine the influence of the other kinases, we analyzed compound mutants, including Prkdc−/−;AtmNes-cre, Prkdc−/−;AtrNes-cre, (Atm;Atr)Nes-cre and Prkdc−/−;(Atm;Atr)Nes-cre to determine how DNA-PKcs and ATM influenced checkpoint regulation. In each case, when ATR was inactivated, we found similar levels of pH3 accumulation at the apical boundary, indicating that the other kinases did not influence the kinetics of accumulation. In Prkdc−/−;AtrNes-cre, we observed less pH3 accumulation, which may reflect increased DNA damage in G1 associated with DNA-PKcs loss leading to apoptosis (Fig. 9C). Thus, ATR alone controls the G2/M checkpoint after IR and contributions of the other kinases to either the G1 or S phase checkpoint is negligible, at least in the neocortex.
Discussion
Here we have used mouse genetics to explore DNA damage signaling within a physiologic context, and identified multiple levels of neuroprotection provided by key DNA damage-responsive kinases: DNA-PKcs, ATM, and ATR. DNA-PKcs prevents DNA damage accumulation, particularly in noncycling cells, ranging from immature progenitors to mature neuronal populations. ATR modulates the cell cycle G2/M checkpoint in proliferating neuroprogenitors, whereas ATM is required for DNA damage-induced apoptosis in immature progenitors. In this manner, each DDR kinase has unique and nonoverlapping essential roles that collectively provide expansive protection against genome instability in the nervous system, thereby forming a broad barrier to neural genome instability (Fig. 10).

DNA-PKcs, ATM, and ATR provide genome maintenance at different stages of neural development. Genome stability is maintained during neural development by independent functions of key DNA damage-response kinases that integrate the maintenance of genome stability. DNA-PKcs, ATM, and ATR can function independently at different stages of neural development to regulate cell cycle control, apoptosis, and DNA repair after genome damage. The available pathways for DNA DSB repair change as cells differentiate, with NHEJ becoming the main pathway for repair of this lesion in nonproliferating cells (i.e., those that are differentiated and mature). HR is operative in cycling cells as it requires a sister chromatid present in the S and G2/M phase of the cell cycle as a repair template. Although this schema outlines independent protein kinase functions relevant to the current study, multiple additional roles for theses kinases (not indicated), especially for ATM, have been established. Notwithstanding this, it is apparent that each kinase has separate independent roles linked to the proliferative and differentiation status of the nervous system.
Analysis of the response of DNA-PKcs-null cortical progenitors to radiation-induced DNA damage revealed a marked sensitivity of nonproliferative progenitors that were undergoing differentiation and migration. In the Prkdc−/− neocortex at E15.5, abundant radiation-induced apoptosis was readily apparent in the IZ and CP. Within the CP, more immature cells (e.g., Tbr1+Brn2+) showed greater hypersensitivity to IR compared with the Tbr1+Ctip2+ population. Albeit limited, DNA-PKcs also protected proliferative progenitors in the VZ after radiation, possibly reflecting the need for repair of DNA damage in G1. The less obvious impact of DNA-PKcs loss in the VZ may relate to the sensitivity of these progenitors to radiation doses we used, as these cells can undergo apoptosis after DNA DSBs introduced by as little as 0.13 Gy of radiation (Hoshino and Kameyama, 1988; Hoshino et al., 1991). Therefore, the consequence of DNA-PKcs loss in this population may be negligible with radiation doses in the range used in our study.
In contrast to other key NHEJ factors, loss of DNA-PKcs had distinctly less physiological impact, especially in the nervous system (Barnes et al., 1998; Gao et al., 1998a; Frank et al., 2000; Gu et al., 2000; Lee et al., 2000). This is somewhat surprising as we found that DNA-PKcs was clearly required to restrain radiation-induced DNA damage accumulation and apoptosis in this tissue. This may reflect the relative dispensability of certain NHEJ components for low-level repair of endogenous DNA lesions (Gu et al., 2000), with XRCC4/LIG4 in conjunction with accessory factors, such as XLF being sufficient for repairing low levels of damage. Importantly, in regions where higher endogenous damage would be expected to occur, such as the rapidly proliferative neural stem and progenitors in the VZ, HR will be intact in Prkdc−/− tissue and able to address replication-associated DNA breaks. Studies involving hypomorphic mutants of Lig4 (Lig4Y288C) have shown that incapacitation of LIG4 impacts the VZ/IZ after low-level radiation exposure (Gatz et al., 2011). Our analysis with Prkdc−/− shows that there is an increase in apoptosis in the VZ of Prkdc−/− tissue, but a more marked sensitivity in the SVZ and noncycling noncycling IZ/CP is readily apparent, consistent with NHEJ being the main DSB repair pathway in noncycling neural cells.
ATR loss resulted in reduced apoptosis in the proliferative cells of the VZ. ATM is a critical signaling intermediate for transducing the DNA damage signal via p53 to activate apoptosis, particularly in the immature but postmitotic cells in the developing nervous system, and its inactivation can lead to a block in apoptosis (McKinnon, 2012). Notably, dual inactivation of ATM and ATR eliminates IR-induced apoptosis in the neocortical VZ, implying that a portion of VZ progenitors may be exiting the cell cycle before commencement of migration. The reduced apoptosis after ATR loss might be seen as counterintuitive to the fact that damaged cells accumulate in M phase in AtrNes-cre neocortex. Possibly, in this situation, cells accumulated in M phase are not competent for apoptosis and are eliminated via other means, such as autophagy.
Congenital human neurologic syndromes resulting from mutations in DNA-PKcs, ATM, or ATR underscore their importance in the nervous system (O'Driscoll et al., 2004; van der Burg et al., 2009; McKinnon, 2012; Woodbine et al., 2013). As the development of the nervous system involves the programmed elimination of cells (Buss et al., 2006; Yamaguchi and Miura, 2015), then genome-damaged progenitors would normally be easily replaced by the overabundance of progenitors produced during development. However, given the neurodevelopmental abnormalities associated with certain genome instability syndromes (e.g., ATR mutations), there is nonetheless clearly an ongoing need for genome surveillance. Our data provide the first coordinated analysis of these three principal DNA damage-responsive kinases, in a disease-relevant setting. Further, our study contrasts other settings where more direct cooperativity between these factors is observed, such as early embryonic lethality observed after ATR loss or dual germline inactivation of ATM and DNA-PKcs (Brown and Baltimore, 2000; de Klein et al., 2000; Gurley and Kemp, 2001). Our findings suggest that DNA-PKcs is important for the prevention of DNA damage in immature noncycling progenitors and the increased genomic damage in this population would result in apoptosis and cell loss contributing to the microcephalic phenotypes present in humans with PRKDC mutation (van der Burg et al., 2009; Woodbine et al., 2013). In the case of ataxia telangiectasia resulting from ATM deficiency, immature progenitors that incur DNA damage would normally undergo apoptosis, but the loss of ATM would result in cell sparing and the inappropriate incorporation of the damaged cell into the nervous system. The compromised G2 checkpoint resulting from ATR deficiency and DNA damage would also likely result in a phenotype reflective of that seen in Seckel syndrome individuals with hypomorphic ATR mutation, where systemic developmental defects are linked to aberrations of proliferation (O'Driscoll et al., 2003; Murga et al., 2009).
Of the three mouse models, only ATR loss effectively reflects the phenotypic severity seen in humans. Although individuals with (germline) inactivating mutations in ATM and DNA-PKcs have profound neurologic issues, murine models of this deficiency fail to show overt neuropathology, possibly reflecting a general higher threshold for genome damage to manifest in the mouse. However, in the case of ATM (Herzog et al., 1998; Shull et al., 2009; Katyal et al., 2014) and shown here for DNA-PKcs loss, there are nonetheless very clear consequences in the mouse nervous system after loss of these factors in the face of DNA damage. Restricting analysis to neural tissues provides a more disease-relevant context for determining functional contributions of these kinases during tissue homeostasis. The fact that neural development proceeds in the absence of all three DNA damage-responsive kinases may reflect a primary role for these factors in preventing stochastic adverse events that impact tissue homeostasis.
Footnotes
P.J.M. was supported by the National Institutes of Health Grants NS-37956 and CA-21765, Cancer Center Support Grant P30 CA21765, and the American Lebanese and Syrian Associated Charities of St. Jude Children's Research Hospital. E.J.B. was supported by the National Institutes of Health Grant AG027376. We thank Dr. Jingfeng Zhao for genotyping and technical help; Animal Resource Center for support with all mouse work; and Prof. Fred Alt for providing Prkdc mutant mice.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Peter J. McKinnon, Department of Genetics, St Jude Children's Research Hospital, Memphis, TN 38105. peter.mckinnon{at}stjude.org





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}