Article Text

Abstract
Objective Antibiotic use is associated with an increased risk of developing multiple inflammatory disorders, which in turn are linked to alterations in the intestinal microbiota. How these alterations in the intestinal microbiota translate into an increased risk for inflammatory responses is largely unknown. Here we investigated whether and how antibiotics promote inflammation via the translocation of live native gut commensal bacteria.
Design Oral antibiotics were given to wildtype and induced mutant mouse strains, and the effects on bacterial translocation, inflammatory responses and the susceptibility to colitis were evaluated. The sources of the bacteria and the pathways required for bacterial translocation were evaluated using induced mutant mouse strains, 16s rRNA sequencing to characterise the microbial communities, and in vivo and ex vivo imaging techniques.
Results Oral antibiotics induced the translocation of live native commensal bacteria across the colonic epithelium, promoting inflammatory responses, and predisposing to increased disease in response to coincident injury. Bacterial translocation resulted from decreased microbial signals delivered to colonic goblet cells (GCs), was associated with the formation of colonic GC-associated antigen passages, was abolished when GCs were depleted and required CX3CR1+ dendritic cells. Bacterial translocation occurred following a single dose of most antibiotics tested, and the predisposition for increased inflammation was only associated with antibiotics inducing bacterial translocation.
Conclusions These findings reveal an unexpected outcome of antibiotic therapy and suggest that bacterial translocation as a result of alterations in the intestinal microflora may provide a link between increasing antibiotic use and the increased incidence of inflammatory disorders.
- ANTIBIOTICS
- BACTERIAL TRANSLOCATION
- GUT IMMUNOLOGY
- INFLAMMATION
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Antibiotic use is associated with an increased risk of developing multiple inflammatory disorders. These inflammatory disorders have been linked to alterations in the gut microbiome. How antibiotic induced alterations in the microbiome result in an increased risk for inflammation is largely unknown.
What are the new findings?
We observed that following oral antibiotic therapy indigenous live commensal gut bacteria are translocated across the epithelium to induce inflammatory responses. Bacteria crossing the epithelium resulted from decreased microbial sensing by goblet cells, was dependent upon the presence of goblet cells and was associated with the formation of goblet cell-associated antigen passages in the colon. Most but not all antibiotics tested induced bacterial translocation, and increased inflammation in response to coincident epithelial injury was only associated with antibiotics inducing bacterial translocation.
How might it impact on clinical practice in the foreseeable future?
These findings identify an unexpected affect of antibiotics that increases the risk for inflammation. Understanding the interactions between the host and specific members of the gut microbiota that restrict bacterial translocation may offer new strategies to avoid some of the untoward outcomes related to antibiotic exposure.
Introduction
Antibiotics were introduced into the medical armamentarium <100 years ago and altered the course of healthcare and humanity by allowing survival from diseases previously believed to be incurable. Since their introduction, medical antibiotic use has increased dramatically in the USA with a particular increase in the use of broad spectrum antibiotics in recent years.1 In addition, low-dose antibiotics have been used for agricultural purposes for >50 years,2 leading a nearly ubiquitous exposure to low levels of antibiotics in food and water for individuals who are not receiving antibiotic therapy for therapeutic purposes3 ,4 Epidemiological studies suggest that this increasing exposure to antibiotics is associated with an increased risk for developing inflammatory diseases including autoimmune disease, inflammatory bowel disease, coeliac disease, food allergy, asthma and obesity.5–11 The spectrum and number of inflammatory disorders associated with antibiotic use and the association of these disorders with a variety of antibiotics suggests that this increased risk is not due to side effects of specific antibiotics, but rather a shared effect across multiple antibiotics impacting a risk for general inflammatory responses. In accordance with this premise, multiple studies have associated alterations in the gut microbiota, including alterations induced by antibiotics, with enhanced inflammatory responses.12–17 Despite these observations, how dysbiosis of the gut microbial community due to antibiotic therapy confers a risk for enhanced inflammatory responses remains largely unknown.
Disruption of the gut microflora with prolonged courses of oral antibiotics prior to the introduction of pathogens is a commonly used technique in enteric infection models.18–21 This manoeuvre provides a niche for the pathogens to occupy within the host, and accordingly antibiotic treatment is closely linked to pathology and dissemination of the enteric pathogens in these models.18 ,19 Recent studies revealed that prolonged antibiotic treatment to disrupt the gut microbiota also resulted in the translocation of gavaged, laboratory adapted, non-pathogenic bacterial species,22 ,23 indicating that bacterial pathogenicity may not be required for epithelial translocation. Whether antibiotic-induced translocation applies to native gut commensals, which may not share properties with laboratory-adapted bacterial strains and which would be suppressed by antibiotic therapy, is unknown. However, these observations raise the possibility that antibiotic therapy might induce the translocation of native commensal gut species providing an unappreciated link between antibiotic therapy, enhanced inflammatory responses and untoward outcomes associated with antibiotic therapy. To this end, we investigated whether and how antibiotic therapy induces the translocation of native gut commensal bacteria and the outcomes associated with antibiotic-induced gut commensal translocation.
Materials and methods
See online supporting supplementary information for details.
Mice
All mice were maintained on the C57BL/6 background and bred in house. Mice were fed routine chow diet and given filtered autoclaved water. See online supplementary table S2 for specifics on mouse strains and their use in this study.
Antibiotic treatment
For continual antibiotic treatment, mice were treated with a combination of antibiotics (ABX): ampicillin (1 g/L), metronidazole (1 g/L), neomycin (1 g/L) and vancomycin (500 mg/L) in drinking water for 14 days. For one-time dosing of a combination of antibiotics, mice were gavaged once with 500 µg ampicillin, 500 µg metronidazole, 500 µg neomycin and 250 µg vancomycin. For one-time dosing of single antibiotics, mice were gavaged once with 500 µg of ampicillin, streptomycin, metronidazole, ciprofloxacin, erythromycin, neomycin or tetracycline, or 250 µg vancomycin, or 1 mg amoxicillin. For subtherapeutic antibiotic therapy, mice were given vancomycin 6 µg/mL in drinking water as previously described.5
Statistical analysis
A Student's t test, a one-way analysis of variance with Dunnett's multiple comparisons, or a Mann–Whitney test was performed using GraphPad Prism (GraphPad Software, San Diego, California, USA). Analysis of the microbial communities was performed with Partek Genomics Suite (Partek, St. Louis, Missouri, USA).
Results
Antibiotic therapy induces translocation of live colonic commensal bacteria to the mesenteric lymph node and induces an inflammatory response
Four days following the administration of a single dose of a combination of oral antibiotics (ABX) to disrupt the gut microbiota, we observed an increase in the inflammatory cytokines CXCL1, interleukin (IL)-17 and interferon (IFN)γ, but not in the immunomodulatory cytokines transforming growth factor-β or IL-10 (figure 1A). This inflammatory response was accompanied by the translocation of live bacteria to the mesenteric lymph nodes (MLNs) in antibiotic-treated mice, but not in vehicle-treated controls (figure 1B); however, we did not observe an increase in the size of the MLNs following antibiotic treatment (not shown). Mass spectrometry analysis revealed the bacteria cultured from the MLN following antibiotic treatment were Enterococcus faecalis and Escherichia coli. While culture-based methods may not identify all organisms present within the MLN, these species are consistent with an origin in the gut microbiota (see online supplementary figure S1). To better identify where the bacteria were translocating within the GI tract, we used a metagenomic approach to characterise the microbial communities present in the small intestine (SI), colon and MLNs following antibiotic treatment. Principal component analysis of the species identified by bacterial 16s rRNA sequencing four days following ABX treatment revealed that the microbial communities from each organ from multiple mice clustered together (figure 1C), while a dendrogram generated by unsupervised hierarchical clustering of bacterial species present in each tissue revealed that the bacteria within the MLNs more closely resembled the colonic community as opposed to that in the SI (figure 1D). Likewise, a Venn diagram of bacterial species that were represented at least once within each respective tissue in four different mice following antibiotic treatment revealed substantial overlap between the species present within the MLNs with those present in the colon, including species shared exclusively between the MLNs and colon, but not the SI. Conversely, we found no species were shared between the MLNs and SI but not the colon (figure 1E). Some species were observed to be present in the MLN but not either the SI or colon, which may be due to the increased depth of sequencing of the MLN when compared with the colon, relative to the diversity of the bacterial community in each organ. Likewise, analysis of the species by the percentage of operational taxonomic units in the major phyla showed a significant difference between the MLN and the SI, but not the MLN and the colon (not shown). The SI and colon predominantly drain into distinct MLNs, a colonic MLN (cMLN) and an SI MLN, which can be identified following the injection of dye into the colonic or SI submucosa (figure 1F). Following treatment with antibiotics, we observed live bacteria could be found in the cMLN and, to a much lesser extent, the SI MLN, while bacteria were not detected in either MLN in untreated mice (figure 1G). Therefore, following antibiotic treatment live commensal bacteria translocate across the colonic epithelium to the MLN, resulting in an inflammatory response.

Antibiotic therapy induces translocation of live colonic commensal bacteria to the mesenteric lymph node (MLN) and induces an inflammatory response. (A) Cytokine production measured by ELISA in MLN four days after vehicle or antibiotic treatment (ABX; single dose of a combination of ampicillin, metronidazole, neomycin and vancomycin). (B) Quantification of live bacteria by colony forming units (CFUs) cultured from the MLNs of mice treated with vehicle ABX four days earlier. (C) Principal component analysis performed on bacterial species identified by 16s rRNA sequencing from MLN, colon contents or small intestine (SI) contents of mice treated with ABX four days earlier. (D) Unsupervised hierarchical cluster of bacterial species present in the SI, colon and MLN of four mice following ABX treatment. (E) Venn diagram showing overlap of species found on average at least once in each organ from four ABX-treated mice. (F) Photomicrographs following subserosal injection of Chicago blue dye into the SI (left panel) or colon (right panel) identifying the MLNs (black boxes), and blue dye within the MLN indicates the MLN drains the respective organ (white arrows). (G) Quantification of live bacteria by CFUs cultured from the SI or colon draining MLNs of mice treated with vehicle treated or ABX four days earlier. Data are presented as the mean±SEM; *p<0.05; ABX, antibiotic treatment detailed in methods; n=3 mice per group (A and F); n=4 mice (C–E); n=10 mice (B and G).
Live commensal bacteria translocation is associated with colonic goblet cells and goblet cell-associated antigen passages
Dendritic cells (DCs) can extend dendrites through the intestinal epithelial layer to sample luminal contents.24–26 This process of DC transepithelial dendrite (TED) extension has largely been observed in the SI as opposed to the colon.24 ,25 In vivo imaging of the colon of CD11cYFP and CX3CR1GFP reporter mice at a time point in which bacterial translocation occurs following treatment with antibiotics did not reveal the presence of TED extension by DCs (see online supplementary figure S2). In addition, we could not find evidence of TED formation by colonic DCs using higher-resolution imaging on fixed tissue sections of colon following antibiotic treatment (figure 2A and online supplementary movie 1). Therefore, the induction of TEDs by DCs following antibiotic treatment appeared to be an unlikely explanation for commensal bacterial translocation across the colonic epithelium. Goblet cells (GCs) have been reported to act as passages to deliver luminal substances across the intestinal epithelium to DCs.27 GC-associated antigen passages (GAPs) were present in the SI, but not the colon at the steady state, and SI GAPs were reported to deliver small soluble antigens, but not inert particles as small as 20 nm. However, it was recently reported that microbial sensing by colonic GCs suppressed GAP formation,28 and that colonic GAPs formed when the microbial load was reduced by antibiotic treatment (ref.28 and see online supplementary figure S2). Mouse atonal homologue 1 (Math1) is a transcription factor required for the development of secretory epithelial cell lineages, which is largely restricted to GCs in the colon (see online supplementary figure S3). We observed that following antibiotic treatment bacteria were not present in the cMLN of mice in which Math1-expressing epithelial cells had been deleted (figure 2B), suggesting that colonic GCs and GAPs might play a role in translocating commensal bacteria. Further, we observed fluorescently labelled E. faecalis, which was isolated from the cMLN following antibiotic treatment (see online supplementary figure S1), preferentially associated with colonic GCs that formed GAPs, which were identified as epithelial cells with a GC morphology acquiring luminal 10kDa dextran, in antibiotic-treated mice (see online supplementary figures 2 and 4). The labelled E. faecalis could be observed within the apical mucin compartment of GCs that had formed GAPs, and near the basolateral surface or within GCs following antibiotic treatment (figure 2C, D). To further evaluate the relationship between GCs and GAPs with commensal bacteria, we sorted GCs and other non-GC intestinal epithelial cells (IECs) from the colons of mice following antibiotic treatment and evaluated the presence of commensal bacteria by culture. Live bacteria were only associated with GCs following antibiotic treatment and were rarely found to be associated with IECs (figure 2E). Mass spectrometry analysis revealed that the cultured bacteria that were associated with GCs were E. coli, which is similar to the live bacteria isolated from the MLN (see online supplementary figure S1). To better understand the relationship between antibiotic therapy, GAP formation and bacterial translocation to the MLN, we evaluated the kinetics of GAP formation and bacterial translocation to the cMLN following continuous antibiotic therapy and withdrawal. Colonic GAPs formed within a few days of starting antibiotic treatment in drinking water ad libitum and remained open for approximately five days following the removal of antibiotics. Live bacteria appeared in the cMLN two days after the withdrawal of antibiotics and remained in the cMLN during the time in which GAPs remained after antibiotic cessation (figure 2F). Disruption of GC microbial sensing has been demonstrated to induce colonic GAPs independent of antibiotic therapy in specific pathogen-free housed mice.28 We observed deletion of myeloid differentiation primary response gene 88 (Myd88) globally or selectively in Math1-expressing cells, which disrupts GC microbial sensing and induces colonic but not SI GAPs,28 resulted in translocation of bacteria to the MLNs in the absence of antibiotic treatment (figure 2G, H), thus alleviating concerns that the translocation was secondary to toxic effects of antibiotics on the epithelium, and demonstrating that loss of Myd88 in Math1-expressing lineages, such as GCs, is sufficient to induce bacterial translocation. Recently, it was reported that blockade of epidermal growth factor receptor (EGFR) or the downstream p42/p44 mitogen-activated protein kinase (MAPK) relieved the repression of colonic GC microbial sensing and allowed colonic GAPs to form.28 We observed pharmacological inhibition of EGFR or p42/p44 MAPK, which has no effect on SI GAPs,28 also resulted in the translocation of live bacteria to the MLN (see online supplementary figure S5), further linking bacterial translocation with the formation of colonic GAPs. Therefore, translocation of live bacteria across the epithelium to the MLN can occur in the absence of antibiotic therapy when GC microbial sensing is disrupted, consistent with the translocation of live native commensal bacteria to the MLNs following antibiotic withdrawal when the microbiota is rebounding yet still transiently altered enough to allow GCs to form GAPs.

Live commensal bacteria translocation is associated with the presence of colonic goblet cells (GCs) and goblet cell-associated antigen passages (GAPs). (A) Confocal image of a single colonic crypt from CX3CR1gfp/+ mice given ABX demonstrating the lack of transepithelial dendrites (TEDs) (epithelial border denoted by a white dotted line). (B) Quantification of live bacteria by colony forming units (CFUs) cultured from the small intestine (SI) or colon draining mesenteric lymph nodes (MLNs) of Math1iΔvil or littermate Math1f/f mice following tamoxifen administration and ABX treatment. (C) Confocal image of a colonic epithelial cells with GC morphology from ABX-treated mice given luminal Enterococcus faecalis (green) and luminal dextran (red) 4 h earlier demonstrating the presence of E. faecalis within the apical mucin compartment of a GC that has taken up dextran, or a GAP. (D) Confocal image of colonic tissue from ABX-treated Math1dtomato reporter mice given luminal E. faecalis (green) 6 h earlier, demonstrating the presence of E. faecalis at the lateral border of a Math1-expressing cells with GC morphology. (E) Quantification of CFUs after plating sorted GCs or non-GC intestinal epithelial cells (IECs) from the colons of ABX-treated or vehicle-treated mice. (F) Graphical representation of GAPs per GC and corresponding CFUs per MLN during continuous ABX therapy and withdrawal. (G) GAPs per GC in the SI and colon and (H) CFUs in the MLNs of wildtype, myeloid differentiation primary response gene 88 (Myd88)−/− and Myd88iΔMath1 mice following treatment with RU486 to delete Myd88 in Math1-expressing cells. Scale bar in A=50 µm; scale bar in C and D = 20 µm. Data are presented as the mean± SEM; *p<0.05; ND, not detected; ns, not significant; n=3 or more mice per group or time point (A) and (C–F). Each symbol represents one mouse (B and H).
Myd88-dependent and Myd88-independent microbial signals regulate the transfer of live bacteria to the MLN following antibiotic therapy
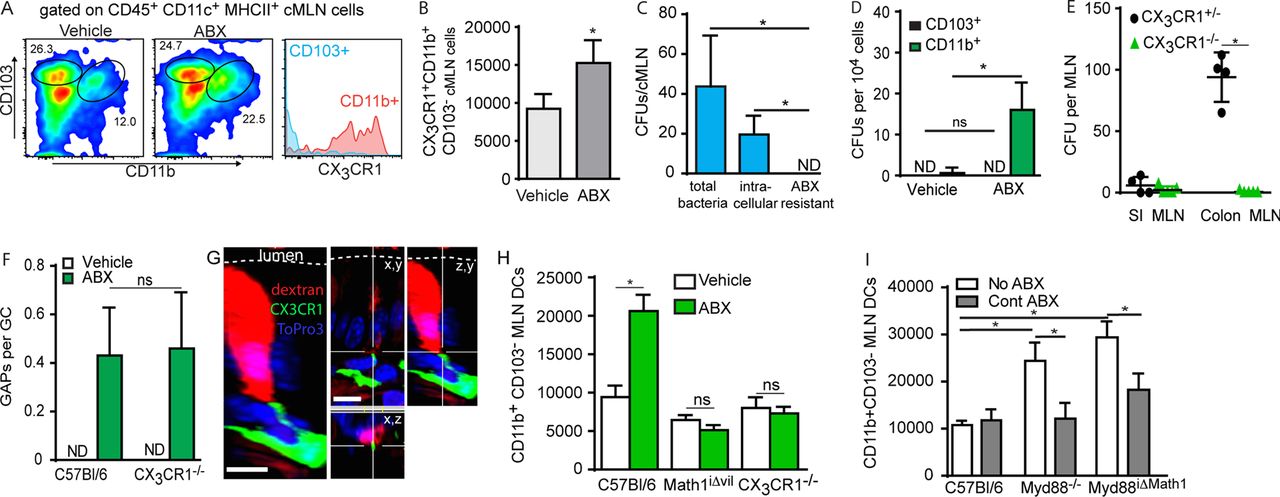
Following antibiotic treatment, CX3CR1+ DCs can carry luminally inoculated exogenous pathogens and non-pathogens to the MLN.23 Consistent with this, we observed that following antibiotic treatment the population of CX3CR1+CD11b+CD103- DCs was significantly increased in the cMLN (figure 3A, B). Additionally following antibiotic treatment, a substantial proportion of the native commensal bacteria in the MLN were intracellular (figure 3C), and culture of sorted cMLN DCs populations isolated after antibiotic treatment revealed that live bacteria were found almost exclusively within the CX3CR1+CD11b+CD103− DCs (figure 3D). Further, we observed translocation of these bacteria to the cMLN was abolished in the absence of CX3CR1 despite no change in the formation of colonic GAPs in CX3CR1−/− mice in response to antibiotic therapy (figure 3E, F). In contrast to what was previously reported for GAPs in the SI at the steady state, we readily observed CX3CR1+ antigen presenting cells (APCs) interacting with colonic GAPs following antibiotic treatment (figure 3G), further supporting the role for CX3CR1+ APCs in acquiring native commensal bacteria and transporting them to the MLN after antibiotic therapy.

Myeloid differentiation primary response gene 88 (Myd88)-dependent and Myd88-independent microbial signals regulate trafficking of live bacteria to the mesenteric lymph node (MLN) by CX3CR1+ dendritic cells (DCs) following antibiotic therapy. (A) Flow cytometry plots showing changes in the populations of CD103+ CD11b− CX3CR1− and CD103− CD11b+ CX3CR1+ DCs (CD45+ CD11c+ MHCII+) in the colonic MLN after vehicle or ABX treatment; histogram gated on the populations (right panel) demonstrates that CD11b+ CD103− DCs are CX3CR1+ and CD11b− CD103+ DCs are CX3CR1−. (B) Absolute number of CD103− CD11b+ CX3CR1+ DCs in the colonic MLN after vehicle or ABX treatment. (C) Quantification live bacteria in the MLN of ABX-treated mice by colony forming units (CFUs) demonstrating the total bacterial load, intracellular bacterial load and antibiotic resistant bacteria determined as outlined in the methods. (D) Quantification of live bacteria by CFUs in the sorted MLN DCs from mice following vehicle or ABX treatment. (E) Quantification of live bacteria by CFUs in the small intestine (SI) and colonic MLNs from CX3CR1−/− and CX3CR1+/− mice following ABX treatment. (F) Graphical representation of goblet cell-associated antigen passages (GAPs) per goblet cell (GC) in wildtype and CX3CR1−/− mice following vehicle or ABX treatment. (G) Confocal image of CX3CR1GFP+ cell interacting with a dextran (red) filled colonic GC following ABX therapy. (H) Absolute number of CD103−CD11b+ MLN DCs in wildtype mice, Math1iΔvil mice given tamoxifen to delete GC, or CX3CR1−/− mice following vehicle or ABX treatment. (I) Absolute number of CD103− CD11b+ MLN DCs in the MLN of wildtype mice, Myd88−/− mice, or Myd88iΔMath1 mice given RU486 to delete Myd88 given no antibiotics or continual ABX therapy in drinking water. Data are presented as the mean±SEM; *p<0.05; ns, not significant; ABX, antibiotics; n=4 or more mice in each group for data.
Antibiotic treatment, or loss of microbial sensing by the global deletion of Myd88, induced CX3CR1+ DCs to carry gavaged exogenous bacteria to the MLN.23 These observations were interpreted to indicate a role for the microbiota and Myd88-dependent microbial sensing restricting DC trafficking to the MLN.23 Therefore, we explored a role for the microbiota in the translocation of bacteria to the MLN, independent of its role in suppressing GAP formation. Similar to the prior studies,23 we observed that following antibiotic treatment the CX3CR1+ MLN DC population, which could also be identified as CD103− CD11b+ DCs, was increased, and that this increase was dependent upon CX3CR1 (figure 3H). In addition, we observed that this increase was ablated when Math1-expressing epithelial cells were depleted, suggesting the increase in MLN CX3CR1+ DCs is also dependent upon GCs and by extension GAPs (figure 3H). This is consistent with the suppression of bacterial transfer to the MLN by the microbiota due to the inhibition of GAP formation and bacterial translocation across the epithelium. We evaluated the population of CX3CR1+ DCs in the MLNs of mice where Myd88 deletion was restricted to Math1-expressing lineages to understand whether the microbiota affected DC trafficking independent of its effect on inhibiting GAP formation. When colonic GAPs were induced by ablating Myd88-dependent, GC-intrinsic, microbial sensing, the CD103− CD11b+, or CX3CR1+, cMLN APC population expanded (figure 3I), indicating that loss of Myd88 signals in Math1-expressing cells was sufficient to induce DC trafficking to the MLN. Interestingly, we found this cMLN DC population was also expanded in mice globally lacking Myd88 and that continuous antibiotic treatment reduced this DC population in mice lacking Myd88 globally, and in mice lacking Myd88 in Math1-expressing lineages (figure 3I). Thus, Myd88-dependent microbial sensing in Math1-expressing cells inhibits translocation of bacteria across the colonic epithelium, and non-Myd88 microbial-mediated events induce CD103− CD11b+ CX3CR1+ DCs to traffic to the MLN.
The enhanced inflammatory responses following antibiotic therapy require GCs and are associated with GAP formation
The presence of bacteria and the associated inflammatory environment we observed within the MLN could result in a predisposition to enhanced and potentially harmful responses to a coincident inflammatory stimuli. Consistent with this, mice with disrupted GC-intrinsic microbial sensing, which results in colonic GAP formation and translocation of commensal bacteria to the MLN, had more severe weight loss and disease in the dextran sodium sulfate (DSS) colitis model when compared with their littermate controls (figure 4A).

Enhanced colitis following antibiotic treatment is associated with goblet cell-associated antigen passages (GAPs) and dependent upon goblet cells (GCs). (A) Weight loss in Myd88iΔMath1 mice given RU486 to delete Myd88 in GCs or littermate controls placed on 2% dextran sodium sulfate (DSS) in drinking water. (B) Quantification of GAPs per GC and live bacteria in the MLNs by colony forming units (CFUs) after a single gavage ABX on day 0. (C) Weight loss in mice placed on 2% DSS in drinking water, after single dose of ABX or vehicle on day 0. (D) Graphical representation of total histology scores and (E) representative images showing increased oedema, ulceration (white arrows) and lymphocyte infiltration (black arrows) in DSS-treated mice given vehicle or ABX as in (C). (F) Weight loss and (G) graphical representation of total histology scores in Math1iΔvil mice given tamoxifen to delete GCs or littermate controls given ABX on day 0 and 1.5% DSS in drinking water. Data are presented as the mean±SEM; *p<0.05; ns, not significant. Scale bar=50 µm. n=5 mice per group (A–E); n=3 mice per group (F and G).
Epithelial recognition of the commensal microbiota plays a role in protection from damage and in epithelial repair.29 To reveal these effects, studies used continual treatment with a combination of antibiotics, which would mask effects arising from bacterial translocation, as translocation occurs following antibiotic withdrawal (figure 2F), and therefore, the detrimental effects of microbial translocation would not be apparent in these studies. We observed the kinetics of colonic GAP formation and bacterial translocation following a single ABX treatment were similar to those found after withdrawal of prolonged antibiotic treatment, with GAPs forming on day 1 and bacteria appearing in the MLN on day 2 (figure 4B). This suggests the period of time following antibiotic therapy in which the gut microbiota is rebounding, but is not yet sufficient to suppress colonic GAP formation, may be a vulnerable time for the development of enhanced inflammatory responses to coincident injury. We observed that mice given ABX treatment and DSS on day 0, to allow translocation of bacteria to occur during epithelial damage, lost more weight (figure 4C), and had worse pathology (figure 4D), with areas of large ulceration and lymphocyte infiltration into the muscle layer when compared with those given DSS alone (figure 4E). Antibiotic treatment did not worsen DSS colitis in Math1iΔvil mice, which lack GCs and GAPs, consistent with the increased severity of colitis seen after ABX treatment being dependent upon GCs and by extension GAPs, and bacterial translocation (figure 4F, G).
Enhanced inflammatory responses only occur following administration of antibiotics that induce GAPs and bacterial translocation
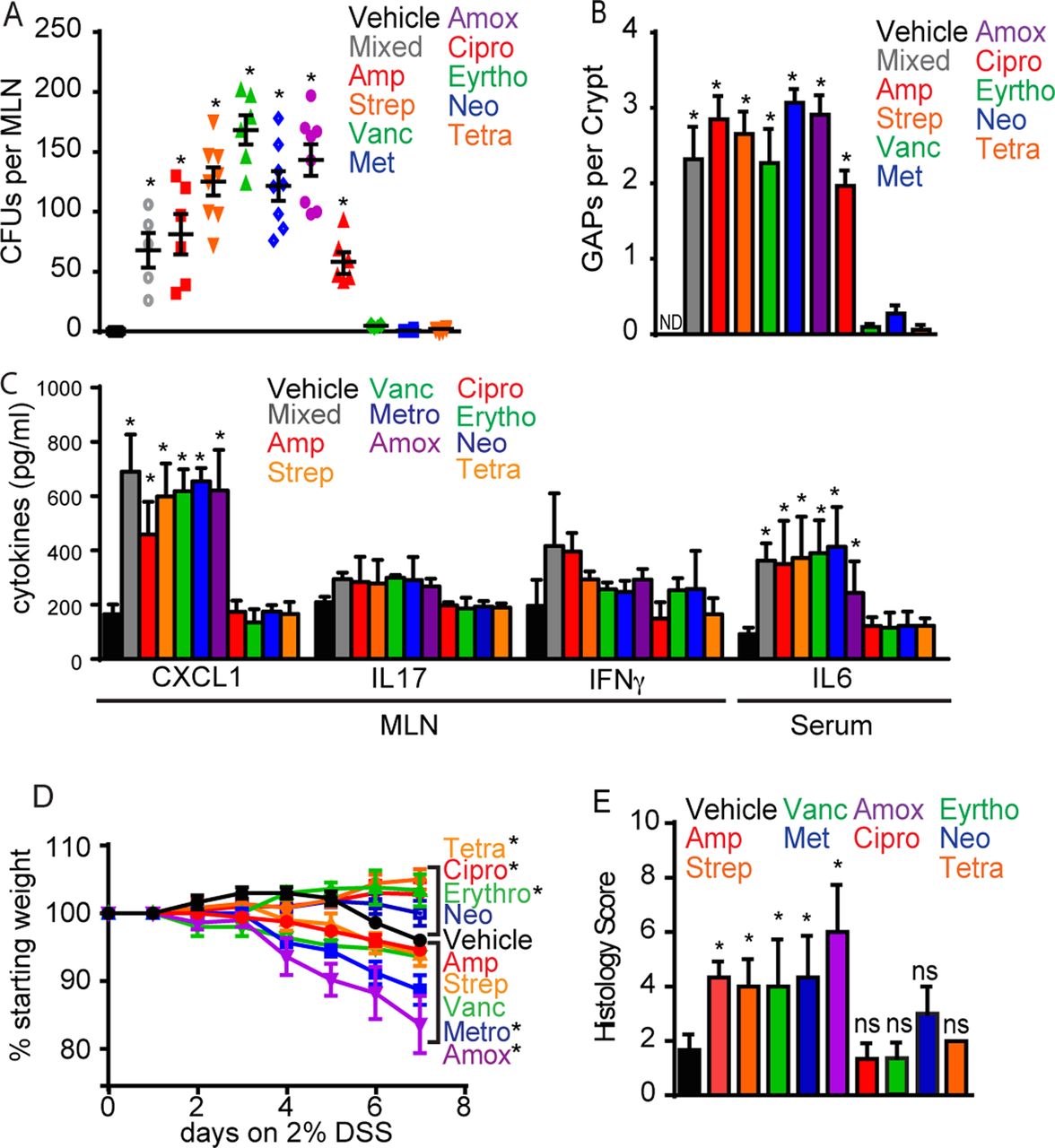
While general antibiotic use has been associated with an increased risk of developing inflammatory diseases, certain classes of antibiotics have been found to have stronger correlations with the disease risk, while others were not associated with an increased risk for disease.8 ,30 We evaluated the effects of multiple commonly used oral antibiotics, with differing mechanisms of action and spectra (see online supplementary table S3), at single clinically relevant doses on the induction of colonic GAPs, the translocation of bacteria to the MLN, the production of inflammatory mediators and on the course of colitis. Bacteria could be found in the cMLN following a single dose of six of the nine antibiotics tested, and this correlated with the induction of colonic GAPs in each case (figure 5A, B). The lack of colonic GAP induction or bacterial translocation was not directly correlated with levels of the colonic bacterial load as measured by quantitative PCR for bacteria 16s rRNA (see online supplementary figure S7), suggesting that the specificity in the bacterial species depleted by the various antibiotics may affect GAP induction and bacterial translocation. In addition, local inflammatory responses, evidenced by significantly increased levels of CXCL1 with slight increases in IL-17 and IFNγ in the cMLN were only seen in mice treated with single doses of antibiotics that induce bacterial translocation (figure 5C). Similar to the observations with mixed antibiotics, we did not see increased production of immunomodulatory cytokines following single antibiotic doses (see online supplementary figure 7B), indicating that these antibiotic treatments were not globally inducing cytokine production. We observed dissemination of bacteria to distant sites following antibiotic therapy (see online supplementary figure S6). Accordingly, only mice treated with antibiotics that induced GAPs and bacterial translocation had systemic inflammatory responses as seen by significantly increased serum IL-6 (figure 5C and online supplementary figure S7). Moreover, when single doses of antibiotics that do and do not promote GAP induction and bacterial translocation were given coincident with DSS-induced epithelial damage, only antibiotics inducing GAP formation and bacterial translocation resulted in worsened disease (figure 5D, E and online supplementary figure S7). Antibiotics given at low doses have been used to promote the growth of farm animals and changes in the gut microbiome due to subtherapeutic antibiotic therapy have been causally linked to obesity,5 which has recently been appreciated as an inflammatory disorder.31 We observed colonic GAP formation and bacterial translocation occurred during continuous subtherapeutic antibiotic therapy (see online supplementary figures 1 and 8), suggesting that in addition to the alteration in the gut microbial community and its metabolic function5 ,32 ,33 bacterial translocation may provide an additional inflammatory stimulus promoting obesity.

Single doses of antibiotics that induce colonic goblet cell-associated antigen passages (GAPs) and bacterial translocation worsen inflammatory responses and colitis in response to coincident epithelial injury. (A) Quantification of live bacteria by colony forming units (CFUs) cultured from the mesenteric lymph node (MLN) and (B) quantification of GAPs per colonic crypt four days after single antibiotic treatment. (C) Quantification of MLN or serum cytokines by ELISA four days after single antibiotic treatment. (D) Weight loss in mice given single antibiotics on day 0 and placed on 2% dextran sodium sulfate (DSS) in drinking water. (E) Graphical representation of total histological scores in mice given single antibiotics on day 0 and given DSS in drinking water. Data are presented as the mean±SEM; *p<0.05; ND, not detected; ns, not significant. n=5 mice per group.
Discussion
Here we have identified a surprising and unappreciated effect of antibiotic use that results in the translocation of live indigenous commensal gut bacteria across the colonic epithelium, resulting in an inflammatory response, and a predisposition to worsened disease in response to coincident injury (figure 6). Confocal imaging and analysis of isolated GCs demonstrated that commensal bacteria were only associated with colonic GCs and within colonic GCs after antibiotic treatment. Further, we observed that antibiotic-induced translocation of gut commensal bacteria was ablated when GCs were absent and translocation of gut commensal correlated with the presence of colonic GAPs. Consistent with our findings, a recent study reported that translocation of exogenous bacteria following antibiotic treatment was independent of disruption of the epithelial barrier or alterations in tight junction permeability.22 While our observations suggest that gut commensals may use GAPs to traverse the epithelium following antibiotic treatment, the specific mechanism and pathways by which commensal gut bacteria traverse the colonic epithelial layer remain to be explored. GAPs in the SI were unable to transport inert particles as small as 20 nm,27 and very few CFUs were found in the SI draining MLN in any of our models, despite continuous GAP formation in the SI, yet here colonic GAPs are associated with the translocation of live native commensal bacteria to colon draining MLN. What accounts for the disparity in these observations is unclear, but may be related to differences in the properties of the SI and colonic GCs as well as differences in the properties and density of the bacteria between the SI and colon, as enteric pathogens have been reported to use SI GCs as a portal of entry.34

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of the antibiotic-induced bacterial translocation and inflammation. (left panel) In the presence of a normal gut microbiota and intact goblet cell (GC) microbial sensing, colonic goblet cell-associated antigen passages (GAPs) and bacterial translocation are inhibited. (middle panel) Following antibiotic therapy, the altered microbiota is no longer able to inhibit GAP formation and bacterial translocation across the epithelium, resulting in low-level inflammation. Coincident epithelial damage leads to increased inflammation and worsened colitis. (right panel) The microbiota has dual and opposite roles in bacterial translocation to distant sites. Myeloid differentiation primary response gene 88 (Myd88)-dependent GC sensing of the microbiota inhibits GAP formation and bacterial translocation across the epithelium, while Myd88-independent microbial signals induce antigen presenting cells (APCs) to migrate to the mesenteric lymph nodes (MLNs).
The microbiota play dual and reciprocal roles in the process of antibiotic-induced commensal bacterial translocation from the intestinal lumen to the MLN. Prior studies demonstrated that GC-intrinsic Myd88-dependent microbial sensing inhibits colonic GAP formation28 and the observations here demonstrate that this role for microbial sensing by GCs extends to bacterial translocation, thus defining a role for the gut microbiota in suppressing bacterial transfer across the epithelium. Our observations and prior studies identified that bacterial trafficking to the MLN is dependent upon CX3CR1+ DCs.23 We observed that when Myd88 was depleted from GCs to allow bacteria to cross the epithelium and be acquired by CX3CR1+ DCs, trafficking of these DCs to the MLN was increased, and conversely, depletion of the microbiota by antibiotics abolished the increased DC trafficking to the MLN. This effect of the microbiota enhancing DC trafficking to the MLN was Myd88 independent as these effects were observed in global as well as cell-type-specific Myd88 knockouts. Thus the microbiota play dual and opposite roles in the translocation of bacteria to the MLN, first inhibiting bacteria crossing the epithelial layer by inhibiting GAP formation in a Myd88-dependent manner, and second by enhancing CX3CR1+ DC trafficking to the MLN in a Myd88-independent manner after GAPs have formed to expose the underlying DCs to luminal substances.
The dual role of the microbiota in bacterial translocation suggests unexpected relationships between antibiotic use, bacterial translocation and enhanced inflammatory responses. We observed that although colonic GAPs were present, bacterial translocation did not occur during continuous high, or therapeutic, doses of antibiotics. This is consistent with continuous therapeutic doses of antibiotics reducing the microbiota to a level that relieves the repression on GAP formation and below the level required to either allow bacteria to cross the epithelium or to deliver the Myd88-independent signals inducing DCs to migrate to the MLN. Bacterial translocation to the MLN occurred several days after the cessation of continuous high-dose antibiotics during a window of time in which the gut microbiota had not returned to a level that was sufficient to suppress GAP formation, yet was sufficient to allow bacteria to cross the epithelium and translocate to the MLN. Consistent with this, we observed that bacterial translocation occurred during continuous low-dose subtherapeutic antibiotic therapy. Thus the most tenuous situations for the translocation of gut bacteria and antibiotic-induced inflammatory responses are paradoxically after cessation of therapy or during exposure to low doses of antibiotics. These observations may indicate antibiotic therapy as a contributor to late-onset neonatal sepsis, as a significant proportion of these children are infected with bacterial species that were already present within the gut and these children received more antibiotics prior to septicaemia.16 In addition, low levels of antibiotics are known to contaminate food and water sources,3 suggesting a role for antibiotics in the dramatic increase in obesity and inflammatory diseases that could be independent of intentional antibiotic therapy.
We observed that a single dose of antibiotics was sufficient to cause a dysbiosis of the gut microbiome, which induced GAP formation and bacterial translocation. While bacterial translocation occurred for a limited time until the dysbiosis was corrected in the normal host, in susceptible hosts antibiotic-induced alterations in the gut microbiota could be a trigger initiating inflammation, which could maintain the dysbiosis of the gut microbiota, thus perpetuating the cycle of bacterial translocation, inflammation and dysbiosis. This could provide a basis for the observation that prior antibiotic therapy is associated with inflammatory bowel disease, a condition known to be associated with alterations of the gut microbiome8 ,30 ,35–37 We evaluated a range of antibiotics with differing spectra and mechanisms of action (see online supplementary table S3). Not all antibiotics, administered at a commonly given dose, resulted in GAP induction and bacterial translocation; however, the increased severity of inflammation in response to epithelial injury was only seen with antibiotics inducing GAPs and bacterial translocation. One exception to the dichotomy we observed amongst the antibiotics tested was ciprofloxacin, which induced GAPs and bacterial translocation, but did not worsen inflammation. This could be related to a previously identified anti-inflammatory property of ciprofloxacin in colitis models.38 Interestingly, the two bacteriostatic antibiotics tested did not induce GAPs, bacterial translocation or inflammation, while all antibiotics inducing GAPs, bacterial translocation and inflammation were bacteriocidal. The antibiotics inducing GAPs, bacterial translocation and increasing severity of colitis targeted Gram-positive species and anaerobes, suggesting that members of Firmicutes, such as Clostridia, might be potential candidates regulating GAP formation. However, given the complexity and interdependence of the members of the gut microbiota, it is likely that antibiotics affect gut bacterial species indirectly, and the relevant bacterial species that are altered and regulate GAPs may not be those anticipated by the antibiotic spectra alone. The observation that not all antibiotics induce bacterial translocation, coupled with the observation that those that do induce bacterial translocation can have this effect at very low doses, implies that inducing bacterial translocation and resulting inflammation is not solely a property of reducing the global gut bacterial load. Interestingly, we observed that a limited number of bacterial species were significantly depleted from the gut microbial community four days after antibiotic therapy when GAPs were present and bacterial translocation to the MLN was high (see online supplementary table S5), making these species candidates for regulating this process. Together this gives promise that understanding the interactions between the host and specific members of the gut microbiota that restrict bacterial translocation may be possible and offer new strategies to avoid some of the untoward outcomes related to antibiotic exposure.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online video
Footnotes
Contributors KAK performed immunofluorescence, GC and GAP quantification, flow cytometry, colitis experiments and data analysis. KAK and DHK performed bacterial quantification and data analysis. KGM performed confocal imaging and data analysis. KAK and RDN conceived of the study, directed the experiment design, analysed the data and wrote the manuscript. All authors reviewed and discussed the manuscript.
Funding Supported by grants DK64798-RDN, AI009550-RDN, DK097317-RDN, DK097893-KAK. The High Speed Cell Sorter Core at the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO, provided flow cytometric cell sorting services. The Siteman Cancer Center is supported in part by NCI Cancer Center Support Grant P30 CA91842. The Speed Congenics Facility of the Rheumatic Diseases Core Center, supported by NIH grant P30AR048335 bred the Myd88−/− mice onto the C57BL/6 background. The two photon imaging was performed at the In Vivo Imaging Core at Washington University School of Medicine. The authors wish to thank Dr. Mark J. Miller for advice and assistance with in vivo imaging. The Washington University Digestive Diseases Research Center Core (DDRCC), supported by NIH grant P30 DK052574 assisted with imaging services.
Competing interests None declared.
Ethics approval Animal procedures and protocols were carried out in accordance with the institutional review board at Washington University School of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The 16s bacterial rRNA sequencing will be made available upon request .