





Abstract
Recently, statins have been shown to have anti-inflammatory effects on lung inflammatory diseases. However, the mechanisms of action of simvastatin in viral pneumonia have yet to be elucidated, although viral infection remains a considerable health threat. In this study, we hypothesised that simvastatin inhibits polyinosinic–polycytidylic acid (poly I:C)-induced airway inflammation, such as RANTES (regulated on activation, normal T-cell expressed and secreted) expression and inflammatory cell recruitment.
In bronchial cells, the effect of simvastatin on poly I:C-induced RANTES expression and signal transducer and activator of transcription (STAT)3-mediated signal transduction was determined using an ELISA and short hairpin (sh)RNA system. In a poly I:C-induced pneumonia mouse model, immunological changes in the lungs after simvastatin inhalation, such as inflammatory cell recruitment and cytokine/chemokine release, were examined.
In poly I:C-stimulated bronchial cells, RANTES secretion was increased by STAT3 activation, and simvastatin suppressed poly I:C-induced STAT3 activation, resulting in inhibition of RANTES expression. In BALB/c mice stimulated with inhaled poly I:C, RANTES expression and neutrophil infiltration into the airway were elevated. However, simvastatin treatment attenuated STAT3 activation, RANTES release and subsequent neutrophilia in the lungs.
These findings suggest that simvastatin inhibits airway inflammation, but there are other mechanisms that need to be fully elucidated.
Statins are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, which catalyses the conversion of HMG-CoA into mevalonate; this is the rate-limiting step in the cholesterol biosynthesis pathway [1]. Statins, widely used as cholesterol-lowering agents, have numerous beneficial pleiotropic anti-inflammatory and immune-modulatory effects with potential clinical applications beyond lipid lowering [2]. More recently, statins have been shown to benefit patients at risk for contracting some types of lung inflammatory disease [3, 4]. Frost et al. [5] reported that statins dramatically reduce the risk of death from chronic obstructive pulmonary disease (COPD) and significantly reduce the risk of death from influenza. Furthermore, clinical studies have suggested that statin use is associated with decreased mortality in patients hospitalised with pneumonia [6].
Pneumonia is the leading cause of death due to infectious disease in industrialised countries. Although bacteria are the most common causes of pneumonia in adults, interestingly, in children, a high rate of co-infections with viruses such as influenza A or B and respiratory syncytial virus (RSV) is observed in pneumococcal pneumonia [7]. Moreover, in recent years, respiratory viruses have also been recognised as a potential common cause of pneumonia in adults, with a prevalence of 2–35% [8].
Despite growing clinical evidence of a role for respiratory viral infections in the pathogenesis of pneumonia, the precise mechanisms of respiratory virus-induced airway inflammation are poorly understood [9]. Double-stranded (ds) RNA, recognised by Toll-like receptor (TLR) 3 within the endocytoplasmic compartment, is a by-product of respiratory viral replication and a representative inflammatory stimulus. Polyinosinic–polycytidylic acid (poly I:C), the synthetic viral dsRNA analogue, is also detected by TLR3, which reportedly participates in the recognition of many viruses [10].
RANTES (regulated on activation, normal T-cell expressed and secreted), a C-C chemokine, is chemotatic for T-lymphocytes, monocytes and eosinophils [11]. Viral infection of bronchial epithelial cells is known to induce RANTES secretion and RANTES is implicated in viral diseases, inducing airway inflammation caused by several viral infections in human and animal models [12, 13]. However, the mechanisms of inducible RANTES gene expression in airway epithelial cells have not yet been fully demonstrated, although RANTES expression may be a major element in the pathogenesis of viral infection [14].
Signal transducer and activator of transcription (STAT)3 plays a potential role in mediating inflammatory responses by inducing gene expression for chemokine, cytokine and inflammatory enzymes [15]. In lung inflammatory disease, STAT3 acts as an epithelial regulator of the allergic response in asthmatic mice models [16]. However, there are few studies on the role of STAT3 in lung inflammatory diseases, particularly viral-induced pneumonia.
In the present study, we hypothesised that poly I:C induces RANTES expression through STAT3 activation in human bronchial epithelial cells. In addition, we investigated whether simvastatin inhibits dsRNA-induced RANTES expression, and examined the anti-inflammatory effect of simvastatin via suppression of RANTES secretion in a dsRNA-induced mouse pneumonia model.
METHODS
Cell culture
Primary normal human bronchial epithelial (NHBE) cells were obtained from Cambrex (Charles City, NJ, USA). Before stimulation, NHBE cells were cultured in bronchial epithelial cell growth medium without hydrocortisone for at least 2 days because of the anti-inflammatory effect of corticosteroids. The human lung epithelial cell line A549 was purchased from the American Type Culture Collection (Manassas, VA, USA). Additional experimental details are provided in the online supplementary material.
ELISA
RANTES concentrations in cell-free supernatants were measured using a specific ELISA kit (R&D Systems, Abingdon, UK), according to the manufacturer's instruction.
Doxycycline-inducible STAT3 short hairpin RNA system
The doxycycline (Dox)-inducible short hairpin (sh)RNA system was designed so that expression of STAT3 shRNA is induced when doxycycline is added to the culture medium. To establish stable cell lines that express the reverse tetracycline/doxycycline responsive transcriptional activator (rtTA), A549 cells were transfected using Effectene reagent (Qiagen, Alameda, CA, USA) with 40 μg pTet-On (Clontech Laboratories Inc., Seoul, Korea) encoding rtTA. After selection with 400 μg·mL−1 G418 for 28 days, resistant clones were selected. Then, a BLOCK-iT inducible H1 RNAi entry vector (Invitrogen, Grand Island, NY, USA) encoding STAT3 shRNA was transfected into the clones. After selection with 400 μg·mL−1 zeocin for 28 days, resistant clones were selected and cultured. The final clones were stably transfected with the rtTA and STAT3 shRNA constructs. Dox was used to silence the STAT3 gene.
Establishment of pneumonia animal model by poly I:C
For testing the anti-inflammatory effect of simvastatin on the poly I:C-induced pneumonia mouse model, we developed an experimental mouse model. 6-week-old BALB/c mice (specific pathogen-free females) were divided into four groups: those intranasally injected with 30 μL PBS (PBS group), 100 μg poly I:C in 30 μL PBS (poly I:C group), 100 μg poly I:C and 30 μg simvastatin in 30 μL PBS (poly I:C + SIM group), and 100 μg poly I:C and 2 μg neutralising RANTES antibody in 30 μL PBS (poly I:C + α-RAN group). All mice were sensitised on days 1, 2, 3 and 4. 2 weeks after the first injection, mice were again challenged (on days 15, 16, 17 and 18) with the same conditions as the primary treatment, except for the doses of poly I:C; this was decreased from 100 μg to 50 μg. All mice were sacrificed the following day (day 19). All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the College of Medicine, Seoul National University Seoul, Republic of Korea.
Bronchoalveolar lavage fluid collection
Bronchoalveolar lavage fluid (BALF) collection was adapted from a method described previously [16]. Levels of several cytokines in BALF were measured with a Bio-Plex 200 system (Bio-Rad, Hercules, CA, USA), according to the manufacturer's instructions. Additional experimental details are provided in the online supplementary material.
Immunohistochemistry
The mice were sacrificed, and their lungs were perfused with PBS. Lungs were inflated and fixed in 4% formaldehyde. Tissues were cut mid-sagittally and embedded in paraffin 24 h after fixation. Serial sections were obtained for histological analysis. Additional experimental details are provided in an online data supplement.
Statistical analysis
Differences between groups were determined using a one-way ANOVA test. Data were expressed as mean±sem. Statistical comparisons were made using t-tests.
RESULTS
Inhibition of RANTES secretion by simvastatin in poly I:C-stimulated lung epithelial cells
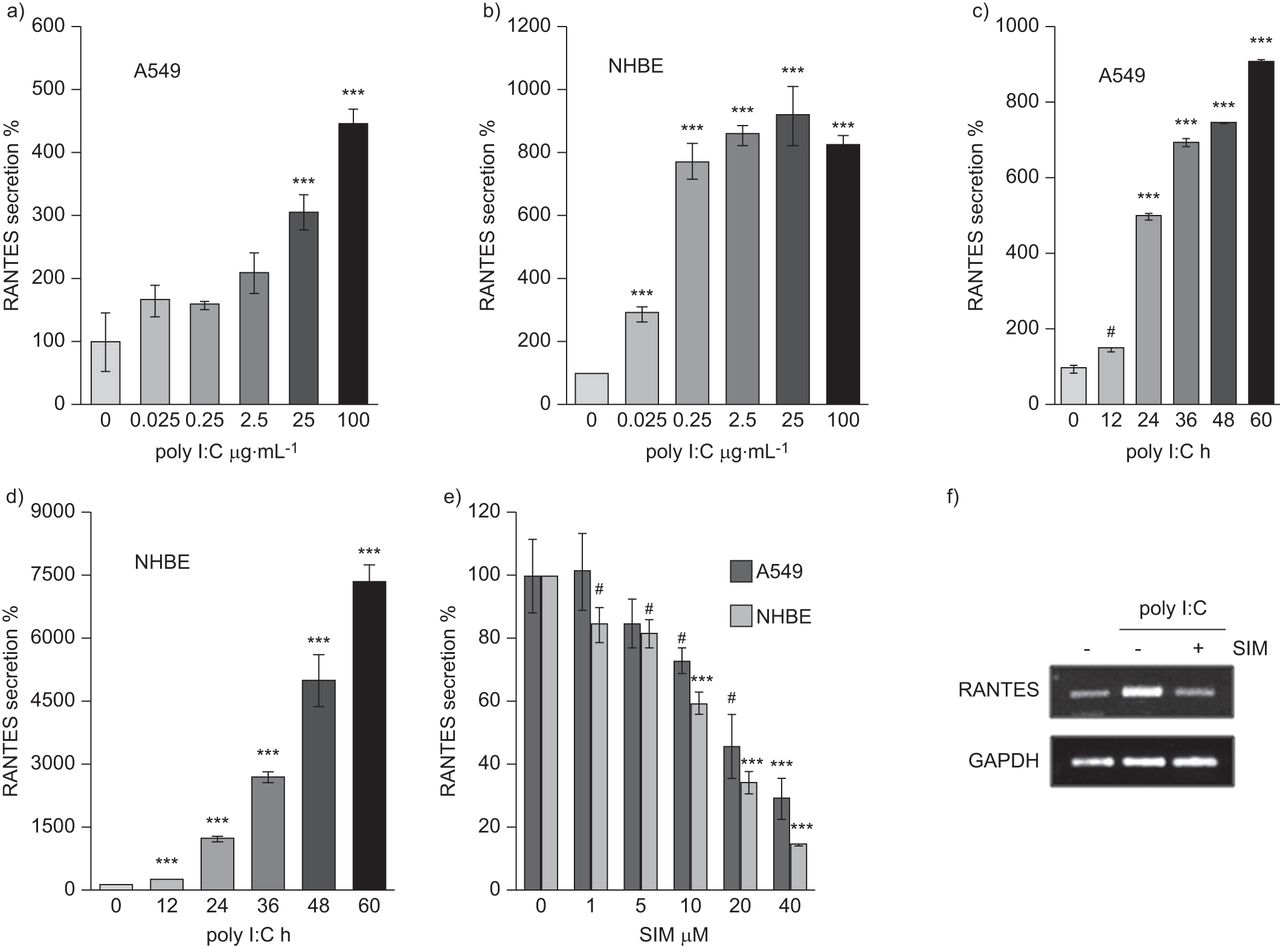
We examined whether poly I:C, a synthetic dsRNA viral component, would induce RANTES secretion in human alveolar epithelial cells (A549) and primary NHBE cells. We treated A549 and NHBE cells with poly I:C at the doses indicated (fig. 1a and b) or for the times indicated (fig. 1c and d). RANTES secretion was clearly induced in a dose-dependent manner and dramatically increased in a time-dependent manner in both types of cells. These findings indicate that poly I:C induced RANTES secretion in both bronchial and alveolar epithelial cells.

Simvastatin (SIM) inhibits polyinosinic–polycytidylic acid (poly I:C)-induced RANTES (regulated on activation, normal T-cell expressed and secreted) secretion in alveolar and bronchial epithelial cells. A549 or normal human bronchial epithelial (NHBE) cells were seeded in 24-well plates. After 24 h, the medium was changed and the experiment performed under the following conditions. a) A549 and b) NHBE cells were treated with poly I:C at indicated doses for 24 h. Then, the culture media were subjected to ELISA assay. c) A549 and d) NHBE cells were treated with 25 μg·mL−1 poly I:C for the indicated times. Then, the media was subjected to ELISA assay. e) A549 or NHBE cells were seeded in 24-well plate. After 24 h, the medium was changed. Cells were pre-treated with SIM at indicated doses for 6 h, and then, 25 μg·mL−1 poly I:C was added to the media. After 24 h, the media was subjected to ELISA assay. f) For detection of RANTES transcripts level, A549 cells were pre-treated with 20 μM simvastatin for 6 h, and then 25 μg·mL−1 poly I:C was added to the media for 12 h. Then, RANTES mRNA levels were detected by RT-PCR analysis, which was performed using the primers as described in the online supplementary material. Data are presented as mean±sem (n=3). #: p<0.005 versus control. ***: p<0.001 versus control.
Next, we determined the effect of simvastatin in dsRNA-induced RANTES secretion on A549 and NHBE cells. Simvastatin suppressed the poly I:C-induced RANTES secretion in both cell types (fig. 1e) and reduced poly I:C-induced RANTES transcript level in A549 cells (fig. 1f). Thus, we concluded that simvastatin inhibits both RANTES mRNA and protein expression in poly I:C-stimulated bronchial and alveolar epithelial cells.
STAT3 mediates poly I:C-induced RANTES secretion in lung epithelial cells
STAT3 is involved in a variety of inflammatory diseases, including those of the lung. Thus, to try to define the role of STAT3 in viral pneumonia, we checked whether STAT3 mediates poly I:C-induced RANTES secretion in bronchial and alveolar epithelial cells. We first developed a Tet-On system for inducible-shRNA expression, as indicated in the Methods section. Prior to examining STAT3-mediated RANTES expression, we checked STAT3 transcript levels induced by Dox in both rtTA stable and rtTA/shSTAT3 stable cell lines (fig. 2a and b). In rtTA stable cells, STAT3 transcripts were constitutively expressed at a basal level independent of Dox dose. In rtTA/shSTAT3 stable cells, however, STAT3 transcript levels were decreased by doxycycline in a dose-dependent manner. Then, we investigated whether STAT3 mediates RANTES secretion in poly I:C-stimulated epithelial cells. After doxycycline treatment, poly I:C-induced RANTES secretion did not decrease in rtTA stable cells, but did decrease in a dose-dependent manner in rtTA/shSTAT3 stable cells (fig. 2c and d). These results indicate that STAT3 gene expression is mediated by poly I:C-induced RANTES secretion in lung epithelial cells.

Polyinosinic–polycytidylic acid (poly I:C)-induced RANTES (regulated on activation, normal T-cell expressed and secreted) secretion through signal transducer and activator of transcription (STAT)3 activation in alveolar epithelial cells. Both a) responsive transcriptional activator (rtTA) and b) rtTA-short hairpin (sh)STAT3 cell lines were incubated for 24 h with doxycycline (Dox) at indicated doses, then STAT3 mRNA levels were detected by RT-PCR analysis, which was performed using the primers as described in the online supplementary material. For the detection of RANTES secretion, c) rtTA and d) rtTA-shSTAT3 cell lines were pre-incubated for 24 h with Dox at indicated doses, then 25 μg·mL−1 poly I:C was added to the medium. After 24 h, the medium was subjected to ELISA assay. e) A549 cells were pre-treated for 2 h with STAT3 inhibitor, S31–201, at indicated doses, then 25 μg·mL−1 poly I:C was added to the media. After 24 h, the media was subjected to ELISA assay. Data are presented as mean±sem (n=3). #: p<0.005 versus control. ***: p<0.001 versus control.
Next, we investigated whether STAT3 inactivation, but not STAT3 gene knockdown, would inhibit poly I:C-induced RANTES secretion. We used the STAT3 inhibitor S31-201, which suppresses STAT3 phosphorylation following its translocation. STAT3 inactivation by S31-201 decreased poly I:C-induced RANTES secretion in lung epithelial cells (fig. 2e). This indicates that not only STAT3 gene silencing but also STAT3 inactivation inhibits poly I:C-induced RANTES production.
Simvastatin suppresses poly I:C-induced STAT3 activation in lung epithelial cells
We found that poly I:C-induced RANTES expression was inhibited by simvastatin and mediated by active STAT3 in lung epithelial cells. Thus, we tested whether simvastatin inhibited STAT3 activation in poly I:C-stimulated epithelial cells. Simvastatin dramatically inhibited phosphorylation of STAT3 expression induced by poly I:C in a dose-dependent manner in A549 (fig. 3a) and NHBE cells (fig. 3b). These results indicate that simvastatin may suppress poly I:C-induced RANTES expression through STAT3 inactivation in lung epithelial cells.

Signal transducer and activator of transcription (STAT)3 activation is involved in the inhibitory effect of simvastatin (SIM) on polyinosinic–polycytidylic acid (poly I:C)-induced RANTES (regulated on activation, normal T-cell expressed and secreted) secretion in alveolar and bronchial epithelial cells. a) A549 or b) normal human bronchial epithelial (NHBE) cells were incubated with 25 μg·mL−1 poly I:C for indicated times. Then, cell lysates were subjected to Western blotting using the antibodies indicated. Next, A549 or NHBE cells were pre-incubated with SIM at indicated doses for 6 h, then 25 μg·mL−1 poly I:C was added to the medium. Next, cell lysates were subjected to Western blot using the antibodies indicated. c) A549 cells were seeded in a 24-well plate. After 24 h, the medium was changed and the experiment performed under the following conditions. Cells were transfected with the plasmids indicated for 12 h. The medium was changed, then cells were pre-treated with 20 μM SIM. After 6 h, 25 μg·mL−1 poly I:C was added to the medium for 24 h. The medium was subjected to ELISA assay. Data are presented as mean±sem (n=3). pY-STAT3: anti phospho-tyrosine STAT3. #: p<0.005 versus control.
Furthermore, we presumed that STAT3 overexpression in poly I:C-treated cells could recover the inhibitory effect of simvastatin on polyI:C-induced RANTES secretion. Thus, we mock transfected or transfected wild-type STAT3 plasmids into A549 cells and then added poly I:C to the media in either the presence or absence of simvastatin (fig. 3c). STAT3 overexpression enhanced RANTES secretion in poly I:C-treated cells. In addition, the decreased RANTES secretion induced by simvastatin was recovered by STAT3 overexpression under poly I:C-treated conditions. These data suggest that STAT3 expression regulates RANTES expression, and that simvastatin may suppress poly I:C-induced RANTES secretion through STAT3 inactivation.
Simvastatin inhibits RANTES secretion through inactivation of AKT and STAT3 in lung epithelial cells
STAT3 can be phosphorylated by several kinases in cytoplasm [17, 18]. Therefore, we investigated which kinase phosphorylates STAT3 in poly I:C-stimulated epithelial cells. We used AG490 as a Janus kinase (JAK)2 inhibitor, LY294002 as an AKT inhibitor, PD098059 as an extracellular signal-regulated kinase inhibitor, and PP2 as an Src inhibitor, to treat NHBE and A549 cells. AG490 (fig. 4a) and LY294002 (fig. 4b) inhibited poly I:C-induced RANTES secretion in a dose-dependent manner in both cell types, but PD098059 (fig. 4c) and PP2 (fig. 4d) did not. Because JAK2 activates STAT3 in response to cytokines or growth factors, we were interested in AKT-activated STAT3 expression in poly I:C-stimulated epithelial cells. Thus, we quantified phospho-AKT levels (the active AKT form) by Western blotting. Poly I:C induced AKT activation after 3 h of poly I:C treatment in A549 (fig. 4e) and 1 h of poly I:C treatment in NHBE cells (fig. 4f). AKT was activated relatively early compared to STAT3 in poly I:C-stimulated epithelial cells, as expected. Moreover, we investigated whether AKT mediates STAT3 activation. Phospho-STAT3 expression was decreased by LY294002 in a dose-dependent manner (fig. 4g). This indicates that active AKT mediates STAT3 activation in poly I:C-stimulated epithelial cells. Furthermore, simvastatin inhibited poly I:C-induced phospho-AKT expression in a dose-dependent manner (fig. 4h). These findings suggest that simvastatin probably inhibits STAT3 activation through AKT dephosphorylation in poly I:C-stimulated epithelial cells.
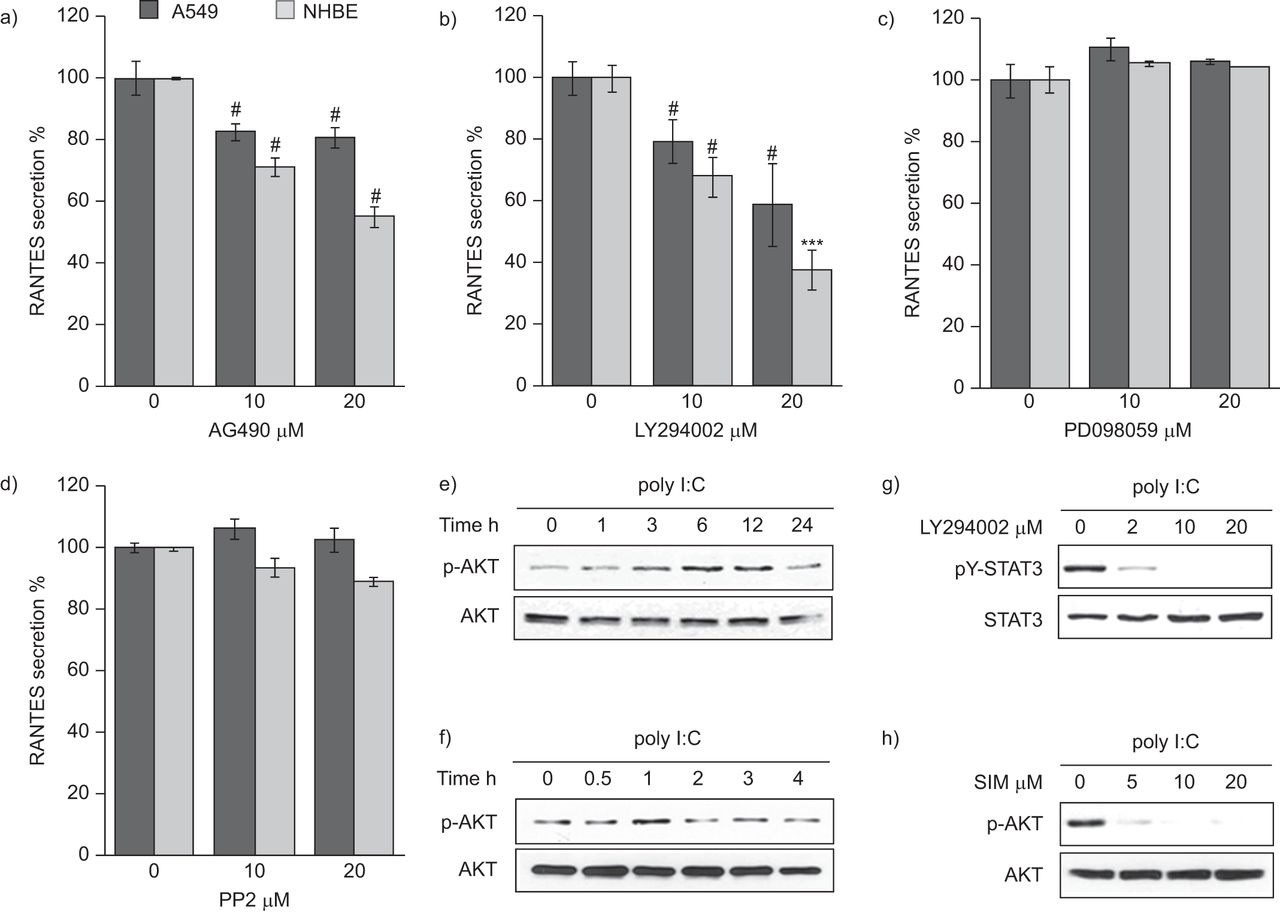
LY294002 inhibits polyinosinic–polycytidylic acid (poly I:C)-induced signal transducer and activator of transcription (STAT)3 activation and simvastatin suppress poly I:C-induced AKT activation in alveolar epithelial cells. A549 or normal human bronchial epithelial (NHBE) cells were seeded in 24-well plates. After 24 h, the medium was changed and the experiment performed under the following conditions. Cells were pre-treated for 2 h with a) AG490 (Janus kinase (JAK)2 inhibitor), b) LY294002 (phosphinositide 3-kinase (PI3K)/AKT inhibitor), c) PD098059 (extracellular signal-regulated kinase (ERK) inhibitor) or d) PP2 (Src inhibitor) at the indicated doses. Then, 25 μg·mL−1 poly I:C was added to the medium for 24 h. The medium was subjected to ELISA assay. Data are presented as mean±sem (n=3). e) A549 and f) NHBE cells were incubated with 25 μg·mL−1 poly I:C for the indicated times. Then, cell lysates were subjected to Western blot using the antibodies indicated. g) NHBE cells were pre-treated with LY294002 at indicated doses for 2 h, then 25 μg·mL−1 poly I:C was added to the medium. After 1 h, cell lysates were subjected to Western blotting using the antibodies indicated. h) NHBE cells were pre-treated with simvastatin (SIM) at the indicated doses for 6 h, then 25 μg·mL−1 poly I:C was added to the media. After 1 h, cell lysates were subjected to Western blotting using the antibodies indicated. RANTES: regulated and normal T-cell expressed and secreted. pY-STAT3: anti-phosphotyrosine STAT3; p-AKT: phospho-AKT. #: p<0.005 versus control. ***: p<0.001 versus control.
Simvastatin alleviates histopathological changes in lung tissues and inhibits STAT3 activation and RANTES secretion by poly I:C in airway epithelial cells
In a poly I:C-induced pneumonia mouse model, simvastatin alleviates histopathological changes in lung tissues and inhibits STAT3 activation and RANTES secretion by poly I:C in airway epithelial cells. Based on the aforementioned data, we investigated whether simvastatin has an anti-inflammatory effect in the lung in vivo. We first developed a poly I:C-induced pneumonia mouse model, as indicated in the Methods section (fig. 5a). After the mice were sacrificed, we detected that lung tissue sections from the poly I:C group showed infiltration of inflammatory cells in the peri-bronchial (large or small airways) and peri-vascular areas (fig. 5b) by haematoxylin and eosin staining. However, simvastatin or RANTES antibody treatment significantly inhibited inflammatory cell infiltration. Thus, we concluded that simvastatin suppresses inflammatory cell recruitment into airways induced by poly I:C.

Simvastatin (SIM) attenuates the infiltration of immune cells into lung tissues in polyinosinic–polycytidylic acid (poly I:C)-inhaled mice. a) Development of the poly I:C-induced pneumonia mice model. Poly I:C was intranasally injected to BALB/c mice in either the presence or absence of SIM or neutralising RANTES (regulated on activation, normal T-cell expressed and secreted) antibody as described in the online supplementary material. b) Lung tissues were fixed with 4% paraformaldehyde, sectioned and stained with haematoxylin and eosin (magnification ×100). Scale bars=100 μm. c) Immunohistochemistry was performed using paraffin-embedded lung tissues and primary anti-phospho-tyrosine signal transducer and activator of transcription (pY-STAT)3 antibody, then diaminobenzidine staining was performed. Arrowheads indicate stained phospho-STAT3 in nucleus (magnification ×400). d) For the detection of RANTES secretion in lung tissues, bronchoalveolar lavage fluid from each group of mice was collected, as described in the online supplementary material. RANTES secretion was determined by ELISA. α-RANTES: anti-RANTES. Data are presented as mean±sem (n=5). ***: p<0.001 versus PBS group.
Next, we sought to confirm whether poly I:C induces STAT3 activation and RANTES secretion in lung epithelial cells in vivo, as detected in vitro. To identify the cell types in which poly I:C-mediated STAT3 activation occurred, immunohistochemistry was performed (fig. 5c). In the PBS group, only baseline STAT3 activation was detected in lung tissue. Poly I:C administration, however, resulted in significant STAT3 activation in the airway epithelium and in the immune cells surrounding the airway. However, as expected, STAT3 activation in the airway epithelium was markedly decreased in the simvastatin-injected group. Furthermore, RANTES secretion in BALF was also increased by poly I:C, but was decreased by simvastatin (fig. 5d). These results show that simvastatin inhibits poly I:C-induced STAT3 activation and RANTES secretion by airway epithelial cells in vivo, as it does in vitro.
To detect further inflammatory effects, we determined whether simvastatin affected several major inflammatory cytokines, including those related to the T-helper cell (Th) type 1 and Th2 responses (fig. S1). In poly I:C mice, simvastatin significantly decreased tumour necrosis factor (TNF)-α, interleukin (IL)-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-12(p70), IL-4 and IL-5 secretion. Thus, simvastatin inhibits inflammatory cell recruitment by suppressing the production of several pro-inflammatory cytokines.
In the poly I:C-induced pneumonia mouse model, simvastatin suppresses neutrophil recruitment in lung tissues
To profile the poly I:C-induced infiltrating immune cells, BALF differential cell counts were performed by Diff-Quik staining. Total cell counts were 10-fold greater in the poly I:C group than in the PBS group; however, the number of total cells and lymphocytes after simvastatin or neutralising RANTES antibody treatment showed decreasing trends (data not shown). In particular, neutrophils were the major infiltrating cell type, and simvastatin and blockade of RANTES markedly reduced the poly I:C-induced neutrophil influx (fig. 6a). To confirm the inhibitory effect of simvastatin on neutrophil recruitment, we performed immunohistochemistry on lung tissue sections (fig. 6b). These results also indicated that administration of simvastatin or neutralising RANTES antibody decreased the number of infiltrating neutrophils in lung tissues by 50% (p<0.001) (fig. 6c).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simvastatin (SIM) inhibits neutrophils recruitment in polyinosinic–polycytidylic acid (poly I:C)-inhaled mice. a) For the profiling of recruited immune cells, bronchoalveolar lavage fluid (BALF) was used in Diff-Quik staining as described in the online supplementary material. The number of neutrophils was counted and data are expressed as the mean±sem (n=5). ***: p<0.001 versus PBS group. b) For the detection of recruited neutrophils, immunohistochemistry was performed using paraffin-embedded lung tissues and primary anti-neutrophil membrane marker antibody, followed by diaminobenzidine staining. Arrowheads indicate recruited neutrophils. Magnification×100. c) The average number of neutrophils in each section are shown. Data are expressed as the mean±sem (n=5). ###: p<0.001 versus PBS or poly I:C-inhaled mice group. α-RAN: anti-regulated on activation, normal T-cell expressed and secreted.
DISCUSSION
In the present study, we demonstrated for the first time that simvastatin inhibits AKT/STAT3-mediated RANTES secretion in response to poly I:C in alveolar and bronchial epithelial cells in vitro. Again, local simvastatin treatment in poly I:C-inhaled mice attenuated the inflammatory response, as indicated by reduced neutrophil recruitment, pro-inflammatory cytokine secretion, RANTES production and STAT3 activation in vivo. These results mean that simvastatin improves poly I:C-induced lung inflammation via suppression of poly I:C/AKT/STAT3/RANTES signalling in airway epithelial cells and subsequent neutrophil infiltration. Furthermore, these results suggest the possibility that simvastatin could inhibit the progression of viral pneumonia in early step of pathogenesis because airway epithelial cells are the primary sites of respiratory viral infection.
In recent years, there has been a considerable number of review papers indicating that statins may have a role in preventing pneumonia, or improving prognosis in hospitalised patients with community-acquired pneumonia [19], and that their use may ameliorate the adverse effects of pneumonia [20]. However, there are few reports of the anti-inflammatory effect of simvastain on virus-induced lung inflammation, such as viral pneumonia. Thus, we wished to assess whether simvastatin can work in a poly I:C-induced pneumonia mice model. We have shown that simvastatin suppressed neutrophilia, resulting from the inhibition of STAT3 activation and RANTES secretion in poly I:C-inhaled mice (figs 5 and 6). In addition to the anti-viral effects of simvastatin demonstrated in this study, there are other reports indicating that simvastatin has antimicrobial activity. For example, simvastatin is protective during Staphylococcus aureus pneumonia [21] and ameliorates acute lung injury in streptococcal infections [22]. A recent study indicates that oral simvastatin at physiologically relevant doses only modestly protects against pneumococcal pneumonia [23]. However, these anti-inflammatory effects are not limited to simvastatin. Rosuvastatin treatment may modestly reduce the incidence of pneumonia [24]. Exposure to atrovastatin was associated with a reduced risk of pneumonia [25]. The lipophilic pitavastatin and the hydrophilic pravastatin also inhibited the inflammatory cytokine production in lipopolysaccharide-stimulated human bronchial epithelial cells [26]. Therefore, these reports have shown that various statins, including simvastatin, may exert the anti-inflammatory effects on inflammatory diseases, such as viral or bacterial pneumonia, although further studies are required to confirm this.
RANTES is known to induce the recruitment of a variety of immune cells into inflamed tissues [27]. In agreement with our data (fig. 5), simvastatin dramatically inhibited the infiltration of immune cells into lung tissues, and blockade of RANTES showed identical results. These results can be explained by the data showing that simvastatin attenuates RANTES secretion via STAT3 inactivation, as we detected in vitro, resulting in a decrease in infiltrating neutrophils. Furthermore, simvastatin and neutralising RANTES antibody decreased pro-inflammatory cytokine production (fig. S1). Thus, induction of the expression of several cytokines (TNF-α, IL-6, GM-CSF and IL-4) by poly I:C is mediated by RANTES, and simvastatin is more likely to inhibit secretion of these cytokines by suppressing RANTES. One of the major cells expressing RANTES is the airway epithelial cell. The mechanism of virus-induced RANTES gene expression has recently been demonstrated. RSV induces RANTES production via the nuclear factor (NF)-κB signalling pathway in human bronchial epithelial cells [28]. Endogenous human RANTES gene transcription is directly induced by interferon regulatory factor (IRF)-3 [29]. dsRNA also activates RANTES gene transcription via NF-κB and IRFs [30]. These previous reports suggest that respiratory viral infections and viral components induce RANTES expression via IRFs and the NF- κB signalling pathway. In this study, we focused on the STAT3 transcription factor to identify the mechanism of simvastatin inhibition of RANTES secretion, because its activation mediates inflammatory gene expression [15]. We provided the first evidence that AKT/STAT3 activation by poly I:C induced RANTES secretion, and that simvastatin inhibited STAT3 activation by inactivating AKT (fig. 4). Therefore, simvastatin suppresses the AKT/STAT3/RANTES signalling pathway in poly I:C-stimulated epithelial cells. However, further studies are needed to fully elucidate the dsRNA-activated AKT/STAT3/RANTES signalling pathway, because few reports have indicated that dsRNA-activated AKT mediates STAT3 activation. We hypothesise that active AKT induces interferon gene expression via the NF-κB and/or IRF signalling pathways, which then activate JAK2-STAT3 signalling by autocrine or paracrine routes. We have more questions about the mechanism by which simvastatin inhibits poly I:C-induced AKT activation. These unknown mechanisms suggest that simvastatin should be investigated in more detail for its usefulness as a drug therapy against viral infections such as pneumonia.
In this study, simvastatin was administered to mice through the nasal route. In animals, intraperitoneal injection is predominantly used in animal testing for the administration of systemic drugs. However, nasal drug administration has been used as an alternative route for the systemic availability of drugs. This method is used to administer drugs that act on the lungs, such as anti-asthmatic drugs, because drugs administered by inhalation can pass through the trachea and into the lungs. Furthermore, the nasal mucosa presents an ideal site for bioadhesive drug delivery systems, and drugs administered via this route generally work quickly [17]. Recently, Xu et al. [18] reported that simvastatin administered by inhalation and intratracheal injection had a more potent effect than that of intraperitoneal injection and gavage. Their results also showed that inhalation delivery routes led to a higher drug concentration in local lung tissue and a lower drug concentration in the plasma than that obtained by gavage. Thus, they suggested that simvastatin is a potential anti-inflammatory drug for airway inflammatory diseases with properties suitable for delivery by inhalation, which will probably reduce the side-effects and increase clinical efficacy, as we expected. However, we used poly I:C to mimic viral pneumonia in this study. It should be noted that our study focused on the anti-inflammatory mechanisms of simvastatin in a poly I:C-induced pneumonia model, which limits potential conclusions regarding the role of simvastatin in the respiratory virus-infected pneumonia model.
In conclusion, simvastatin and neutralising RANTES antibody dramatically inhibited poly I:C-induced neutrophil recruitment in the lungs. In addition, we found that simvastatin inhibits STAT3-mediated RANTES secretion, resulting in the inhibition of neutrophilia in poly I:C-induced pneumonia. It is important to understand the pathogenesis of viral pneumonia and develop a specific and useful anti-viral or anti-inflammatory drug against individual inflammatory conditions. Thus, based on the data in the present study, we suggest that simvastatin should be considered a new drug candidate for viral pneumonia, particularly in neutrophilic inflammation by viral component, although the details of the mechanism for inhibiting airway inflammation by simvastatin would be fully elucidated.
Footnotes
For editorial comments see page 1010.
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MESF) (grant numbers 2010–0027827, 2011–0010571 and 2011–0030739).
Statement of Interest
None declared.
- Received March 24, 2012.
- Accepted July 7, 2012.
- ©ERS 2013
REFERENCES










