
Surveillance of SARS-CoV-2 in Frankfurt am Main from October to December 2020 Reveals High Viral Diversity Including Spike Mutation N501Y in B.1.1.70 and B.1.1.7

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and RT-qPCR-Testing
2.2. Cell Culture and Viral Outgrowth Assay
2.3. RNA Isolation and Confirmatory RT-qPCR
2.4. NGS Sequencing of SARS-CoV-2 Genomes
2.5. Bioinformatics
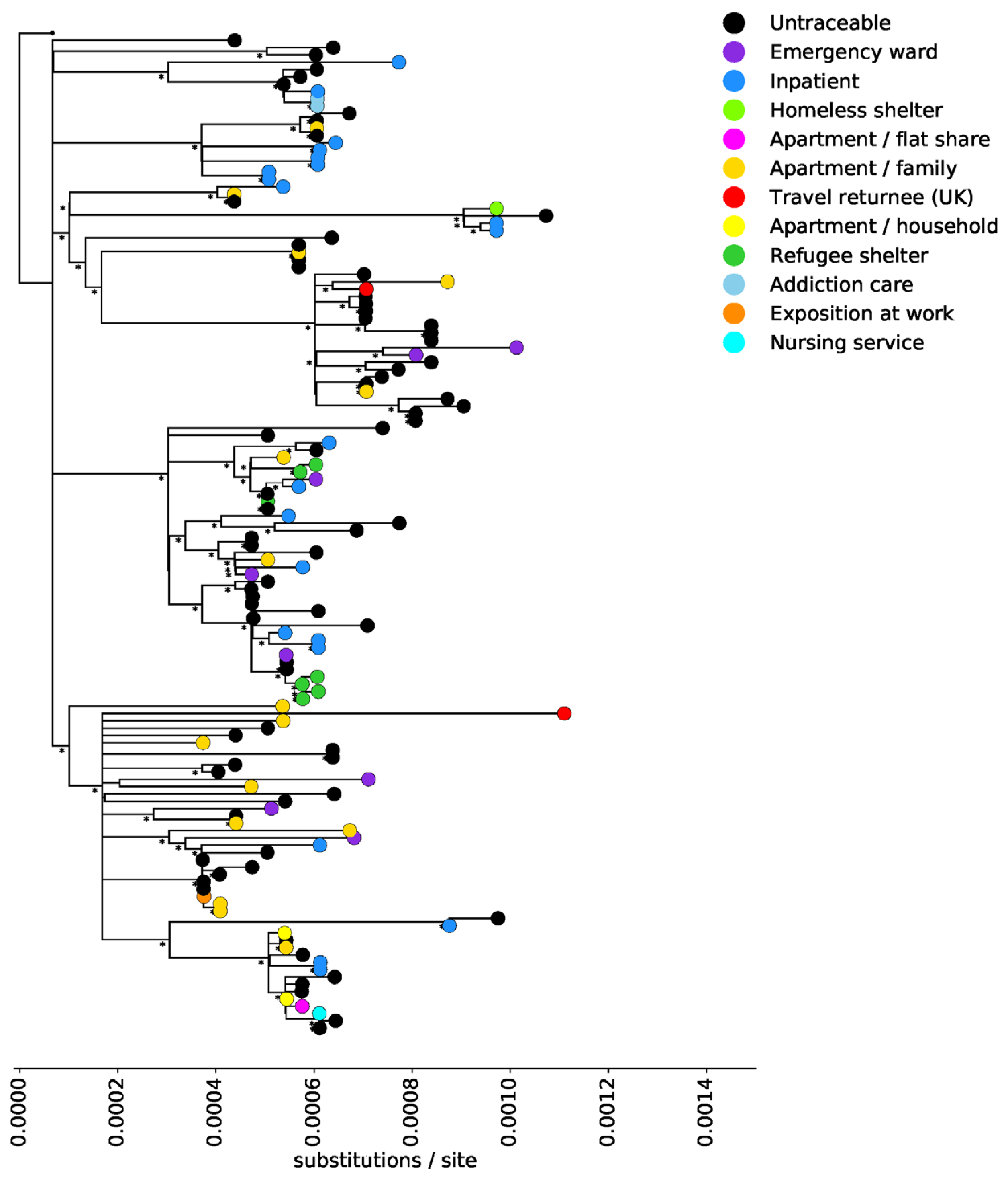
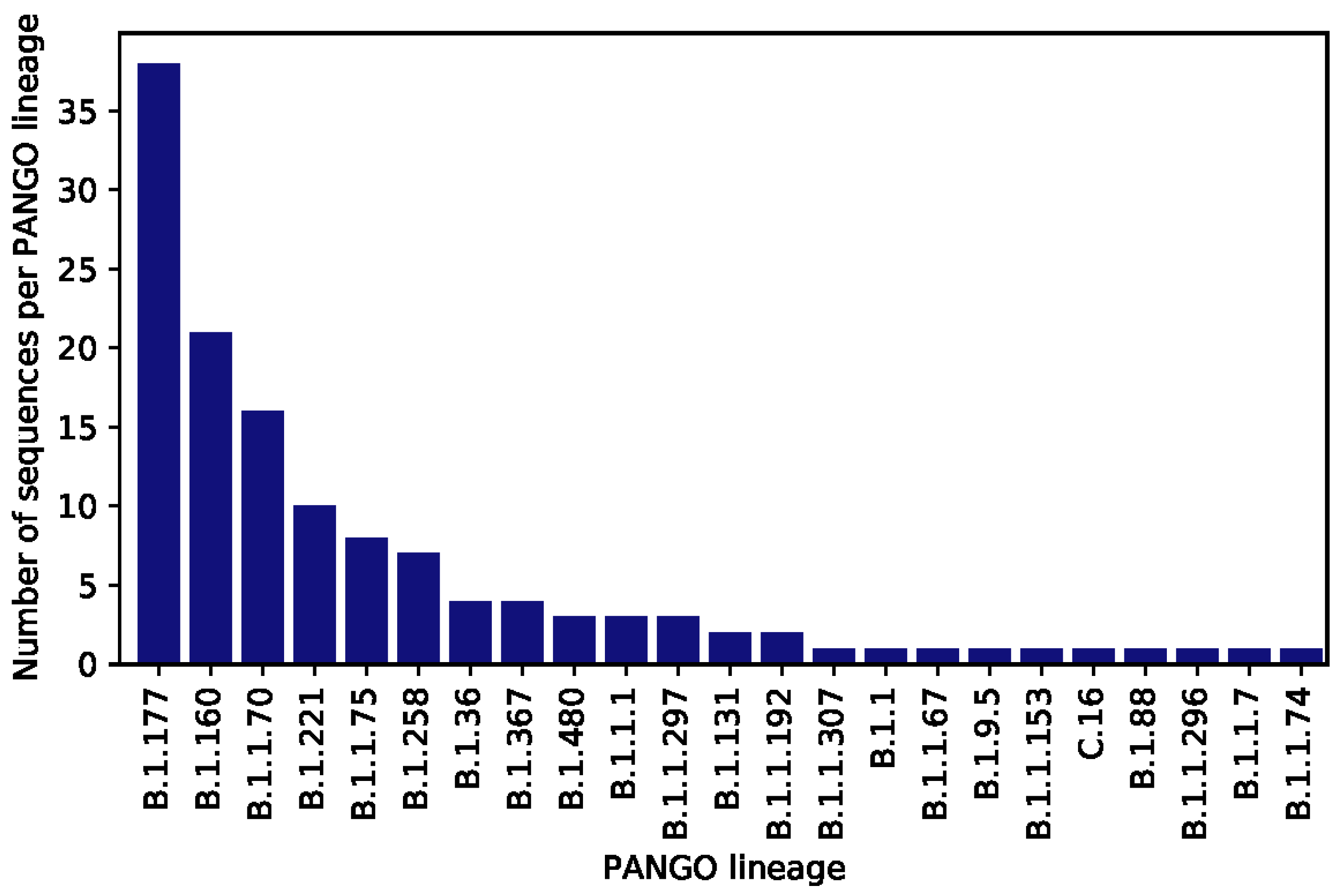
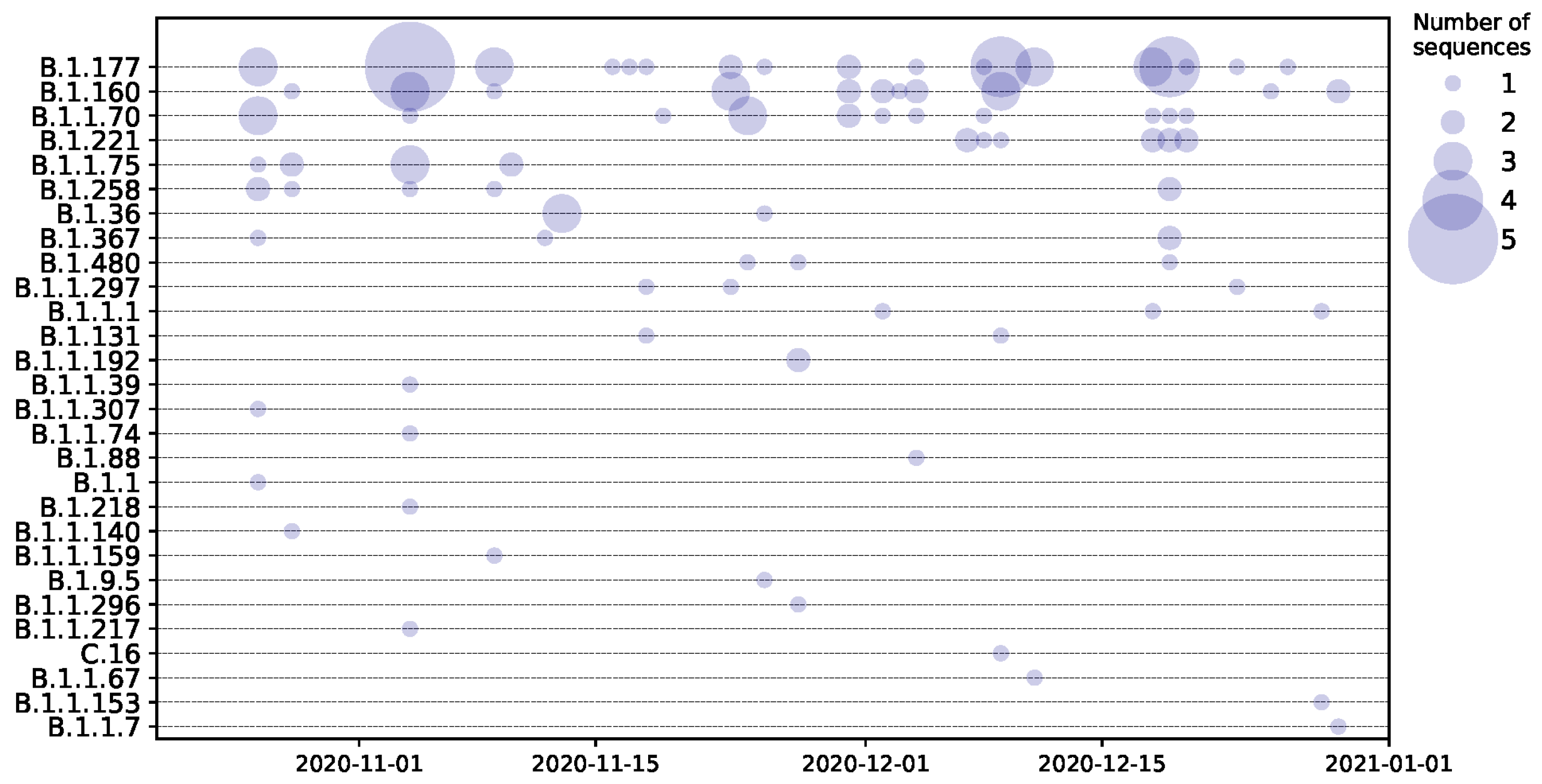
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Zahradník, J.; Marciano, S.; Shemesh, M.; Zoler, E.; Chiaravalli, J.; Meyer, B.; Dym, O.; Elad, N.; Schreiber, G. SARS-CoV-2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. medRxiv 2021. [Google Scholar] [CrossRef]
- Kemp, S.; Harvey, W.; Lytras, S.; Carabelli, A.; Robertson, D.; Gupta, R. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion H69/V70. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral Spike vaccines. bioRxiv 2021. [Google Scholar] [CrossRef]
- Muik, A.; Wallisch, A.-K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Sarkar, R.; Türeci, Ö.; Dormitzer, P.R.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021. [Google Scholar] [CrossRef]
- Du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021. [Google Scholar] [CrossRef] [PubMed]
- Westhaus, S.; Widera, M.; Rabenau, H.F.; Hoehl, S.; Bojkova, D.; Cinatl, J.; Ciesek, S. Evaluation of stability and inactivation methods of SARS-CoV-2 in context of laboratory settings. bioRxiv 2020. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. PCR Protocol—World Health Organization; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Muller, N.; Kunze, M.; Steitz, F.; Saad, N.J.; Muhlemann, B.; Beheim-Schwarzbach, J.I.; Schneider, J.; Drosten, C.; Murajda, L.; Kochs, S.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Outbreak Related to a Nightclub, Germany, 2020. Emerg. Infect. Dis. 2020, 27, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Capoferri, A.A.; Katusiime, M.G.; Patro, S.C.; Kearney, M.F. Low genetic diversity may be an Achilles heel of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 24614–24616. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Vilar, S.; Isom, D.G. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. medRxiv 2021. [Google Scholar] [CrossRef]
- Lemmermann, N.; Lieb, B.; Laufs, T.; Renzaho, A.; Runkel, S.; Kohnen, W.; Linke, M.; Gerber, S.; Schweiger, S.; Michel, A.; et al. SARS-CoV-2 genome surveillance in Mainz, Germany, reveals convergent origin of the N501Y spike mutation in a hospital setting. medRxiv 2021. [Google Scholar] [CrossRef]
- Widera, M.; Wilhelm, A.; Hoehl, S.; Pallas, C.; Kohmer, N.; Wolf, T.; Rabenau, H.F.; Corman, V.; Drosten, C.; Vehreschild, M.J.; et al. Bamlanivimab does not neutralize two SARS-CoV-2 variants carrying E484K in vitro. medRxiv 2021. [Google Scholar] [CrossRef]
- Wang, P.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; Graham, B.S.; et al. Increased Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to Antibody Neutralization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.; Hahn, A.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and B.1.1.248: Escape from therapeutic antibodies and antibodies induced by infection and vaccination. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Deletion/Mutation | Number of Sequences | PANGO Lineage | Sequence ID |
|---|---|---|---|
| 9 bp deletion in ORF1ab (amino acid position 141–143), 7 bp deletion in ORF7a which truncates ORF7a at amino acid position 108 | 1 | B.1.160 | ChVir21561 |
| Deletion of amino acid position 69/70 in S | 17 | B.1.1.70, B.1.258 | ChVir21551, ChVir21580, ChVir21582, ChVir21585, ChVir21586, ChVir21588, ChVir21589, ChVir21591, ChVir21596, ChVir21597, ChVir21598, ChVir21606, ChVir21609, ChVir21618, ChVir21619, ChVir21621, ChVir21626 |
| Deletion of amino acid positions 141-144 in S | 1 | B.1.1.153 | ChVir22027 |
| Deletion of amino acid position 210 in S | 4 | B.1.36 | ChVir21555, ChVir21563, ChVir21571, ChVir21603 |
| N501Y substitution in S | 1 | B.1.1.70 | ChVir21997 |
| 4 bp deletion in ORF3a, which truncates ORF3a at amino acid position 259 | 3 | B.1.160, B.1.1.67 | ChVir21565, ChVir21625, ChVir21632, ChVir22011 |
| 1 bp deletion in ORF3a, which truncates ORF3a at amino acid position 259 | 1 | B.1.160 | ChVir21550 |
| Deletion of amino acid position 58 in ORF8 | 1 | B.1.258 | ChVir21586 |
| 12 bp deletion in 3p UTR | 1 | B.1.258 | ChVir21502 |
| 41 bp deletion in the 3’ UTR | 4 | B.1.221 | ChVir21994, ChVir21995, ChVir21996, ChVir22006 |
| Substitutions typical of B.1.1.7, including a deletion of amino acid position 69/70 and N501Y in S | 1 | B.1.1.7 | ChVir22031 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Widera, M.; Mühlemann, B.; Corman, V.M.; Toptan, T.; Beheim-Schwarzbach, J.; Kohmer, N.; Schneider, J.; Berger, A.; Veith, T.; Pallas, C.; et al. Surveillance of SARS-CoV-2 in Frankfurt am Main from October to December 2020 Reveals High Viral Diversity Including Spike Mutation N501Y in B.1.1.70 and B.1.1.7. Microorganisms 2021, 9, 748. https://doi.org/10.3390/microorganisms9040748
Widera M, Mühlemann B, Corman VM, Toptan T, Beheim-Schwarzbach J, Kohmer N, Schneider J, Berger A, Veith T, Pallas C, et al. Surveillance of SARS-CoV-2 in Frankfurt am Main from October to December 2020 Reveals High Viral Diversity Including Spike Mutation N501Y in B.1.1.70 and B.1.1.7. Microorganisms. 2021; 9(4):748. https://doi.org/10.3390/microorganisms9040748
Chicago/Turabian StyleWidera, Marek, Barbara Mühlemann, Victor M. Corman, Tuna Toptan, Jörn Beheim-Schwarzbach, Niko Kohmer, Julia Schneider, Annemarie Berger, Talitha Veith, Christiane Pallas, and et al. 2021. "Surveillance of SARS-CoV-2 in Frankfurt am Main from October to December 2020 Reveals High Viral Diversity Including Spike Mutation N501Y in B.1.1.70 and B.1.1.7" Microorganisms 9, no. 4: 748. https://doi.org/10.3390/microorganisms9040748