
Mammalian DNA Polymerase Kappa Activity and Specificity


Department of Chemistry & Chemical Biology, Northeastern University, Boston, MA 02115, USA
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(15), 2805; https://doi.org/10.3390/molecules24152805
Submission received: 8 July 2019
/
Revised: 27 July 2019
/
Accepted: 30 July 2019
/
Published: 1 August 2019
(This article belongs to the Special Issue Structural and Functional Aspects of DNA Polymerases)
Abstract
:DNA polymerase (pol) kappa is a Y-family translesion DNA polymerase conserved throughout all domains of life. Pol kappa is special6 ized for the ability to copy DNA containing minor groove DNA adducts, especially N2-dG adducts, as well as to extend primer termini containing DNA damage or mismatched base pairs. Pol kappa generally cannot copy DNA containing major groove modifications or UV-induced photoproducts. Pol kappa can also copy structured or non-B-form DNA, such as microsatellite DNA, common fragile sites, and DNA containing G quadruplexes. Thus, pol kappa has roles both in maintaining and compromising genomic integrity. The expression of pol kappa is altered in several different cancer types, which can lead to genome instability. In addition, many cancer-associated single-nucleotide polymorphisms have been reported in the POLK gene, some of which are associated with poor survival and altered chemotherapy response. Because of this, identifying inhibitors of pol kappa is an active area of research. This review will address these activities of pol kappa, with a focus on lesion bypass and cellular mutagenesis.
1. Introduction
DNA is under constant threat of damage from both external sources, such as UV light and environmental pollution, and internal sources, such as reactive oxygen species. Cells have several responses to process the damage, such as repairing or bypassing the damage. Bypassing damage by copying damaged DNA is the mechanism of Y-family DNA polymerases (pols) in a process known as translesion synthesis (TLS) [1]. Y-family DNA polymerases insert nucleotides opposite damaged bases, and then the same or a different Y-family polymerase extends the new primer terminus past the damage in the extension step of TLS (Figure 1) [1]. Y-family DNA polymerases have larger, more accommodating active sites than replicative DNA polymerases [2,3], though they are less processive than replicative polymerases; for example, human pol kappa (κ) incorporates ~20–30 nucleotides per binding event [4]. Y-family DNA polymerases are found in all domains of life and are specialized to bypass particular types of damage. Mammals have four Y-family DNA polymerases: pol η, pol ι, pol κ, and Rev1 [4]. Pol κ differs from the other four mammalian Y-family pols in that its orthologs exist in bacteria and archaea [5,6,7,8]. Pol κ, like its Escherichia coli counterpart DinB, processes DNA adducts mainly at the N2 position of guanine in an error-free manner [6,9,10,11,12,13,14,15,16,17]. Mammalian DNA polymerase κ will be the subject of this review.
2. Structure
Y-family DNA polymerases are composed of the domains that replicative DNA polymerases also typically contain—the thumb, the palm, and the finger domains—but they also contain a fourth, unique domain called the little finger or polymerase-associated domain (PAD) that makes contacts with the major groove of DNA (Figure 2) [2,18,19,20]. DNA pol κ also has a fifth, unique domain at the N-terminus called the N-clasp [21]. The N-clasp has been observed to interact with DNA and to stabilize the ternary complex polymerase bound with DNA and deoxyribonucleotide triphosphate (dNTP) through contacts with the catalytic core, the little finger, and the DNA duplex region [22]. Although human DNA pol κ contains 870 amino acid residues, it was determined through primer extension assays using protein truncations that residues 19–526 are sufficient to maintain wild type-like polymerization activity [21]. It also has been observed that truncated constructs of the protein are more stable than the full-length protein [6] and, thus, are more commonly used in experiments. Removal of the N-clasp significantly reduces activity and, because of its interactions with the DNA, has implications for lesion bypass. Until recently, the crystal structures of pol κ lacked sufficient resolution to identify interactions between the N-clasp residues and other parts of the complex. However, a 2.0-Å resolution crystal structure was reported in 2018 that shows structural characteristics and interactions crucial for our understanding of the role of the N-clasp [23]. This crystal structure shows that the N-terminal residues are well-ordered and interact with the N-clasp, fingers, and thumb domains. The electron density around R18 and K25 is weaker than other residues, which the authors argue demonstrates high flexibility [23]. The active site contains many ordered water molecules engaged in an extensive network of hydrogen bonds, especially with the highly conserved residues R18, R149, and K25. K25 was identified as having a key role in activity through primer extension assays and kinetics assays. Pol κ K25 is at an analogous position and proposed to function similarly to a conserved lysine residue in the O-helix of pol I that makes contacts with phosphates on the incoming nucleotide [23]; pol I K758 is involved in dNTP binding through a conformational change in the fingers domain. However, while Y-family pols do not undergo a similar conformational change in the fingers domain as A-family DNA pols [24,25,26,27,28], this lysine residue K25 still acts as a proton donor to facilitate the formation of pyrophosphate and the reaction to form the phosphodiester bond [23].
3. Regulation
Mouse pol κ expression is regulated by p53 and UV or doxorubicin DNA damage, whereas human pol κ is not [29]. The promoter region of human pol κ harbors binding sites for stimulating protein-1 (SP1) and cyclic AMP-responsive element binding protein (CREB) that positively regulate pol κ expression [30]. Human pol κ expression is increased by exposure to benzo[a]-pyrene diol epoxide (BPDE) [31], and human, rat, and mouse pol κ expression is dependent on the aryl hydrocarbon receptor [32,33]. In addition, human pol κ is upregulated in lung cancers and gliomas, and in some cases this is correlated with genomic instability, while pol κ is downregulated in colorectal cancer [30,34,35,36,37].
In addition to two proliferating cell nuclear antigen (PCNA)-interacting peptides (PIP-boxes) and a Rev1-interacting region [38], pol κ contains two ubiquitin-binding zinc finger (UBZ) domains (Figure 2). The UBZ domains can bind monoubiquitylated PCNA, but upon binding ubiquitin, they undergo a conformational change to inhibit the interaction of Y-family polymerases with Ub-PCNA or other ubiquitylated proteins. This inhibition prevents the polymerase from targeting damaged DNA, so it can no longer participate in TLS [2]. Cells harboring a pol κ variant with a stronger p21-derived PIP box obviated the UBZ domain for pol κ-dependent genome instability [39]. Studies of mouse pol κ have revealed that the UBZ domains are required for pol κ to form nuclear foci after UV exposure [40], presumably reflecting their importance for recruitment of pol κ to sites of DNA replication.
4. Fidelity and Selectivity
Because Y-family pols lack an intrinsic proofreading domain and can incorporate nucleotides opposite damaged DNA bases that block high-fidelity pols, the Y-family pols are often associated with high error rates leading to increased mutagenesis [2]. However, pol κ is the most faithful of the Y-family pols with an error rate 10−3 to 10−4 on undamaged templates [4,41,42]. Studies of members of other DNA polymerase families crystallized in several stages of the catalytic cycle have revealed an important conformational change that serves as a fidelity checkpoint in which the fingers domain transitions from open to closed upon binding the dNTP [24]. The crystal structures of pol κ generally show a large conformational change upon binding DNA, but comparison of crystal structures of binary versus ternary structures are relatively similar regardless of dNTP identity [21,22]. However, kinetics, thermal stability assays, and hydrogen–deuterium exchange-mass spectrometry (HDXMS) experiments indicate that pol κ adopts a specific, active conformation only when the correct or preferred substrates are bound, i.e., undamaged DNA or N2-furfuryl-dG (minor groove damage)-containing templates and the correct incoming dNTP [43,44]. The palm domain in DNA polymerases is associated with catalysis [2]; however, regions in the palm, thumb, and little finger domain were observed to participate in this conformational change. These regions are distally located to the active site, and some do not make contact with the incoming dNTP [43]. This work not only demonstrates the importance of more distal regions of the protein and their ability to discriminate between the correct and incorrect incoming nucleotide but also provides evidence for a fidelity checkpoint in pol κ.
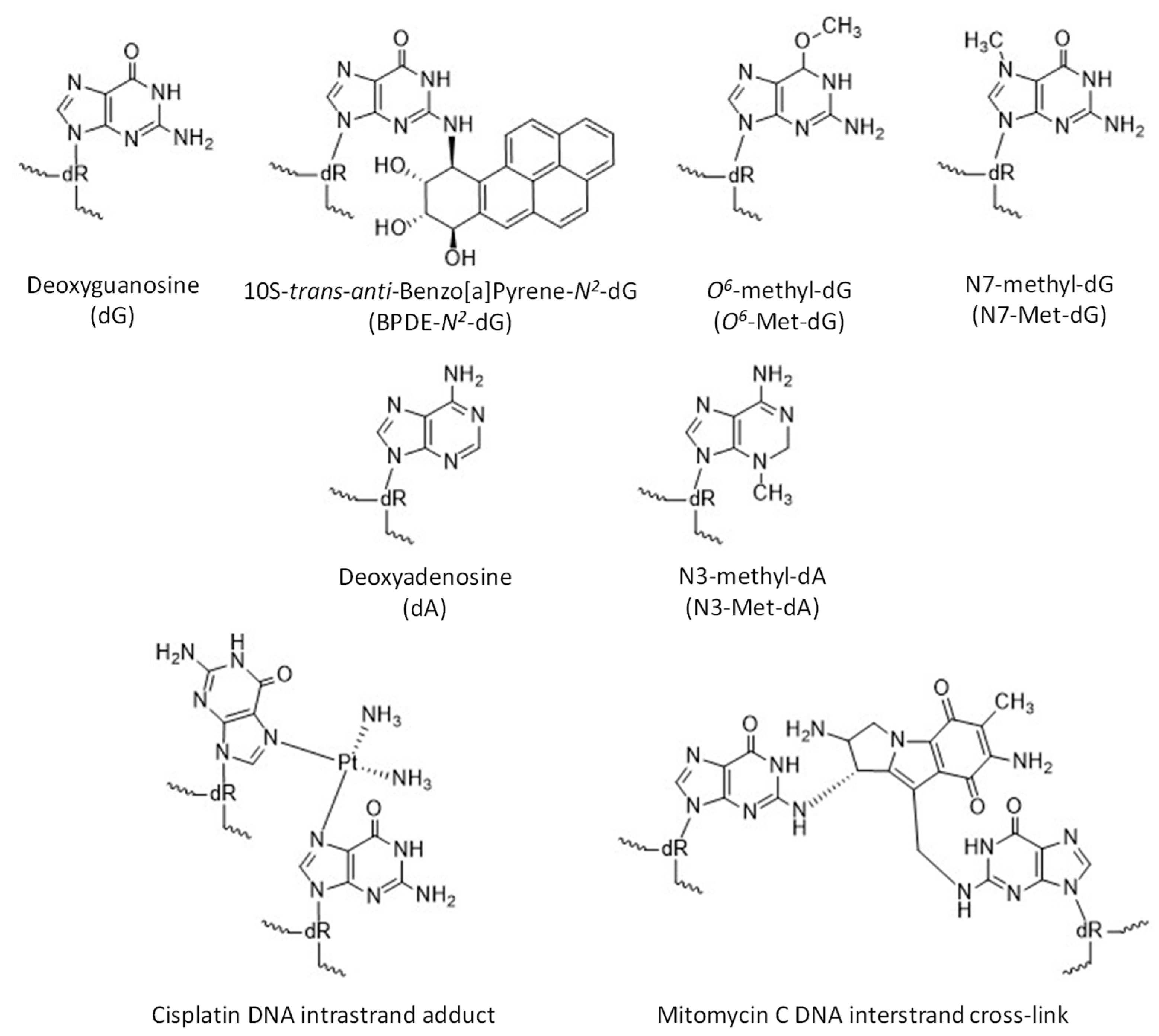
Pol κ is observed to bypass several types of damage at the minor groove of DNA and, generally, is inhibited by major groove DNA adducts (Figure 3). Pol κ has been observed to bypass thymine glycol in an error-free manner [45] and etheno-deoxyadenosine, albeit poorly [46]. Pol κ can also copy DNA containing abasic sites by incorporating dA across from the lesion, though with low efficiency [17,47]. Pol κ is inhibited by the major groove adducts BPDE-N6-dA [14], N6-furfuryl-dA [48], and bulky O6-alkyl-dG modifications [49,50]. Pol κ can replicate DNA templates containing various bulky and non-bulky lesions, such as BPDE-N2-dG [10,14,16,17,51,52,53,54,55], N2-(1-carboxyethyl)-dG [56,57], 8-oxo-dG [17], N2-alkyl-dG [10,58], O2- and some O4-alkyl-dT adducts [59,60,61,62], thymine glycol [45], DNA-peptide crosslinks, and intrastrand adducts and interstrand crosslinks (ICL) formed between purine bases and cisplatin [6,10,17,63,64,65,66,67,68,69,70].
Bypass of 2-acetylaminofluorene-dG can be either error-free or error-prone [16,17,71]. Although pol κ is blocked by the UV-induced T-T cyclobutane pyrimidine dimer (CPD) and the (6-4) photoproduct lesions, the enzyme can extend past a dG inserted opposite the 3′T in the T-T dimer [72]. Pol κ is also inhibited by several major groove purine adducts, such as N6-furfuryl-dA and etheno-dA [48]. The use of isosteric base pairs that lack hydrogen bonding capabilities revealed that pol κ requires hydrogen bonding for efficient primer extension, unlike some replicative DNA polymerases [73]. The 3-aza-dG base analog, missing only the minor groove hydrogen bond acceptor N3, was replicated with reasonable efficiency by pol κ [73].
Examination of the structures of pol κ and comparison to sequences of other Y-family DNA polymerases have provided insights into pol κ preferences for certain lesions. Pol κ was shown to be tolerant to changes in its active site loop comprising residues 127–141 [74]. Notably, pol κ V130I had relaxed discrimination against the major groove adduct N6-furfuryl-dA, which is somewhat analogous to the DinB R35A mutation, indicating the importance of this region for lesion selectivity [74]. Within the same active site loop, pol κ M135 is suggested to be too bulky to accommodate T-T CPD lesions whereas human pol η, which can bypass T-T CPDs, has a glycine in a similar position; this could be a way to select DNA lesions so that the correct polymerase works on its associated adducts [9]. Similarly, analysis of inhibitor binding to pol κ suggests that the M135 and A151 side chains limit the orientation of the nascent base pair in the active site [75].
The ability to discriminate between ribonucleotides (rNTPs) and deoxyribonucleotides (dNTPs) is important to prevent ribonucleotide incorporation into the extending primer strand as this could lead to genomic instability [6,76]. Because rNTP concentrations are as much as 1000-fold higher in the cell than dNTP concentrations [77,78], polymerases must have a way to discriminate against rNTPs. Through the alignment of many Y-family pols, site-directed mutagenesis, and primer extension assays, the residue Y112 was determined to be the steric gate in pol κ as the alanine and valine variants both incorporated rNTPs [6]. However, these variants incorporate rNTPs less efficiently than dNTPs [6]. HDXMS experiments showed that pol κ adopts a substrate-specific conformational change when the correct substrate dNTP is bound versus the incorrect substrate rNTP [76]. The polymerase assumes a similar conformation when an rNTP is bound, but to a lesser extent, which can help to explain the decrease in activity when only rNTPs are present [76].
4.1. Role in TLS Extension Step
During TLS, a Y-family polymerase bypasses the damage and then either the same polymerase extends past the damage or another Y-family polymerase is recruited to extend past the damage so that a replicative polymerase can subsequently continue DNA synthesis [1,2]. Most eukaryotic TLS follows this two-step response, in which pol κ is generally thought to be specialized for the extension step, especially in extending mispaired primer termini [2,72,79,80,81]. While pol κ has a misincorporation frequency of 10−3 to 10−4 on undamaged templates [41], it also extends from mispaired primer termini with a greater proficiency at about 10−1 to 10−2 [72,81]. It has been suggested that mispaired primer termini are preferred over matched primer termini as substrates for pol κ as these would be encountered during the TLS extension step [82]. Pol κ can also generate single-base frame-shifts through template–primer misalignment wherein the wrong nucleotide is incorporated followed by a realignment of the primer on the template to loop out the mispaired base on the template (Figure 1) [47,81]. Taken together, these observations indicate a high tolerance of pol κ for noncanonical primer termini.
4.2. Pol Kappa Specificity and Cellular Functions
Benzo[a]pyrene. One of the most well-characterized bulky adducts, 10S-trans-anti-benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE-N2-dG) (Figure 3), is derived from an environmental mutagen, B[a]P, present in tobacco smoke and combustion products of fossil fuel [65]. Replicative polymerases stall at BPDE-N2-dG, which is predominantly repaired through nucleotide excision repair (NER). However, if not repaired, the lesion leads to G to T transversion mutations in mammalian cells [83]. Pol κ catalyzes the insertion of a nucleotide opposite BPDE-N2-dG as well as the extension past this lesion with the highest efficiency and fidelity of all known DNA polymerases [14,16,51,52,53,54,55,84]. Other TLS polymerases bypass this lesion by misincorporation of dA [85,86,87,88]. The open catalytic core of pol κ easily accommodates the bulky BPDE-N2-dG adduct oriented towards the minor groove, which pairs with dC in a Watson–Crick base pair with an extra H-bond between O2-dC and O7 from the BPDE-N2-dG to allow accurate insertion [23,89,90]. Comparable conformations of ternary complexes with and without adducted DNA templates have revealed unique interactions between the core and little finger domains mediated by the N-clasp domain, resulting in a partial coverage of the DNA duplex minor groove [89]. Moreover, both adducted and normal DNA substrates typically retain standard B-form, contributing to catalysis by pol κ. The 5ʹ orientation of the BPDE ring together with two stable water molecules provide coverage at the side of the minor groove in the active site and thus prevent formation of a wobble base pair and misinsertions during the extension step beyond the BPDE-N2-dG adduct [90]. Consequently, pol κ incorporates the next nucleotide after the lesion with higher fidelity than after a non-modified dG as observed in a primer extension assay [90].
Development of Nalm-6 human pre-B acute lymphoblastic leukemia cell lines (Nalm-6 cell lines), possessing high efficiency for gene targeting, has enabled control of pol κ expression to determine cell survival and the mutation spectrum after exposure to a mutagenic agent as well as to quantify mutation frequencies after replication and repair of a synthetic plasmid carrying a site-specific B[a]P lesion [91,92,93]. The mutation frequency of pol κ-deficient Nalm-6 cells treated with BPDE was increased in the supF gene forward mutation assays with a predominant mutation being that of dA [91]. Mutagenesis induced by BPDE was elevated in catalytically dead (CD) and knockout (KO) pol κ Nalm-6 cells as well as in mouse embryonic stem cells, confirming the catalytic role of pol κ in suppressing mutations by BPDE [6,11,13,16,92,94]. In mice with wild-type (polk+/+) or a catalytically inactive variant (polk−/−) of pol κ, mutagenesis induced by B[a]P was found to be insignificant in the colon, although spontaneous mutation frequency increased significantly in polk−/− mice at old age [95,96,97]. Thus, pol κ appears to catalyze TLS across endogenously generated DNA adducts caused by oxidative damage and alkylating agents in an error-free manner. Hakura et al. showed that B[a]P was not carcinogenic in mice when delivered alone [96]. Mice exposed simultaneously to B[a]P and dextran sulfate sodium (DSS), which causes inflammation in the colon, developed tumors. However, no significant difference was identified in the number of mice with tumors or tumors per mouse between polk+/+ and polk−/− mice. Lipid peroxidation-induced adducts such as 1,N6-etheno-2′-deoxyadenosine, 8-hydroxypropano-2′-deoxyguanosine, and heptanone-etheno-2′-deoxycytidine were detected, but the BPDE-dG adduct was not. Thus, pol κ suppresses mutagenicity triggered by inflammation in the colon and lung of mice.
Phenylalanine 171 (F171) in the palm domain has been recognized as a biologically relevant non-catalytic residue via its interactions with the BPDE-N2-dG [54,89,90,91]. Replacing phenylalanine with an amino acid having a smaller side chain (F171A) resulted in more efficient bypass of BPDE-N2-dG in primer extension assays, suggesting that F171 inhibits pol κ activity [54]. Human cells expressing the F171A variant exhibited a significantly lower mutation frequency compared to cells expressing wild-type pol κ, further supporting the notion that interactions between F171 and the BPDE ring are energetically unfavorable [91]. Crystal structures show the phenyl ring of F171 in an altered conformation, moving away from the active site to accommodate the BPDE ring in the insertion stage complex [89,90].
Crosslinks. Special attention has been dedicated to the platinum-based anticancer drug cisplatin (CP) and the antitumor drug and antibiotic mitomycin C (MMC), of which the reactive species covalently bind DNA via formation of intrastrand adducts involving adjacent dGdG, dAdG, and nonadjacent bases dGdNdG and interstrand crosslinks (ICL) between two guanines in dGdC and dCdG sequences targeted by cisplatin and mitomycin C, respectively (Figure 3) [98,99,100]. ICLs represent the smallest part of the total CP- and MMC-induced modifications in a cell. Nevertheless, this type of DNA lesion is the most carcinogenic because the separation of leading and lagging strands during DNA replication results preferentially in double-strand breaks (DSBs), which can induce apoptosis [101]. Intrastrand adducts are removed mostly through the NER pathway while a complex mechanism using components of NER, homologous recombination (HR), and TLS pathways is required for ICLs repair [102,103,104].
Biochemical experiments have shown that the intrastrand adduct induced by cisplatin (3′-dG-Pt-dG-5′) substantially inhibits pol κ, which otherwise inserts a correct nucleotide easily opposite a native template at the first insertion step at 3′-dG [42,71,105]. However, the subsequent insertion and extension steps at dG-5′ are catalyzed with increased efficiency comparable to normal DNA replication. Pol κ is the most accurate relative to polymerases η and ι and predominantly incorporates correct nucleotides in insertion and extension steps [23]. Furthermore, pol κ can carry out strand displacement synthesis and can fully extend the primer having inserted a dNTP opposite cisplatin-interstrand crosslinked guanine [106]. Pol κ activity was characterized by low fidelity and high extension efficiency of interstrand-crosslink bypass, which depends on the conformation of the DNA duplex and the linker length [106].
Crystallographic studies have revealed a structural basis for human pol κ to bypass cisplatin intrastrand lesions as an efficient and accurate extender [23]. The analysis has suggested that pol κ accommodates lesions without a significant conformational change of its active site. Decreased efficiency could be due to deviation of the DNA damaged template from its protein-free conformation, resulting in a steric clash between the adduct and protein residues. Extensive interactions observed between the DNA template and the N-clasp, fingers, and little finger domains stabilize the ternary complex in a catalytically active conformation and thus orient nascent nucleotides for proper Watson–Crick base pairing.
Involvement of pol κ in the replication of DNA modified by CP was revealed in a study with human cells [107]. By using plasmids carrying a specific single-site lesion and knocking down TLS polymerases, pol κ activity in combination with pol ζ resulted in error-prone bypass past CP adducts. Research focused on the regulation of pol κ function in eukaryotic cells demonstrated the involvement of pol κ in DNA repair pathways induced by CP and MMC treatment [108,109,110]. TLS across ICLs formed on oligodeoxynucleotides with acrolein-derived crosslinks and on plasmids with trimethylene crosslinks demonstrated that human pol κ carried out accurate incorporation opposite the crosslinked guanine and extended the primer beyond the lesion [111]. Cell survival was adversely affected in pol κ-depleted human cells and mice following mitomycin C exposure, thus confirming the biological role of pol κ in tolerating the N2-N2-guanine ICLs in the minor groove [93,97,111]. Takeiri proposed a model mechanism of pol κ recruitment to the intrastrand adducts in vivo, wherein active pol κ suppresses point mutations by error-free DNA synthesis and prevents the formation of DSBs. In the absence of active pol κ, other TLS enzymes are recruited, resulting in higher mutation frequencies observed in mice with inactive pol κ. A similar model has been proposed for the role of pol κ in MMC-induced interstrand crosslinks in human cells based on findings from cytotoxic and genotoxic responses to MMC, mutation spectrum, and chromosome analysis [93].
Pol κ is an efficient DNA polymerase on templates with other lesions exhibiting cytotoxic, mutagenetic, and carcinogenic properties caused by exogenous and endogenous alkylating agents (e.g., S-adenosylmethionine) [67]. Many anticancer drugs (e.g., methyl methanesulfonate, MMS) have been developed as methylating agents because of their ability to trigger cell apoptosis [101,112,113]. The most prevalent and biologically relevant alkylated nucleobases are N3-methyladenine (N3-Met-dA), N7-methylguanine (N7-Met-dG), and O6-methylguanine (O6-Met-dG) (Figure 3) [114]. Base excision repair (BER) is the major pathway in the elimination of N-alkylpurines yielding an apurinic site (AP) as a byproduct, while O6-Met-dG is directly reversed by the action of O6-methylguanine DNA methyltransferase [115,116,117]. Moreover, methylpurine bases are unstable adducts in basic conditions undergoing either spontaneous depurination or imidazole ring opening to generate 5-N-methyl-2,6-diamino-4-hydroxyformamidopyrimidine (Met-FaPy) giving rise to G to T transversion mutations [118,119]. Overexposure to alkylating agents results in clustered abasic DNA lesions, defined as two or more damaged sites within 1.5 helical turns [120,121,122]. The repair mechanism of clustered lesions is a complex cellular process with escalated occurrence of deleterious double-strand breaks.
The initial evidence of pol κ involvement in TLS of the N3-Met-dA lesion comes from in vivo studies due to complications accompanying chemical synthesis of modified nucleotides. Analysis from survival assays on mice, chicken, and yeast cells missing essential repair proteins indicated that pol κ enhanced MMS resistance of Rev1-, Rev3-, and Rad30-deficient cells [123,124]. Additional studies supported a hypothesis that pol κ accomplishes its replication activity of methylated adducts in coordination with the Rev3 and Rev1/pol ζ pathways [125,126]. Primer extension experiments using a DNA template with 3-deaza-3-Met-dA (a stable analog of N3-methyl adduct) demonstrated that pol κ is moderately accurate and more efficient in the insertion of dT opposite undamaged dA than opposite N3-Met-dA [127,128]. Overall translesion synthesis decreased in human cells with pol κ removed by siRNA knockdown to a similar extent measured in cells depleted of both pol κ and pol ι, suggesting that pol κ plays an active role in extension beyond adducted DNA replicated by pol ι [128]. Human pol κ follows the A-rule while replicating a DNA template with an abasic site and requires dT as the next template base for further efficient extension [17,129,130,131].
5. Roles of Pol κ Beyond Translesion Synthesis
In addition to lesion bypass, pol κ contributes to other aspects of DNA metabolism. Human pol κ has roles in microsatellite stability [132,133] and replication of non-B DNA structures, including common fragile sites, G4 structures, and others [134,135,136]. In mouse cells, pol κ contributes to repair synthesis during nucleotide excision repair (NER) of UV-induced damage [137]. In human cells, pol κ also contributes to repair synthesis in NER and is recruited to repair sites by interactions with ubiquitylated PCNA via the pol κ UBZ domain [138].
Human pol κ colocalizes with PCNA in the nucleus, which increases with replication stress induced by treatment with hydroxyurea, UV, cis-Pt [139], or B[a]P [140]. The E3 ubiquitin ligase Rad18 is responsible for B[a]P-induced PCNA ubiquitylation and recruitment of pol κ [140]. Analogously, loss of the deubiquitinase USP1, which removes ubiquitin from PCNA, leads to pol κ-dependent genomic instability and reduction of replication fork speed, likely because of inappropriate engagement of pol κ in DNA replication [39]. Furthermore, pol κ is implicated in the Chk1 checkpoint response and recovery of replication from the stress of limiting dNTPs; pol κ interacts with the 9-1-1 checkpoint clamp as part of this response [141]. Another report indicates that pol κ has no effect on Chk1 activation but does play a role in replication fork restart under conditions of limiting dNTPs, dependent on the Fanconi Anemia pathway [142]. Taken together, these studies implicate pol κ in DNA metabolism beyond translesion synthesis.
6. Cancer-Associated Single-Nucleotide Polymorphisms (SNPs) in Human Pol κ
Numerous cancer-associated SNP variants have been identified in the POLK gene, which could impact pol κ function in several different ways. Mutations that decrease the activity of pol κ would render cells more sensitive to DNA damaging agents, including some chemotherapeutics. Mutations that alter the fidelity of pol κ will modulate the genomic (in)stability of tumors. Pol κ SNP variants with improved lesion bypass capability could play a role in resistance to DNA-damaging chemotherapy. The extension activity of pol κ can lead to mutations being fixed in the genome, and thus, increased primer extension activity can increase the mutation load if pol κ extends mismatched primer termini. Characterized pol κ mutations that have been reported in the literature are summarized in Table 1. Notably, the SNP variants associated with early-onset prostate cancer exhibit decreased primer extension activity in biochemical assays [143]. Some of the known SNPs modulate chemotherapy responses and cancer risk [144,145], suggesting the potential utility of drugs targeting pol κ [146].
7. Inhibitors
Pol κ is an attractive target for cancer therapies because it has been correlated with a shorter survival time in patients with glioblastoma as well as poor chemotherapy responses in some cases [92,144,145]. A high-throughput method to characterize the mechanism and inhibition of Y-family polymerases was developed through the use of a real-time fluorescent reporter [151]. This method can be used to monitor the inhibition of the polymerases by a small molecule in the presence of damaged template or damaged nucleotides [151]. The method was used to screen a library of ~16,000 bioactive molecules; the top 60 hits were then validated by primer extension with undamaged DNA [152]. From the top 60 hits, three compounds were studied further due to their specificity toward pol κ and one compound was observed to reduce XP-V cell resistance to UV light, which suggested in-cell inhibition of pol κ [152]. Another of the three compounds, MK886, was further studied through docking experiments and enzyme inhibition assays to determine its mechanism of action [153]. The results of these studies indicated that the mechanism by which the small molecule MK886 inhibits pols ι, η, and κ is different because the molecule can bind to different pockets identified through docking [153]. However, the small molecule results suggest that derivatization can lead to more potent and specific inhibitors, especially of pol κ. Recently an indole-aminoguanidine (IAG) scaffold-based inhibitor was identified to inhibit pol κ [75]. Mass spectrometry analysis of chemical footprinting reactions with an arginine-reactive probe localized binding sites for IAG-derived molecules in the fingers and little finger domains and in the N-clasp. Thus, it was concluded that the inhibitor disrupts interactions among these domains and alters normal N-clasp positioning, causing inhibition of pol κ activity [75]. Because the N-clasp is unique to pol κ and this molecule targets this domain, the molecule is attractive for further study, especially in chemotherapy-resistant cancers [75].
8. Conclusions
DNA pol κ plays a multitude of roles in DNA metabolism. Because pol κ is implicated in both cancer development and chemotherapy resistance, understanding the activity and specificity of pol κ, its SNPs, and the resulting variants can lead to new methods of detection and personalized medicine using inhibitors that target pol κ specifically. In the twenty years since pol κ was first discovered, many distinguishing characteristics have been identified, including its structure and unique domains, its preferred substrates, and disease-related mutations and patient outcomes. However, there are still several outstanding questions that can help guide the pursuit of understanding. Most biochemical work to date has been carried out with the polymerase core of pol κ; do other regions of the protein play roles in lesion specificity or extension activity? More studies with full-length pol κ need to be carried out in order to determine the contributions of less-characterized regions to the activity of pol κ. In addition, the effects on pol κ activity of the interactions of pol κ with its protein partners, including PCNA, Rev1, and others, should be probed in greater depth.
It is suggested that pol κ has a major role as an extender in TLS; what are the relative contributions to insertion or extension, and is this specific to each lesion? The search for specific inhibitors of pol κ and related SNPs is currently being pursued, especially ones that work in the cellular environment; is it possible to target specific SNPs? Addressing these questions will be important in understanding the basic biochemistry of pol κ as well as realizing the potential to improve cancer treatments.
Funding
Research in the Beuning laboratory is supported by the National Institutes of Health (R01GM123239 to P.J.B.) and the National Science Foundation (MCB-1615946 to P.J.B.). V.E.C. was also funded by a Project SEED scholarship from the American Chemical Society.
Acknowledgments
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell. Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.F.; Woodgate, R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boudsocq, F.; Woodgate, R.; Yang, W. Crystal Structure of a Y-Family DNA Polymerase in Action: A Mechanism for Error-Prone and Lesion-Bypass Replication. Cell 2001, 107, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [PubMed]
- Gruz, P.; Pisani, F.M.; Shimizu, M.; Yamada, M.; Hayashi, I.; Morikawa, K.; Nohmi, T. Synthetic activity of Sso DNA polymerase Y1, an archaeal DinB-like DNA polymerase, is stimulated by processivity factors proliferating cell nuclear antigen and replication factor C. J. Biol. Chem. 2001, 276, 47394–47401. [Google Scholar] [CrossRef] [PubMed]
- Niimi, N.; Sassa, A.; Katafuchi, A.; Gruz, P.; Fujimoto, H.; Bonala, R.R.; Johnson, F.; Ohta, T.; Nohmi, T. The steric gate amino acid tyrosine 112 is required for efficient mismatched-primer extension by human DNA polymerase kappa. Biochemistry 2009, 48, 4239–4246. [Google Scholar] [CrossRef]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Wagner, J.; Gruz, P.; Kim, S.R.; Yamada, M.; Matsui, K.; Fuchs, R.P.; Nohmi, T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell 1999, 4, 281–286. [Google Scholar] [CrossRef]
- Chandani, S.; Jacobs, C.; Loechler, E.L. Architecture of Y-family DNA polymerases relevant to translesion DNA synthesis as revealed in structural and molecular modeling studies. J. Nucleic Acids 2010, 2010, 784081. [Google Scholar] [CrossRef]
- Choi, J.Y.; Angel, K.C.; Guengerich, F.P. Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase kappa. J. Biol. Chem. 2006, 281, 21062–21072. [Google Scholar] [CrossRef]
- Huang, X.; Kolbanovskiy, A.; Wu, X.; Zhang, Y.; Wang, Z.; Zhuang, P.; Amin, S.; Geacintov, N.E. Effects of base sequence context on translesion synthesis past a bulky (+)-trans-anti-B[a]P-N2-dG lesion catalyzed by the Y-family polymerase pol kappa. Biochemistry 2003, 42, 2456–2466. [Google Scholar] [CrossRef]
- Jarosz, D.F.; Godoy, V.G.; DeLaney, J.C.; Essigmann, J.M.; Walker, G.C. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 2006, 439, 225–228. [Google Scholar] [CrossRef]
- Ogi, T.; Shinkai, Y.; Tanaka, K.; Ohmori, H. Polkappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. PNAS 2002, 99, 15548–15553. [Google Scholar] [CrossRef]
- Rechkoblit, O.; Zhang, Y.; Guo, D.; Wang, Z.; Amin, S.; Krzeminsky, J.; Louneva, N.; Geacintov, N.E. trans-Lesion synthesis past bulky benzo[a]pyrene diol epoxide N2-dG and N6-dA lesions catalyzed by DNA bypass polymerases. J. Biol. Chem. 2002, 277, 30488–30494. [Google Scholar] [CrossRef]
- Shen, X.; Sayer, J.M.; Kroth, H.; Ponten, I.; O’Donnell, M.; Woodgate, R.; Jerina, D.M.; Goodman, M.F. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J. Biol. Chem. 2002, 277, 5265–5274. [Google Scholar] [CrossRef]
- Suzuki, N.; Ohashi, E.; Kolbanovskiy, A.; Geacintov, N.E.; Grollman, A.P.; Ohmori, H.; Shibutani, S. Translesion synthesis by human DNA polymerase kappa on a DNA template containing a single stereoisomer of dG-(+)- or dG-(-)-anti-N(2)-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene). Biochemistry 2002, 41, 6100–6106. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, M.; Rechkoblit, O.; Taylor, J.S.; Geacintov, N.E.; Wang, Z. Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic. Acids Res. 2000, 28, 4138–4146. [Google Scholar] [CrossRef] [Green Version]
- Boudsocq, F.; Kokoska, R.J.; Plosky, B.S.; Vaisman, A.; Ling, H.; Kunkel, T.A.; Yang, W.; Woodgate, R. Investigating the role of the little finger domain of Y-family DNA polymerases in low fidelity synthesis and translesion replication. J. Biol. Chem. 2004, 279, 32932–32940. [Google Scholar] [CrossRef]
- Pata, J.D. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta 2010, 1804, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.C.; Jackson, M.A.; Pata, J.D. Y-family polymerase conformation is a major determinant of fidelity and translesion specificity. Structure 2013, 21, 20–31. [Google Scholar] [CrossRef]
- Uljon, S.N.; Johnson, R.E.; Edwards, T.A.; Prakash, S.; Prakash, L.; Aggarwal, A.K. Crystal Structure of the Catalytic Core of Human DNA Polymerase Kappa. Structure 2004, 12, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Lone, S.; Townson, S.A.; Uljon, S.N.; Johnson, R.E.; Brahma, A.; Nair, D.T.; Prakash, S.; Prakash, L.; Aggarwal, A.K. Human DNA polymerase kappa encircles DNA: Implications for mismatch extension and lesion bypass. Mol. Cell 2007, 25, 601–614. [Google Scholar] [CrossRef]
- Jha, V.; Ling, H. 2.0 A resolution crystal structure of human polkappa reveals a new catalytic function of N-clasp in DNA replication. Sci Rep. 2018, 8, 15125. [Google Scholar] [CrossRef]
- Joyce, C.M.; Benkovic, S.J. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry 2004, 43, 14317–14324. [Google Scholar] [CrossRef]
- Berezhna, S.Y.; Gill, J.P.; Lamichhane, R.; Millar, D.P. Single-molecule Forster resonance energy transfer reveals an innate fidelity checkpoint in DNA polymerase I. J. Am. Chem. Soc. 2012, 134, 11261–11268. [Google Scholar] [CrossRef]
- Hohlbein, J.; Aigrain, L.; Craggs, T.D.; Bermek, O.; Potapova, O.; Shoolizadeh, P.; Grindley, N.D.; Joyce, C.M.; Kapanidis, A.N. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nat. Commun. 2013, 4, 2131. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, P.J.; Allen, W.J.; Sisamakis, E.; Kalinin, S.; Felekyan, S.; Widengren, J.; Waksman, G.; Seidel, C.A. dNTP-dependent conformational transitions in the fingers subdomain of Klentaq1 DNA polymerase: Insights into the role of the “nucleotide-binding” state. J. Biol. Chem. 2013, 288, 13575–13591. [Google Scholar] [CrossRef]
- Santoso, Y.; Joyce, C.M.; Potapova, O.; Le Reste, L.; Hohlbein, J.; Torella, J.P.; Grindley, N.D.; Kapanidis, A.N. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. PNAS 2010, 107, 715–720. [Google Scholar] [CrossRef]
- Velasco-Miguel, S.; Richardson, J.A.; Gerlach, V.L.; Lai, W.C.; Gao, T.; Russell, L.D.; Hladik, C.L.; White, C.L.; Friedberg, E.C. Constitutive and regulated expression of the mouse Dinb (Polkappa) gene encoding DNA polymerase kappa. DNA Repair (Amst) 2003, 2, 91–106. [Google Scholar] [CrossRef]
- Lemee, F.; Bavoux, C.; Pillaire, M.J.; Bieth, A.; Machado, C.R.; Pena, S.D.; Guimbaud, R.; Selves, J.; Hoffmann, J.S.; Cazaux, C. Characterization of promoter regulatory elements involved in downexpression of the DNA polymerase kappa in colorectal cancer. Oncogene 2007, 26, 3387–3394. [Google Scholar] [CrossRef]
- Zhu, H.; Fan, Y.; Shen, J.; Qi, H.; Shao, J. Characterization of human DNA polymerase kappa promoter in response to benzo[a]pyrene diol epoxide. Environ. Toxicol. Pharmacol. 2012, 33, 205–211. [Google Scholar] [CrossRef]
- Brauze, D.; Rawluszko, A.A. The effect of aryl hydrocarbon receptor ligands on the expression of polymerase (DNA directed) kappa (Polkappa), polymerase RNA II (DNA directed) polypeptide A (PolR2a), CYP1B1 and CYP1A1 genes in rat liver. Environ. Toxicol. Pharmacol. 2012, 34, 819–825. [Google Scholar] [CrossRef]
- Ogi, T.; Mimura, J.; Hikida, M.; Fujimoto, H.; Fujii-Kuriyama, Y.; Ohmori, H. Expression of human and mouse genes encoding polkappa: Testis-specific developmental regulation and AhR-dependent inducible transcription. Genes Cells 2001, 6, 943–953. [Google Scholar] [CrossRef]
- Bavoux, C.; Leopoldino, A.M.; Bergoglio, V.; Jiyang, O.W.; Ogi, T.; Bieth, A.; Judde, J.G.; Pena, S.D.; Poupon, M.F.; Helleday, T.; et al. Up-regulation of the error-prone DNA polymerase {kappa} promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 2005, 65, 325–330. [Google Scholar]
- Wang, J.; Kawamura, K.; Tada, Y.; Ohmori, H.; Kimura, H.; Sakiyama, S.; Tagawa, M. DNA Polymerase k, Implicated in Spontaneous and DNA Damage-induced Mutagenesis, Is Overexpressed in Lung Cancer. Cancer Res. 2001, 61, 5366–5369. [Google Scholar]
- Wang, H.; Wu, W.; Wang, H.W.; Wang, S.; Chen, Y.; Zhang, X.; Yang, J.; Zhao, S.; Ding, H.F.; Lu, D. Analysis of specialized DNA polymerases expression in human gliomas: Association with prognostic significance. Neuro Oncol. 2010, 12, 679–686. [Google Scholar] [CrossRef]
- Wang, Y.; Seimiya, M.; Kawamura, K.; Yu, L.; Ogi, T.; Takenaga, K.; Shishikura, T.; Nakagawara, A.; Sakiyama, S.; Tagawa, M.; et al. Elevated expression of DNA polymerase kappa in human lung cancer is associated with p53 inactivation: Negative regulation of POLK promoter activity by p53. Int. J. Oncol. 2004, 25, 161–165. [Google Scholar]
- Xie, W.; Yang, X.; Xu, M.; Jiang, T. Structural insights into the assembly of human translesion polymerase complexes. Protein Cell 2012, 3, 864–874. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.J.; Colnaghi, L.; Huang, T.T. Dysregulation of DNA polymerase kappa recruitment to replication forks results in genomic instability. EMBO J. 2012, 31, 908–918. [Google Scholar] [CrossRef]
- Guo, C.; Tang, T.S.; Bienko, M.; Dikic, I.; Friedberg, E.C. Requirements for the interaction of mouse Polkappa with ubiquitin and its biological significance. J. Biol. Chem. 2008, 283, 4658–4664. [Google Scholar] [CrossRef]
- Johnson, R.E.; Prakash, S.; Prakash, L. The human DINB1 gene encodes the DNA polymerase Pol theta. PNAS 2000, 97, 3838–3843. [Google Scholar] [CrossRef]
- Ohashi, E.; Bebenek, K.; Matsuda, T.; Feaver, W.J.; Gerlach, V.L.; Friedberg, E.C.; Ohmori, H.; Kunkel, T.A. Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. J. Biol. Chem. 2000, 275, 39678–39684. [Google Scholar] [CrossRef]
- Nevin, P.; Lu, X.; Zhang, K.; Engen, J.R.; Beuning, P.J. Noncognate DNA damage prevents the formation of the active conformation of the Y-family DNA polymerases DinB and DNA polymerase kappa. FEBS J. 2015, 282, 2646–2660. [Google Scholar] [CrossRef]
- Zhao, L.; Pence, M.G.; Eoff, R.L.; Yuan, S.; Fercu, C.A.; Guengerich, F.P. Elucidation of kinetic mechanisms of human translesion DNA polymerase kappa using tryptophan mutants. FEBS J. 2014, 281, 4394–4410. [Google Scholar] [CrossRef]
- Fischhaber, P.L.; Gerlach, V.L.; Feaver, W.J.; Hatahet, Z.; Wallace, S.S.; Friedberg, E.C. Human DNA polymerase kappa bypasses and extends beyond thymine glycols during translesion synthesis in vitro, preferentially incorporating correct nucleotides. J. Biol. Chem. 2002, 277, 37604–37611. [Google Scholar] [CrossRef]
- Levine, R.L.; Miller, H.; Grollman, A.; Ohashi, E.; Ohmori, H.; Masutani, C.; Hanaoka, F.; Moriya, M. Translesion DNA synthesis catalyzed by human pol eta and pol kappa across 1,N6-ethenodeoxyadenosine. J. Biol. Chem. 2001, 276, 18717–18721. [Google Scholar] [CrossRef]
- Wolfle, W.T.; Washington, M.T.; Prakash, L.; Prakash, S. Human DNA polymerase kappa uses template-primer misalignment as a novel means for extending mispaired termini and for generating single-base deletions. Genes Dev. 2003, 17, 2191–2199. [Google Scholar] [CrossRef]
- Walsh, J.M.; Ippoliti, P.J.; Ronayne, E.A.; Rozners, E.; Beuning, P.J. Discrimination against major groove adducts by Y-family polymerases of the DinB subfamily. DNA Repair (Amst) 2013, 12, 713–722. [Google Scholar] [CrossRef]
- Choi, J.Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef]
- Du, H.; Wang, P.; Li, L.; Wang, Y. Repair and translesion synthesis of O (6)-alkylguanine DNA lesions in human cells. J. Biol. Chem. 2019, 294, 11144–11153. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Guo, D.; Rechkoblit, O.; Wang, Z. Activities of human DNA polymerase kappa in response to the major benzo[a]pyrene DNA adduct: Error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair (Amst) 2002, 1, 559–569. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Guo, C. Effects of the N terminus of mouse DNA polymerase kappa on the bypass of a guanine-benzo[a]pyrenyl adduct. J. Biochem. 2016, 159, 471–479. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Tang, T.S.; Zhang, H.; Wang, Z.; Friedberg, E.; Yang, W.; Guo, C. Variants of mouse DNA polymerase kappa reveal a mechanism of efficient and accurate translesion synthesis past a benzo[a]pyrene dG adduct. PNAS 2014, 111, 1789–1794. [Google Scholar] [CrossRef]
- Sassa, A.; Niimi, N.; Fujimoto, H.; Katafuchi, A.; Gruz, P.; Yasui, M.; Gupta, R.C.; Johnson, F.; Ohta, T.; Nohmi, T. Phenylalanine 171 is a molecular brake for translesion synthesis across benzo[a]pyrene-guanine adducts by human DNA polymerase kappa. Mutat. Res. 2011, 718, 10–17. [Google Scholar] [CrossRef]
- Zhao, L.; Washington, M.T. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes (Basel) 2017, 8, 24. [Google Scholar] [CrossRef]
- Yuan, B.; Cao, H.; Jiang, Y.; Hong, H.; Wang, Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2’-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. PNAS 2008, 105, 8679–8684. [Google Scholar] [CrossRef]
- Wu, J.; Wang, P.; Li, L.; Williams, N.L.; Ji, D.; Zahurancik, W.J.; You, C.; Wang, J.; Suo, Z.; Wang, Y. Replication studies of carboxymethylated DNA lesions in human cells. Nucleic Acids Res. 2017, 45, 7276–7284. [Google Scholar] [CrossRef] [Green Version]
- Gowda, A.S.; Lee, M.; Spratt, T.E. N2 -Substituted 2’-Deoxyguanosine Triphosphate Derivatives as Selective Substrates for Human DNA Polymerase kappa. Angew. Chem. Int. Ed. Engl. 2017, 56, 2628–2631. [Google Scholar] [CrossRef]
- Andersen, N.; Wang, J.; Wang, P.; Jiang, Y.; Wang, Y. In-vitro replication studies on O(2)-methylthymidine and O(4)-methylthymidine. Chem. Res. Toxicol. 2012, 25, 2523–2531. [Google Scholar] [CrossRef]
- Andersen, N.; Wang, P.; Wang, Y. Replication across regioisomeric ethylated thymidine lesions by purified DNA polymerases. Chem. Res. Toxicol. 2013, 26, 1730–1738. [Google Scholar] [CrossRef]
- Gowda, A.S.P.; Spratt, T.E. DNA Polymerases eta and zeta Combine to Bypass O-2-[4-(3-Pyridyl)-4-oxobutyl]thymine, a DNA Adduct Formed from Tobacco Carcinogens. Chem. Res. Toxicol. 2016, 29, 303–316. [Google Scholar] [CrossRef]
- Wilson, K.A.; Holland, C.D.; Wetmore, S.D. Uncovering a unique approach for damaged DNA replication: A computational investigation of a mutagenic tobacco-derived thymine lesion. Nucleic Acids Res. 2019, 47, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Zang, H.; Angel, K.C.; Kozekov, I.D.; Goodenough, A.K.; Rizzo, C.J.; Guengerich, F.P. Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem. Res. Toxicol. 2006, 19, 879–886. [Google Scholar] [CrossRef]
- Wickramaratne, S.; Boldry, E.J.; Buehler, C.; Wang, Y.C.; Distefano, M.D.; Tretyakova, N.Y. Error-prone translesion synthesis past DNA-peptide cross-links conjugated to the major groove of DNA via C5 of thymidine. J. Biol. Chem. 2015, 290, 775–787. [Google Scholar] [CrossRef]
- Basu, A.K.; Pande, P.; Bose, A. Translesion Synthesis of 2’-Deoxyguanosine Lesions by Eukaryotic DNA Polymerases. Chem. Res. Toxicol. 2017, 30, 61–72. [Google Scholar] [CrossRef]
- Roy, U.; Scharer, O.D. Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair (Amst) 2016, 44, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, A.V.; Bondarenko, K.A.; Makarova, A.V. Non-bulky Lesions in Human DNA: The Ways of Formation, Repair, and Replication. Acta Nat. 2017, 9, 12–26. [Google Scholar] [CrossRef]
- Yagi, T.; Fujikawa, Y.; Sawai, T.; Takamura-Enya, T.; Ito-Harashima, S.; Kawanishi, M. Error-Prone and Error-Free Translesion DNA Synthesis over Site-Specifically Created DNA Adducts of Aryl Hydrocarbons (3-Nitrobenzanthrone and 4-Aminobiphenyl). Toxicol. Res. 2017, 33, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Kino, K.; Hirao-Suzuki, M.; Morikawa, M.; Sakaga, A.; Miyazawa, H. Generation, repair and replication of guanine oxidation products. Genes Environ. 2017, 39, 21. [Google Scholar] [CrossRef]
- Yockey, O.P.; Jha, V.; Ghodke, P.P.; Xu, T.; Xu, W.; Ling, H.; Pradeepkumar, P.I.; Zhao, L. Mechanism of Error-Free DNA Replication Past Lucidin-Derived DNA Damage by Human DNA Polymerase kappa. Chem. Res. Toxicol. 2017, 30, 2023–2032. [Google Scholar] [CrossRef]
- Gerlach, V.L.; Feaver, W.J.; Fischhaber, P.L.; Friedberg, E.C. Purification and characterization of pol kappa, a DNA polymerase encoded by the human DINB1 gene. J. Biol. Chem. 2001, 276, 92–98. [Google Scholar] [CrossRef]
- Washington, M.T.; Johnson, R.E.; Prakash, L.; Prakash, S. Human DINB1-encoded DNA polymerase kappa is a promiscuous extender of mispaired primer termini. PNAS 2002, 99, 1910–1914. [Google Scholar] [CrossRef]
- Wolfle, W.T.; Washington, M.T.; Kool, E.T.; Spratt, T.E.; Helquist, S.A.; Prakash, L.; Prakash, S. Evidence for a Watson-Crick hydrogen bonding requirement in DNA synthesis by human DNA polymerase kappa. Mol. Cell Biol. 2005, 25, 7137–7143. [Google Scholar] [CrossRef]
- Antczak, N.M.; Packer, M.R.; Lu, X.; Zhang, K.; Beuning, P.J. Human Y-Family DNA Polymerase kappa Is More Tolerant to Changes in Its Active Site Loop than Its Ortholog Escherichia coli DinB. Chem. Res. Toxicol. 2017. [Google Scholar] [CrossRef]
- Ketkar, A.; Maddukuri, L.; Penthala, N.R.; Reed, M.R.; Zafar, M.K.; Crooks, P.A.; Eoff, R.L. Inhibition of Human DNA Polymerases Eta and Kappa by Indole-Derived Molecules Occurs through Distinct Mechanisms. ACS Chem. Biol. 2019, 14, 1337–1351. [Google Scholar] [CrossRef]
- Nevin, P.; Engen, J.R.; Beuning, P.J. Steric gate residues of Y-family DNA polymerases DinB and pol kappa are crucial for dNTP-induced conformational change. DNA Repair (Amst) 2015, 29, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Ribonucleotide discrimination by translesion synthesis DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 382–402. [Google Scholar] [CrossRef]
- Haracska, L.; Prakash, L.; Prakash, S. Role of human DNA polymerase kappa as an extender in translesion synthesis. PNAS 2002, 99, 16000–16005. [Google Scholar] [CrossRef]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef]
- Carlson, K.D.; Johnson, R.E.; Prakash, L.; Prakash, S.; Washington, M.T. Human DNA polymerase kappa forms nonproductive complexes with matched primer termini but not with mismatched primer termini. PNAS 2006, 103, 15776–15781. [Google Scholar] [CrossRef]
- Dong, H.; Bonala, R.R.; Suzuki, N.; Johnson, F.; Grollman, A.P.; Shibutani, S. Mutagenic potential of benzo[a]pyrene-derived DNA adducts positioned in codon 273 of the human P53 gene. Biochemistry 2004, 43, 15922–15928. [Google Scholar] [CrossRef]
- Choi, J.Y.; Guengerich, F.P. Analysis of the Effect of Bulk at N2-Alkylguanine DNA Adducts on Catalytic Efficiency and Fidelity of the Processive DNA Polymerases Bacteriophage T7 Exonuclease- and HIV-1 Reverse Transcriptase. J. Biol. Chem. 2004, 279, 19217–19229. [Google Scholar] [CrossRef] [Green Version]
- Chiapperino, D.; Kroth, H.; Kramarczuk, I.H.; Sayer, J.M.; Masutani, C.; Hanaoka, F.; Jerina, D.M.; Cheh, A.M. Preferential misincorporation of purine nucleotides by human DNA polymerase eta opposite benzo[a]pyrene 7,8-diol 9,10-epoxide deoxyguanosine adducts. J. Biol. Chem. 2002, 277, 11765–11771. [Google Scholar] [CrossRef]
- Frank, E.G.; Sayer, J.M.; Kroth, H.; Ohashi, E.; Ohmori, H.; Jerina, D.M.; Woodgate, R. Translesion replication of benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyadenosine and deoxyguanosine by human DNA polymerase iota. Nucleic Acids Res. 2002, 30, 5284–5292. [Google Scholar] [CrossRef] [Green Version]
- Klarer, A.C.; Stallons, L.J.; Burke, T.J.; Skaggs, R.L.; McGregor, W.G. DNA polymerase eta participates in the mutagenic bypass of adducts induced by benzo[a]pyrene diol epoxide in mammalian cells. PLoS ONE 2012, 7, e39596. [Google Scholar] [CrossRef]
- Xie, Z.; Braithwaite, E.; Guo, D.; Zhao, B.; Geacintov, N.E.; Wang, Z. Mutagenesis of benzo[a]pyrene diol epoxide in yeast: Requirement for DNA polymerase zeta and involvement of DNA polymerase eta. Biochemistry 2003, 42, 11253–11262. [Google Scholar] [CrossRef]
- Jha, V.; Bian, C.; Xing, G.; Ling, H. Structure and mechanism of error-free replication past the major benzo[a]pyrene adduct by human DNA polymerase kappa. Nucleic Acids Res. 2016, 44, 4957–4967. [Google Scholar] [CrossRef]
- Jha, V.; Ling, H. Structural basis of accurate replication beyond a bulky major benzo[a]pyrene adduct by human DNA polymerase kappa. DNA Repair (Amst) 2017, 49, 43–50. [Google Scholar] [CrossRef]
- Sassa, A.; Suzuki, T.; Kanemaru, Y.; Niimi, N.; Fujimoto, H.; Katafuchi, A.; Gruz, P.; Yasui, M.; Gupta, R.C.; Johnson, F.; et al. In vivo evidence that phenylalanine 171 acts as a molecular brake for translesion DNA synthesis across benzo[a]pyrene DNA adducts by human DNA polymerase kappa. DNA Repair (Amst) 2014, 15, 21–28. [Google Scholar] [CrossRef]
- Kanemaru, Y.; Suzuki, T.; Niimi, N.; Gruz, P.; Matsumoto, K.; Adachi, N.; Honma, M.; Nohmi, T. Catalytic and non-catalytic roles of DNA polymerase kappa in the protection of human cells against genotoxic stresses. Environ. Mol. Mutagen. 2015, 56, 650–662. [Google Scholar] [CrossRef]
- Kanemaru, Y.; Suzuki, T.; Sassa, A.; Matsumoto, K.; Adachi, N.; Honma, M.; Numazawa, S.; Nohmi, T. DNA polymerase kappa protects human cells against MMC-induced genotoxicity through error-free translesion DNA synthesis. Genes Environ. 2017, 39, 6. [Google Scholar] [CrossRef]
- Avkin, S.; Goldsmith, M.; Velasco-Miguel, S.; Geacintov, N.; Friedberg, E.C.; Livneh, Z. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: The role of DNA polymerase kappa. J. Biol. Chem. 2004, 279, 53298–53305. [Google Scholar] [CrossRef]
- Masumura, K.; Toyoda-Hokaiwado, N.; Niimi, N.; Gruz, P.; Wada, N.A.; Takeiri, A.; Jishage, K.I.; Mishima, M.; Nohmi, T. Limited ability of DNA polymerase kappa to suppress benzo[a]pyrene-induced genotoxicity in vivo. Environ. Mol. Mutagen. 2017, 58, 644–653. [Google Scholar] [CrossRef]
- Hakura, A.; Sui, H.; Sonoda, J.; Matsuda, T.; Nohmi, T. DNA polymerase kappa counteracts inflammation-induced mutagenesis in multiple organs of mice. Environ. Mol. Mutagen. 2019, 60, 320–330. [Google Scholar] [CrossRef]
- Takeiri, A.; Wada, N.A.; Motoyama, S.; Matsuzaki, K.; Tateishi, H.; Matsumoto, K.; Niimi, N.; Sassa, A.; Gruz, P.; Masumura, K.; et al. In vivo evidence that DNA polymerase kappa is responsible for error-free bypass across DNA cross-links induced by mitomycin C. DNA Repair (Amst) 2014, 24, 113–121. [Google Scholar] [CrossRef]
- Bargonetti, J.; Champeil, E.; Tomasz, M. Differential toxicity of DNA adducts of mitomycin C. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef]
- Rycenga, H.B.; Long, D.T. The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr. Opin. Pharmacol. 2018, 41, 20–26. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Szikriszt, B.; Poti, A.; Pipek, O.; Krzystanek, M.; Kanu, N.; Molnar, J.; Ribli, D.; Szeltner, Z.; Tusnady, G.E.; Csabai, I.; et al. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 2016, 17, 99. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef]
- Jha, V.; Ling, H. Structural Basis for Human DNA Polymerase Kappa to Bypass Cisplatin Intrastrand Cross-Link (Pt-GG) Lesion as an Efficient and Accurate Extender. J. Mol. Biol. 2018, 430, 1577–1589. [Google Scholar] [CrossRef]
- Ho, T.V.; Guainazzi, A.; Derkunt, S.B.; Enoiu, M.; Scharer, O.D. Structure-dependent bypass of DNA interstrand crosslinks by translesion synthesis polymerases. Nucleic Acids Res. 2011, 39, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Enoiu, M.; Jiricny, J.; Scharer, O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012, 40, 8953–8964. [Google Scholar] [CrossRef]
- Williams, H.L.; Gottesman, M.E.; Gautier, J. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell 2012, 47, 140–147. [Google Scholar] [CrossRef]
- Zhuo, M.; Gorgun, M.F.; Englander, E.W. Translesion Synthesis DNA Polymerase Kappa Is Indispensable for DNA Repair Synthesis in Cisplatin Exposed Dorsal Root Ganglion Neurons. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Minko, I.G.; Yamanaka, K.; Kozekov, I.D.; Kozekova, A.; Indiani, C.; O’Donnell, M.E.; Jiang, Q.; Goodman, M.F.; Rizzo, C.J.; Lloyd, R.S. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 2008, 21, 1983–1990. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef]
- Boysen, G.; Pachkowski, B.F.; Nakamura, J.; Swenberg, J.A. The formation and biological significance of N7-guanine adducts. Mutat. Res. 2009, 678, 76–94. [Google Scholar] [CrossRef] [Green Version]
- Klapacz, J.; Pottenger, L.H.; Engelward, B.P.; Heinen, C.D.; Johnson, G.E.; Clewell, R.A.; Carmichael, P.L.; Adeleye, Y.; Andersen, M.E. Contributions of DNA repair and damage response pathways to the non-linear genotoxic responses of alkylating agents. Mutat. Res. Rev. Mutat. Res. 2016, 767, 77–91. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb Perspect Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Yi, C.; He, C. DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol. 2013, 5, a012575. [Google Scholar] [CrossRef]
- Gates, K.S. Structural biology: FaPy lesions and DNA mutations. Nat. Chem. Biol. 2013, 9, 412–414. [Google Scholar] [CrossRef]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef]
- Banerjee, S.; Chakraborty, S.; Jacinto, M.P.; Paul, M.D.; Balster, M.V.; Greenberg, M.M. Probing Enhanced Double-Strand Break Formation at Abasic Sites within Clustered Lesions in Nucleosome Core Particles. Biochemistry 2017, 56, 14–21. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res. 2011, 711, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Yu, S.L.; Prakash, S.; Prakash, L. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol. Cell Biol. 2007, 27, 7198–7205. [Google Scholar] [CrossRef]
- Takenaka, K.; Ogi, T.; Okada, T.; Sonoda, E.; Guo, C.; Friedberg, E.C.; Takeda, S. Involvement of vertebrate Polkappa in translesion DNA synthesis across DNA monoalkylation damage. J. Biol. Chem. 2006, 281, 2000–2004. [Google Scholar] [CrossRef]
- Takenaka, K.; Miki, Y. Introduction and characterization of a polymerase-dead point mutation into the POLK gene in vertebrates. FEBS Lett 2009, 583, 661–664. [Google Scholar] [CrossRef]
- Wit, N.; Buoninfante, O.A.; van den Berk, P.C.; Jansen, J.G.; Hogenbirk, M.A.; de Wind, N.; Jacobs, H. Roles of PCNA ubiquitination and TLS polymerases kappa and eta in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015, 43, 282–294. [Google Scholar] [CrossRef]
- Plosky, B.S.; Frank, E.G.; Berry, D.A.; Vennall, G.P.; McDonald, J.P.; Woodgate, R. Eukaryotic Y-family polymerases bypass a 3-methyl-2’-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008, 36, 2152–2162. [Google Scholar] [CrossRef]
- Yoon, J.H.; Roy Choudhury, J.; Park, J.; Prakash, S.; Prakash, L. Translesion synthesis DNA polymerases promote error-free replication through the minor-groove DNA adduct 3-deaza-3-methyladenine. J. Biol. Chem. 2017, 292, 18682–18688. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, E.; Ogi, T.; Kusumoto, R.; Iwai, S.; Masutani, C.; Hanaoka, F.; Ohmori, H. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000, 14, 1589–1594. [Google Scholar]
- Sherrer, S.M.; Fiala, K.A.; Fowler, J.D.; Newmister, S.A.; Pryor, J.M.; Suo, Z. Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 2011, 39, 609–622. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lim, S.; Kim, E.J.; Jo, A.; Guengerich, F.P. Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J. Mol. Biol. 2010, 404, 34–44. [Google Scholar] [CrossRef]
- Hile, S.E.; Eckert, K.A. DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res. 2008, 36, 688–696. [Google Scholar] [CrossRef]
- Hile, S.E.; Wang, X.; Lee, M.Y.; Eckert, K.A. Beyond translesion synthesis: Polymerase kappa fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012, 40, 1636–1647. [Google Scholar] [CrossRef]
- Barnes, R.P.; Hile, S.E.; Lee, M.Y.; Eckert, K.A. DNA polymerases eta and kappa exchange with the polymerase delta holoenzyme to complete common fragile site synthesis. DNA Repair (Amst) 2017, 57, 1–11. [Google Scholar] [CrossRef]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef]
- Eddy, S.; Tillman, M.; Maddukuri, L.; Ketkar, A.; Zafar, M.K.; Eoff, R.L. Human Translesion Polymerase kappa Exhibits Enhanced Activity and Reduced Fidelity Two Nucleotides from G-Quadruplex DNA. Biochemistry 2016, 55, 5218–5229. [Google Scholar] [CrossRef]
- Ogi, T.; Lehmann, A.R. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 2006, 8, 640–642. [Google Scholar] [CrossRef]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef]
- Bergoglio, V.; Bavoux, C.; Verbiest, V.; Hoffmann, J.S.; Cazaux, C. Localisation of human DNA polymerase kappa to replication foci. J. Cell Sci. 2002, 115, 4413–4418. [Google Scholar] [CrossRef]
- Bi, X.; Barkley, L.R.; Slater, D.M.; Tateishi, S.; Yamaizumi, M.; Ohmori, H.; Vaziri, C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol. Cell Biol. 2006, 26, 3527–3540. [Google Scholar] [CrossRef]
- Betous, R.; Pillaire, M.J.; Pierini, L.; van der Laan, S.; Recolin, B.; Ohl-Seguy, E.; Guo, C.; Niimi, N.; Gruz, P.; Nohmi, T.; et al. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef]
- Tonzi, P.; Yin, Y.; Lee, C.W.T.; Rothenberg, E.; Huang, T.T. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. Elife 2018, 7. [Google Scholar] [CrossRef]
- Yadav, S.; Mukhopadhyay, S.; Anbalagan, M.; Makridakis, N. Somatic Mutations in Catalytic Core of POLK Reported in Prostate Cancer Alter Translesion DNA Synthesis. Hum. Mutat. 2015, 36, 873–880. [Google Scholar] [CrossRef]
- Dai, Z.J.; Liu, X.H.; Ma, Y.F.; Kang, H.F.; Jin, T.B.; Dai, Z.M.; Guan, H.T.; Wang, M.; Liu, K.; Dai, C.; et al. Association Between Single Nucleotide Polymorphisms in DNA Polymerase Kappa Gene and Breast Cancer Risk in Chinese Han Population: A STROBE-Compliant Observational Study. Medicine (Baltimore) 2016, 95, e2466. [Google Scholar] [CrossRef]
- Shao, M.; Jin, B.; Niu, Y.; Ye, J.; Lu, D.; Han, B. Association of POLK polymorphisms with platinum-based chemotherapy response and severe toxicity in non-small cell lung cancer patients. Cell Biochem. Biophys. 2014, 70, 1227–1237. [Google Scholar] [CrossRef]
- Tonzi, P.; Huang, T.T. Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair (Amst) 2019, 78, 20–26. [Google Scholar] [CrossRef]
- Song, I.; Kim, E.J.; Kim, I.H.; Park, E.M.; Lee, K.E.; Shin, J.H.; Guengerich, F.P.; Choi, J.Y. Biochemical characterization of eight genetic variants of human DNA polymerase kappa involved in error-free bypass across bulky N(2)-guanyl DNA adducts. Chem. Res. Toxicol. 2014, 27, 919–930. [Google Scholar] [CrossRef]
- Antczak, N.M.; Walker, A.R.; Stern, H.R.; Leddin, E.M.; Palad, C.; Coulther, T.A.; Swett, R.J.; Cisneros, G.A.; Beuning, P.J. Characterization of Nine Cancer-Associated Variants in Human DNA Polymerase kappa. Chem. Res. Toxicol. 2018, 31, 697–711. [Google Scholar] [CrossRef]
- Kim, J.K.; Yeom, M.; Hong, J.K.; Song, I.; Lee, Y.S.; Guengerich, F.P.; Choi, J.Y. Six Germline Genetic Variations Impair the Translesion Synthesis Activity of Human DNA Polymerase kappa. Chem. Res. Toxicol. 2016, 29, 1741–1754. [Google Scholar] [CrossRef]
- Han, J.; Haiman, C.; Niu, T.; Guo, Q.; Cox, D.G.; Willett, W.C.; Hankinson, S.E.; Hunter, D.J. Genetic variation in DNA repair pathway genes and premenopausal breast cancer risk. Breast Cancer Res. Treat. 2009, 115, 613–622. [Google Scholar] [CrossRef]
- Dorjsuren, D.; Wilson, D.M., 3rd; Beard, W.A.; McDonald, J.P.; Austin, C.P.; Woodgate, R.; Wilson, S.H.; Simeonov, A. A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases. Nucleic Acids Res. 2009, 37, e128. [Google Scholar] [CrossRef]
- Yamanaka, K.; Dorjsuren, D.; Eoff, R.L.; Egli, M.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Lloyd, R.S. A comprehensive strategy to discover inhibitors of the translesion synthesis DNA polymerase kappa. PLoS ONE 2012, 7, e45032. [Google Scholar] [CrossRef]
- Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Yamanaka, K.; Banerjee, S.; Egli, M.; Choi, J.Y.; Lloyd, R.S.; Eoff, R.L. Leukotriene biosynthesis inhibitor MK886 impedes DNA polymerase activity. Chem. Res. Toxicol. 2013, 26, 221–232. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef]
Figure 1.
(a) Primer extension by translesion synthesis showing both the insertion and extension steps. The dot indicates the site of a lesion. (b) Direct extension (top) versus a one-nucleotide deletion resulting from looping a template base out of the DNA helix.
Figure 1.
(a) Primer extension by translesion synthesis showing both the insertion and extension steps. The dot indicates the site of a lesion. (b) Direct extension (top) versus a one-nucleotide deletion resulting from looping a template base out of the DNA helix.

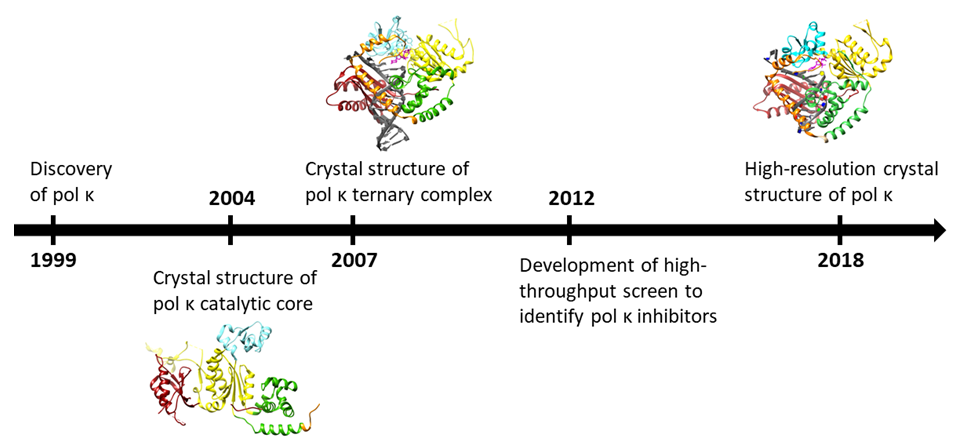
Figure 2.
(a) Diagram of the domains of human pol κ: In addition to the polymerase domains, PCNA-interacting peptide (PIP) regions, Rev1-interacting region (RIR), and ubiquitin-binding zinc finger (UBZ) domains are shown. (b) The structure of the polymerase domain of human pol κ with domains colored as in Figure 2a (PDB ID: 6CST) [23]. (c) Close-up view of the structure of pol κ highlighting the active site residues in contact with the incoming nucleotide. The catalytic residues D107, D198, and E199 are shown in yellow sticks with red oxygen atoms; others are shown as sticks colored by the domain as in Figure 2a,b. Metal ions are shown as green spheres.
Figure 2.
(a) Diagram of the domains of human pol κ: In addition to the polymerase domains, PCNA-interacting peptide (PIP) regions, Rev1-interacting region (RIR), and ubiquitin-binding zinc finger (UBZ) domains are shown. (b) The structure of the polymerase domain of human pol κ with domains colored as in Figure 2a (PDB ID: 6CST) [23]. (c) Close-up view of the structure of pol κ highlighting the active site residues in contact with the incoming nucleotide. The catalytic residues D107, D198, and E199 are shown in yellow sticks with red oxygen atoms; others are shown as sticks colored by the domain as in Figure 2a,b. Metal ions are shown as green spheres.

Figure 3.
Structures of undamaged dG and dA and examples of damaged DNA bases.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
POLK single nucleotide polymorphisms.
| AA | ID | Effects | Domain | Tumor Site | References |
|---|---|---|---|---|---|
| L21F | rs3104729 | 30-fold decrease in incorporation opposite N2-CH2-(9-anthracenyl)-dG (N2-CH2-Anth-dG) | N-clasp | Prostate | [147,148] |
| E29K | Decreased insertion opposite abasic site (2–20×) | N-clasp | Prostate, early onset | [143] | |
| I39T | rs3094258 | Similar activity to WT with several types of DNA damage | N-clasp | Prostate, Melanoma | [147,148] |
| T44M | Lesion-specific reduction in activity; reduced activity with N2-CH2-Anth-dG, O6-Me-dG and abasic sites | N-clasp | [149] | ||
| S137S | Synonymous | Fingers | Prostate | [143] | |
| G154E | COSM3856305 | Decreased activity opposite model abasic site; pathogenic | Fingers | Prostate early onset, stomach | [143] |
| F155S | Decreased activity on model abasic site | Fingers | Prostate | [143] | |
| P169T | rs148385845 | Slight decrease in activity on undamaged DNA | Fingers | Lung a | [148] |
| F171F | Synonymous | Palm | Prostate | [143] | |
| D189G | rs111689950 | Impaired for extension step of TLS | Palm | [147] | |
| F192C | rs150515841 | Slight increase in activity with N2-furfuryl-dG-containing templates | Palm | [148] | |
| T205I | Palm | Prostate, early onset | [143] | ||
| R219I | rs3104717 | Slight decrease in activity | Palm | Prostate | [147,148] |
| R219X | rs3094265 b | Inactive | Palm | [147] | |
| R246X | COSM3073601 | 5-10-fold less active with 8-oxo-dG-, N2-CH2-Anth-dG-, O6-Me-dG- and abasic-containing templates | Stomach | [149] | |
| E292K | rs142203892 | Similar activity as WT | Palm | [148] | |
| R298H | rs151251843 | Less active than WT on several different lesions | Palm | Large intestine a | [148,149] |
| A329A | rs3213801 | Synonymous, not meaningfully associated with breast cancer risk; more likely to respond to Pt-based chemotherapy | Palm | Breast | [144,145] |
| E419G | rs111584802 | 20-fold decrease in kcat/Km on dG and 670-fold decrease on N2-CH2-Anth-dG, extension defect | Little finger c | [147] | |
| E419E | Synonymous | Little finger | Prostate | [143] | |
| S423R | rs35257416 COSM6752124 | 1.6-fold more efficient than WT, 2-fold increased DNA binding affinity, pathogenic | Little finger | Melanoma, large intestine | [147,148] |
| A428A | COSM1070129 | Synonymous | Little finger | Endometrium, Prostate | [143] |
| E430K | Little finger | Prostate | [143] | ||
| E430G | Low activity on AP site | Little finger | Prostate | [143] | |
| Y432S | rs77612491 | Less active on undamaged and damaged DNA, extension defect, decreased DNA binding affinity | Little finger | Melanoma | [147,148] |
| L442F | Low activity on AP site | Little finger | Prostate, early onset | [143] | |
| Q447Q | Synonymous | Little finger | Prostate | [143] | |
| E449K | Low activity on AP site, low fidelity | Little finger | Prostate | [143] | |
| K461E | Little finger | Prostate | [143] | ||
| A471V | rs149894654 | Moderate decrease in activity | Little finger | [149] | |
| T473A | rs186798689 | Decreased activity on undamaged and damaged DNA | Little finger | [149] | |
| I487T | Little finger | Prostate | [143] | ||
| R512W | Decreased activity on undamaged and damaged DNA | Little finger | [149] | ||
| S528N | Prostate | [143] | |||
| D551N | Prostate | [143] | |||
| K564K | Synonymous | Prostate | [143] | ||
| D581N | Prostate | [143] | |||
| S678F | Prostate | [143] | |||
| L731F | Prostate | [143] | |||
| P861P | Prostate | [143] | |||
| D866E | Prostate | [143] | |||
| intron | rs10077427 | Contributes to breast cancer risk, more likely to have progesterone receptor-positive tumors; Decreased progression-free survival with Pt-based chemotherapy | Breast | [144,145] | |
| intron | rs5744533 | Contributes to breast cancer risk, protective in postmenopausal women, no correlation with clinical phenotypes; More likely to respond to Pt-based chemotherapy | Breast | [144,145] | |
| intron | rs3756558 | Breast | [150] |
a 1000 genomes data; b Withdrawn from ClinVar in 2015; c In eukaryotic Y-family polymerases, the little finger domain is also referred to as the Polymerase-Associated Domain (PAD).
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stern, H.R.; Sefcikova, J.; Chaparro, V.E.; Beuning, P.J. Mammalian DNA Polymerase Kappa Activity and Specificity. Molecules 2019, 24, 2805. https://doi.org/10.3390/molecules24152805
AMA Style
Stern HR, Sefcikova J, Chaparro VE, Beuning PJ. Mammalian DNA Polymerase Kappa Activity and Specificity. Molecules. 2019; 24(15):2805. https://doi.org/10.3390/molecules24152805
Chicago/Turabian StyleStern, Hannah R., Jana Sefcikova, Victoria E. Chaparro, and Penny J. Beuning. 2019. "Mammalian DNA Polymerase Kappa Activity and Specificity" Molecules 24, no. 15: 2805. https://doi.org/10.3390/molecules24152805