





Abstract
Despite evidence that transmission is driving an extensively drug-resistant TB (XDR-TB) epidemic, our understanding of where and between whom transmission occurs is limited. We sought to determine whether there was genomic evidence of transmission between individuals without an epidemiologic connection.
We conducted a prospective study of XDR-TB patients in KwaZulu-Natal, South Africa, during the 2011–2014 period. We collected sociodemographic and clinical data, and identified epidemiologic links based on person-to-person or hospital-based connections. We performed whole-genome sequencing (WGS) on the Mycobacterium tuberculosis isolates and determined pairwise single nucleotide polymorphism (SNP) differences.
Among 404 participants, 123 (30%) had person-to-person or hospital-based links, leaving 281 (70%) epidemiologically unlinked. The median SNP difference between participants with person-to-person and hospital-based links was 10 (interquartile range (IQR) 8–24) and 16 (IQR 10–23), respectively. The median SNP difference between unlinked participants and their closest genomic link was 5 (IQR 3–9) and half of unlinked participants were within 7 SNPs of at least five participants.
The majority of epidemiologically-unlinked XDR-TB patients had low pairwise SNP differences with at least one other participant, consistent with transmission. These data suggest that much of transmission may result from casual contact in community settings between individuals not known to one another.
Abstract
Much of XDR-TB transmission may arise from casual contact between individuals not known to one another http://ow.ly/4Wqt30lnHtp
Introduction
Drug-resistant tuberculosis (TB) is increasing in many countries and threatens to reverse recent gains in TB control [1, 2]. Treatment for drug-resistant TB involves complex, toxic and costly regimens, and is associated with high mortality and poor outcomes [2]. In light of these challenges, prevention of drug-resistant disease is critical in order to reduce morbidity and mortality. Despite increasing evidence that transmission is driving the spread of drug-resistant TB in many parts of the world [3–9], our understanding of where and between whom transmission is occurring remains limited. These gaps in our knowledge preclude our ability to design effective interventions to halt transmission and prevent new cases of drug-resistant TB.
Historically, contact investigations have been used to characterise TB outbreaks and chains of transmission. Following the advent of molecular genotyping, Mycobacterium tuberculosis strain-level data have supplemented and enhanced contact investigations, such that cases with similar strains are presumed to be part of a transmission network [10]. However, numerous studies of TB transmission have found that epidemiologic and genotypic data do not always align. For example, a connection to a close contact could be identified for only 9–30% of genotypically-linked individuals across a range of settings [11–14], suggesting that transmission may arise from casual contact between individuals not known to one another. Similarly, household contact studies in high-incidence settings have found that up to half of secondary TB cases are not genotypically linked to their presumed index case [11, 15]. These findings likely reflect the overwhelming burden of disease and multitude of transmission opportunities in areas where TB is prevalent. This discordance between epidemiologic and genotypic data among close contacts also suggests that additional tools, beyond contact investigations and conventional genotyping, may be required to identify the majority of transmission events, particularly in high-burden settings.
Whole-genome sequencing (WGS) enables a more precise delineation of genetic differences between TB strains by examining greater than 90% of the M. tuberculosis genome, compared to less than 1% with traditional genotyping. WGS identifies the sequential accumulation of single nucleotide polymorphism (SNP) differences and facilitates the construction of transmission chains, rather than simply clusters of related cases as identified through genotyping. Furthermore, individuals with few SNP differences between their M. tuberculosis strains may represent a transmission link [16, 17].
We recently demonstrated that 69–92% of extensively drug-resistant TB (XDR-TB) in KwaZulu-Natal province, South Africa can be attributed to person-to-person transmission of drug-resistant strains, rather than acquisition of resistance in the setting of prior TB treatment [7]. We found epidemiologic links through either close contact or hospitalisations for 30% of XDR-TB cases, while the epidemiologic source was not identified for the remaining 70% of cases. These findings are similar to estimates from previous population-based transmission studies, which have inferred community-based transmission from the absence of genotypic or genomic transmission links between close contacts [11–15]. However, prior studies have not closely examined genomic transmission links for individuals without an epidemiologic link. In this study, we utilise WGS to identify potential transmission links between cases without close contact or overlapping hospitalisation. By integrating epidemiologic and genomic data, we demonstrate how WGS can improve our ability to identify XDR-TB transmission events occurring as a result of casual contact between individuals not known to one another. A better understanding of community-based transmission will inform targeted efforts to interrupt transmission and accelerate ongoing efforts to reduce the global burden of TB.
Methods
Study setting and population
This study was conducted in KwaZulu-Natal province, which has a population of 10.3 million persons and the highest rates of TB (1076 cases per 100 000 population) and HIV (16.9% prevalence) in South Africa [18, 19]. Patients with culture-confirmed XDR-TB residing in KwaZulu-Natal were recruited into the Transmission of HIV-Associated XDR Tuberculosis (TRAX) study from 2011 to 2014 [7]. The primary objective of the TRAX study was to determine the proportion of patients who had acquired XDR-TB (i.e. secondary to inadequate treatment) versus transmitted XDR-TB.
XDR-TB cases were identified through the single reference laboratory conducting drug-susceptibility testing for all public healthcare facilities in KwaZulu-Natal. Given the large number of XDR-TB cases diagnosed annually in the province, a convenience sample of all diagnosed patients were screened for enrolment. Using the reference laboratory database, age, sex and district were compared between enroled and unenroled participants to determine the representativeness of the study sample. Written informed consent was obtained from all participants, or from the next of kin of deceased or severely ill participants. Interviews were conducted to collect participant sociodemographics, TB and HIV history, and location and duration of all hospitalisations in the preceding 5 years. Social network interviews elicited information about close contacts from home, work and other locations where participants spent at least 2 h·week−1 during the preceding 5 years. Full details of the clinical, social network and laboratory methods have been described previously [7].
Laboratory methods
The diagnostic XDR-TB isolate was obtained for all participants and recultured on Löwenstein–Jensen slants. Sequencing material was obtained by performing population sweeps of culture plates. Genomic DNA extraction and insertion sequence IS6110-based restriction fragment length polymorphism (RFLP) genotyping were performed according to standard methods [20]. Isolates underwent paired-end WGS and sequencing libraries were prepared using Nextera DNA kits (Illumina, San Diego, CA, USA). Raw paired-end sequencing reads were generated on the Illumina-MiSeq platform (Illumina, San Diego, CA, USA) and aligned to the H37Rv reference genome (NC_000962.2) using the Burrows–Wheeler aligner [21]. All isolates had reads covering >99% of the reference genome and the lowest mean coverage depth for any isolate was 15X. SNPs were detected using standard pairwise resequencing techniques (Samtools version 0.1.19) against the reference and filtered for quality, read consensus (>75% for the alternate allele) and proximity to indels (less than 50 base-pairs from any indel). SNPs at or within 50 base-pairs of hypervariable Pro-Pro-Glu (PPE)/Pro-Glu (PE) gene families, repeat regions and mobile elements were also excluded [22].
Social network analysis
We analysed social network interview data to identify epidemiologic links between participants. Person-to-person links were defined as two participants who named each other or named a common intermediary. We identified hospital-based epidemiologic links if there were overlapping dates of hospital admission when at least one participant was in a “vulnerable period,” defined as at least 1 month prior to the collection of their XDR-TB diagnostic specimen. Some participants had multiple person-to-person and/or hospital-based links. Participants without a person-to-person or hospital-based link were considered “unlinked.” Sociodemographic, clinical and social network data for linked and unlinked participants were compared using a Chi-squared or Kruskal–Wallis test.
Whole-genome sequence analysis
WGS data for linked and unlinked participants was compared and, as we were interested in the ability of WGS to provide further discrimination beyond genotyping, we focused on SNP differences between participants with a matching RFLP pattern (defined as within one band). Thus, for linked participants, we identified those who had a matching RFLP pattern with their epidemiologic link and then determined pairwise SNP differences between those individuals. If a participant had multiple epidemiologic links with a matching RFLP pattern, we selected the link with the fewest pairwise SNP differences (i.e. the “closest”) in order to focus on the link most likely to reflect transmission.
For unlinked participants, we identified their closest genomic link within the study cohort using pairwise SNP differences. With this approach, unlinked participants had the opportunity to be connected to all other study participants, which would increase their probability of having a genomic link with a low SNP difference by chance alone. In order to account for this possibility, we conducted a sensitivity analysis to evaluate whether unlinked participants had multiple genomic connections at SNP thresholds consistent with transmission, which would support the likelihood of transmission with at least one other study participant with whom they did not have an epidemiologic link. For each unlinked participant we determined the number of other participants within five, seven, or 10 SNPs.
Ethical considerations
The study was approved by the Institutional Review Boards of Emory University, Albert Einstein College of Medicine and the University of KwaZulu-Natal, and by the US Centers for Disease Control and Prevention.
Results
Study population
Between May 2011 and August 2014, 1027 patients were diagnosed with culture-confirmed XDR-TB in KwaZulu-Natal (figure 1). Study staff approached and screened 521 culture-confirmed XDR-TB patients and 404 (78%) were eligible and consented to study enrolment. Participants were enroled from each of KwaZulu-Natal's 11 districts and were representative, by age, sex and geography, of all XDR-TB cases diagnosed province-wide during the study period (p=0.52, 0.76 and 0.70, respectively). Of the participants, 234 (58%) were female and 311 (77%) were infected with HIV. The median age was 34 years (interquartile range (IQR) 28–43) (table 1).

Flow diagram for participant enrolment. XDR-TB: extensively drug-resistant TB; TRAX: the Transmission of HIV-Associated XDR Tuberculosis study; KZN: KwaZulu-Natal.
Sociodemographic and clinical characteristics of participants by presence of epidemiologic links
Social interactions and mobility
We explored participants' social interactions and mobility in order to gain insight into their opportunities for exposure to and transmission of TB. Participants named a total of 2901 close contacts from their homes, workplaces and other community locations, with a median of seven contacts named per participant (IQR 4–10) (table 2). Work outside the home was reported by 123 participants (30%) and 129 participants (32%) reported spending >2 h·week−1 in a community congregate location (e.g. churches, bars and hair salons). Forty-six participants (12%) reported using public transport for more than 1 h·day−1 over the year prior to enrolment. In the 5 years prior to their XDR-TB diagnosis, 298 participants (74%) reported at least one hospitalisation, with 86 (29%) reporting two or more hospitalisations. These hospitalisations occurred at 53 different hospitals, with a median admission of 3 months (IQR 2–5).
Social interactions and mobility of participants by presence of epidemiologic links
Epidemiologic links
There were 59 participants (15%) with a person-to-person link to at least one other study participant. The majority of links (84%) were to household members, although links also included coworkers (7%) and other individuals in the community (9%). Among participants hospitalised during the vulnerable period prior to their XDR-TB diagnosis, 72 (18%) overlapped with another study participant and had a hospital-based link. Participants overlapped with a median of three other participants (IQR 1–18). In total, epidemiologic links were identified for 123 participants (30%), of whom eight (2%) had both a person-to-person and a hospital-based link.
The remaining 281 participants (70%) were epidemiologically “unlinked.” These unlinked participants were not significantly different from linked participants with regards to their sociodemographic and clinical characteristics, with the exception of being slightly older and less likely to report a cough (table 1). Unlinked participants also did not differ in the number of close contacts they named, whether they lived in an urban or rural area, or in the number of their hospitalisations (table 2).
Genomic links
IS6110 RFLP genotyping was completed on the M. tuberculosis isolates of 386 participants (96%) and WGS was successful in 342 isolates (85%) (see supplementary table S1 for participant characteristics by SNPs and supplementary table S2 for RFLP clusters). These included 41 of 59 participants (69%) with a person-to-person link, 58 of 72 participants (81%) with a hospital-based link and 243 of 281 unlinked participants (86%) (figure 2).

Flow diagram for availability of whole-genome sequencing (WGS) data and median single nucleotide polymorphism (SNP) differences for participants with and without epidemiologic links. RFLP: restriction fragment length polymorphism; IQR: interquartile range.
Among participants with person-to-person links, 29 of 41 (71%) had a matching RFLP pattern with their epidemiologic link and their median pairwise SNP difference was 10 (IQR 8–24) (figure 2). Among participants with hospital-based epidemiologic links, 37 (64%) had a matching RFLP pattern and the median pairwise SNP difference to their closest hospital-based link was 16 (IQR 10–23). Among epidemiologically-unlinked participants, the median pairwise difference to their closest pair was 5 SNPs (IQR 3–9); thus, half of unlinked participants were within 5 SNPs of at least one other study participant. (See supplementary table S3 for SNP differences according to history of MDR-TB.)
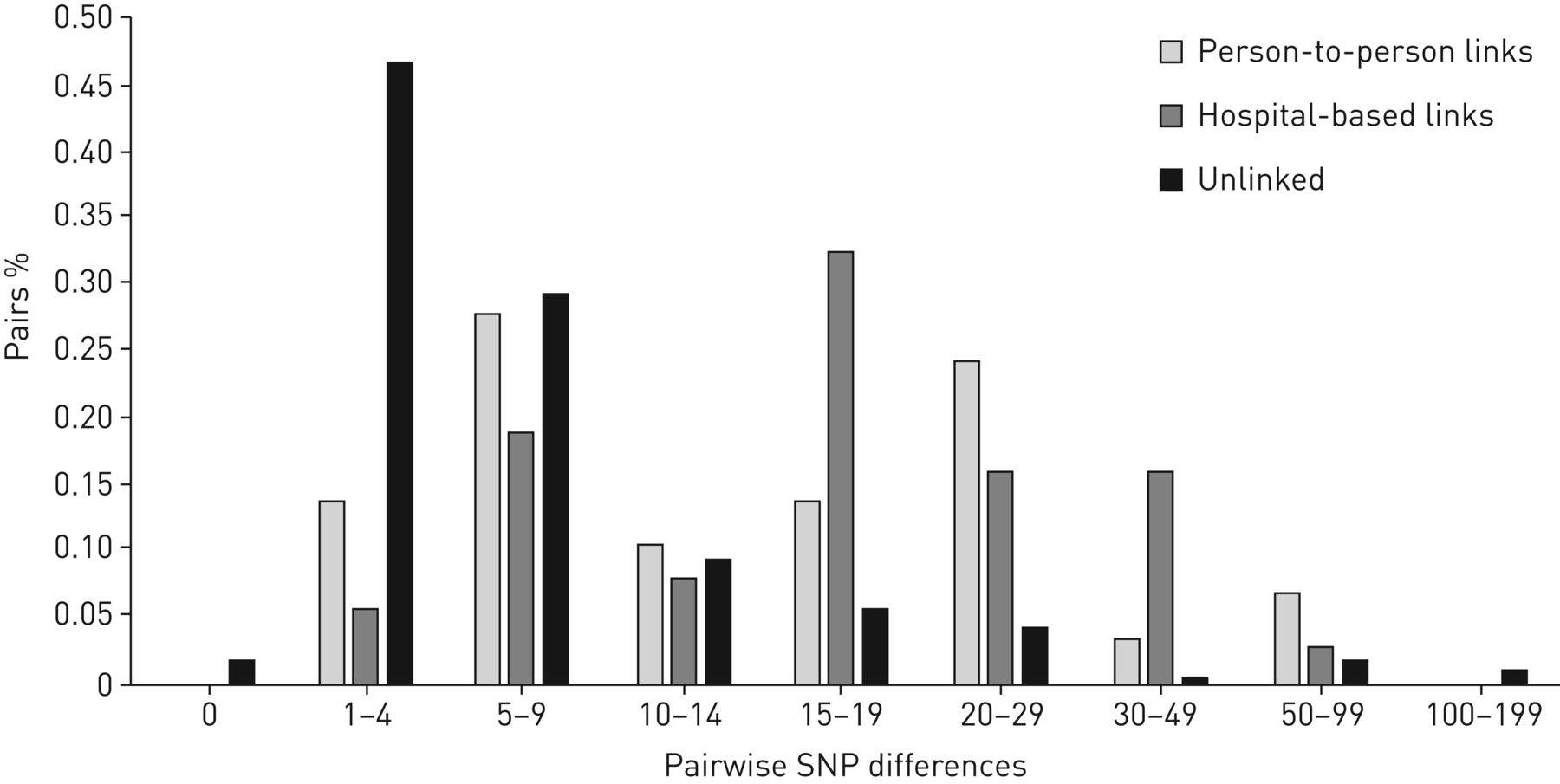
The distribution of SNP differences provided further support for potential transmission events among unlinked participants (figure 3). SNP differences for unlinked participants peaked at 1–4 SNPs (47%), with an additional 29% of participants with 5–9 SNP differences. The M. tuberculosis strain for five unlinked participants was genomically identical to another participant (i.e. 0 SNP differences) and 18 unlinked participants had an M. tuberculosis strain that was only 1 SNP different from another participant. In contrast, SNP differences for participants with person-to-person and hospital-based links had a bimodal distribution, with one peak at 5–9 SNPs and the other at >15 SNPs. This bimodal distribution precluded the identification of a distinct SNP threshold for transmission, as has been reported by other studies [11, 23]. Nonetheless, this distribution supports the likelihood that the majority of unlinked participants had a transmission link within the study sample, since 78% of them were within 10 SNPs of at least one other participant.
{kind=link}
{kind=link}
{kind=link}
Distribution of pairwise single nucleotide polymorphism (SNP) differences for participants with a matching restriction fragment length polymorphism (RFLP) pattern with their name-based epidemiologic link (n=29), a hospital-based epidemiologic link (n=37) and without an epidemiologic link (i.e. unlinked participants, n=240).
To further examine potential transmission events among unlinked participants, we examined the number of genomic connections each unlinked participant had below SNP thresholds previously put forth as suggestive of transmission [23–26]. A total of 192 unlinked participants (79%) were connected to at least one other participant by 10 or fewer SNPs and the median number of connections at this threshold was with 29 participants (IQR 1–80) (table 3). With a threshold of ≤7 SNPs, 173 unlinked participants (71%) were linked to at least one other participant and the median number of connections at this threshold was with five other participants (IQR 0–24). Finally, at a threshold of ≤5 SNPs, 143 unlinked participants (59%) were genomically linked with at least one other participant and the median number of connections was with one other participant (IQR 0–9). Thus, nearly 60% of participants without an epidemiologic link had a genomic link consistent with transmission at the relatively stringent threshold of 5 SNPs and many participants had multiple links at this threshold.
Number of epidemiologically unlinked participants (n=243) with genomic links to other study participants at thresholds of five, seven and 10 single nucleotide polymorphisms (SNPs)
Discussion
South Africa is facing an epidemic of multidrug-resistant TB (MDR-TB) and XDR-TB driven by transmission of drug-resistant strains. The predominant role of transmission in drug-resistant TB epidemics has now been demonstrated in a number of settings and modelling data suggest that transmission will fuel global increases in MDR-TB and XDR-TB over the coming decades [3–6, 27]. Unfortunately, our ability to design effective public health interventions to halt transmission is hindered by our limited understanding of it. In this study, we integrated epidemiologic and genomic data in the largest cohort of XDR-TB patients to date and identified genomic links suggestive of transmission for the majority of epidemiologically-unlinked participants.
Study participants had many opportunities for XDR-TB transmission, with multiple social contacts, frequenting of community congregate locations, hospitalisations and use of public transport. In focusing on epidemiologically-unlinked participants, we were surprised to find that the majority of them had SNP differences similar to and even lower than those between epidemiologically-linked participants. In fact, we found that many unlinked participants were connected to several study participants by only a few SNPs. These findings suggest that many of these participants have transmission links that would not be identified by traditional contact investigations or hospital infection control programs but, rather, arise from casual, community-based contact. These links provide empiric evidence for transmission between casual contacts, supporting the need for future studies to characterise and quantify TB transmission in community locations.
Our findings extend those of several previous studies that have hypothesised about the role of community transmission after finding that close contacts account for a minority of transmission in high-burden settings [11, 15, 28–30]. In Malawi, the use of population-based WGS revealed that only 9.4% of transmission occurred between close contacts [11]. Studies from China and a small study from South Africa also found that a high proportion of genomically-linked cases did not have epidemiologic links [23, 30, 31]. Our study, however, provides genomic evidence for transmission between epidemiologically-unlinked individuals, with multiple genomic links for the majority of these patients strengthening the likelihood of casual contact as a driver of transmission.
Nearly one-third of epidemiologically-linked participants in our study had differing RFLP patterns from one another, which is consistent with previous studies where 39–62% of household contacts had different strains [11, 12, 15]. Furthermore, SNP differences among epidemiologically-linked participants demonstrated a bimodal pattern, with half of pairs having a SNP difference below 10–12 SNPs and the other half having SNP differences in the 15–50 SNP range, even when they shared an RFLP pattern. These findings, similar to those reported in other settings [11, 15], suggest that only a small proportion of secondary cases were attributable to close contact with a known index case. The remaining secondary cases were likely infected with XDR-TB from someone other than a close contact. Thus, in a high burden setting such as South Africa, the presence of an epidemiologic link may be more representative of shared risk factors for exposure than a true transmission link.
WGS has been used in various settings to identify a SNP threshold indicative of transmission [16, 17], with low SNP differences between individuals with an epidemiologic link considered a “gold standard” for transmission in several studies [11, 23]. However, in our study, many epidemiologically-linked participants had SNP differences above previously proposed thresholds (e.g. 0–12 SNPs) [17] and neither epidemiologically-linked nor epidemiologically-unlinked participants had a SNP distribution with a clear transition point below which transmission could be deemed probable. The challenge of defining SNP thresholds is further highlighted by a recent study of a large TB outbreak in London, where nearly 60% of strains over a 14-year period differed by zero or one SNP and the maximum number of SNPs between 344 patients was five [26]. Further research is needed to elucidate how M. tuberculosis mutation rates vary according to pathogen, host and epidemiologic factors.
There are limitations to the interpretation of WGS data, many of which are not specific to this study but represent broader challenges for genomic epidemiology. For example, although mutation rates have been characterised in laboratory settings, it remains difficult to estimate mutation rates from clinical and epidemiologic data given the variable latency period of TB and inherent uncertainty about when an individual may have been infected, particularly in a high-burden setting. Similarly, there is a growing literature demonstrating within-host variability of M. tuberculosis strains, both at a single time point and over time [17, 32, 33]. The clinical significance of this variability, how it is affected by host factors such as HIV co-infection and any potential impact on transmission, remain unclear at present. The impact of clonal M. tuberculosis strains, such as the LAM4/KZN strain in KwaZulu-Natal [34], on transmission dynamics is also not clear. Nevertheless, the diversity of SNP differences in this cohort indicates that WGS has adequate specificity to differentiate potential transmission events, even in the presence of an endemic strain.
Incomplete capture limits our ability to identify all transmission events and, while our study was not designed to capture all XDR-TB cases during the study period, it is possible that greater sampling would have increased the number of epidemiologic links and impacted our estimates of SNP differences between participants. However, studies with more complete sampling worldwide (e.g. the United States, Spain, The Netherlands and Malawi) have still found a high proportion of cases without epidemiologic links [11, 12, 14, 35]. Moreover, we found that the majority of unlinked participants had multiple genomic links, even at stringent SNP thresholds. Thus, our findings likely represent a minimum estimate of the proportion of transmission attributable to casual contact.
The ongoing transmission of drug-resistant TB poses a grave threat to global TB control. Our ability to halt this growing epidemic will hinge upon the design of targeted interventions to interrupt transmission. We found many opportunities for casual contact and transmission among individuals with XDR-TB. The majority of these individuals were epidemiologically unlinked yet had genomic evidence of transmission with other study participants, highlighting the potentially substantial contribution of casual contact to the XDR-TB epidemic in KwaZulu-Natal. While contact investigations and infection control programs to prevent nosocomial transmission have proven benefit as a fundamental pillar of TB control activities, further investigation of transmission through casual contact in community-based settings must be undertaken to determine how and where to intervene and augment our existing approaches for TB control.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00246-2018_Supplement
Acknowledgements
We are grateful to the study team at the University of KwaZulu-Natal for their tireless efforts in data collection, record abstraction, participant recruitment and interviews. We thank the participants and their families who consented to participate in this study.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: None declared.
Support statement: This study was primarily funded by grant R01AI089349 (PI N.R. Gandhi) from the US National Institute of Allergy and Infectious Diseases (NIAID)/National Institutes of Health (NIH). It was also supported in part by NIH/NIAID grants R01AI087465 (PI N.R. Gandhi), K23AI083088 (PI J.C.M. Brust), K23AI134182 (PI S.C. Auld) and K24AI114444 (PI N.R. Gandhi), by Emory CFAR P30AI050409 (PI J. Curran), by Einstein CFAR P30AI051519 (PI H. Goldstein), by Einstein/Montefiore ICTR UL1 TR001073 (PI H. Shamoon) and by NIH/National Heart, Lung, and Blood Institute (NHLBI) grant T32 HL116271 (PI D. Guidot). The findings and conclusions in this manuscript are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention or the US Department of Health and Human Services. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received February 6, 2018.
- Accepted August 8, 2018.
- Copyright ©ERS 2018
References










