Familial Arrhythmogenic Cardiomyopathy: Clinical Determinants of Phenotype Discordance and the Impact of Endurance Sports

 , and
, and
Abstract
:1. Introduction
2. Experimental Section
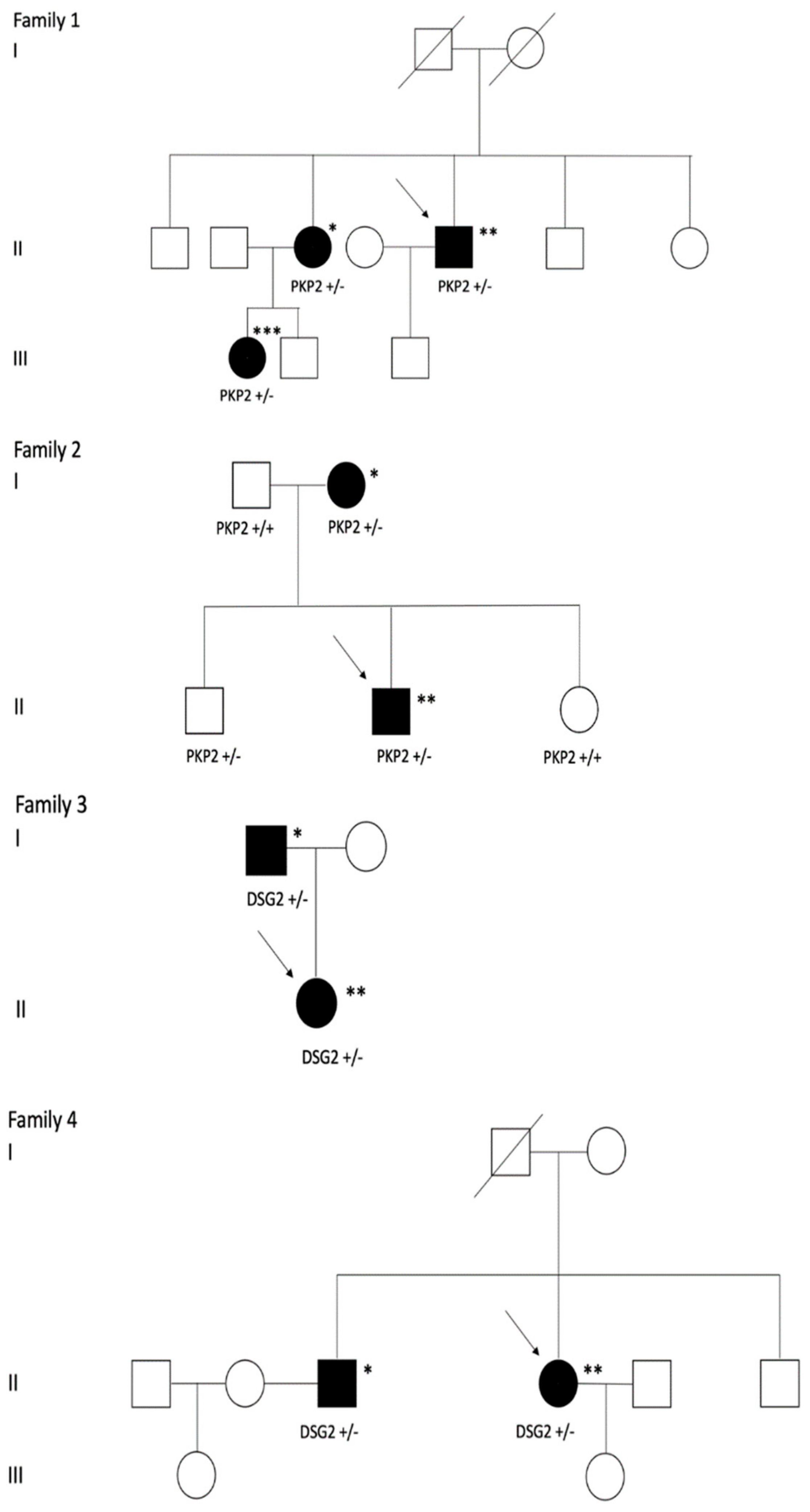
3. Results
4. Discussion
4.1. Classical ARVC vs. Biventricular ACM
4.2. Early Onset ACM vs. No Disease
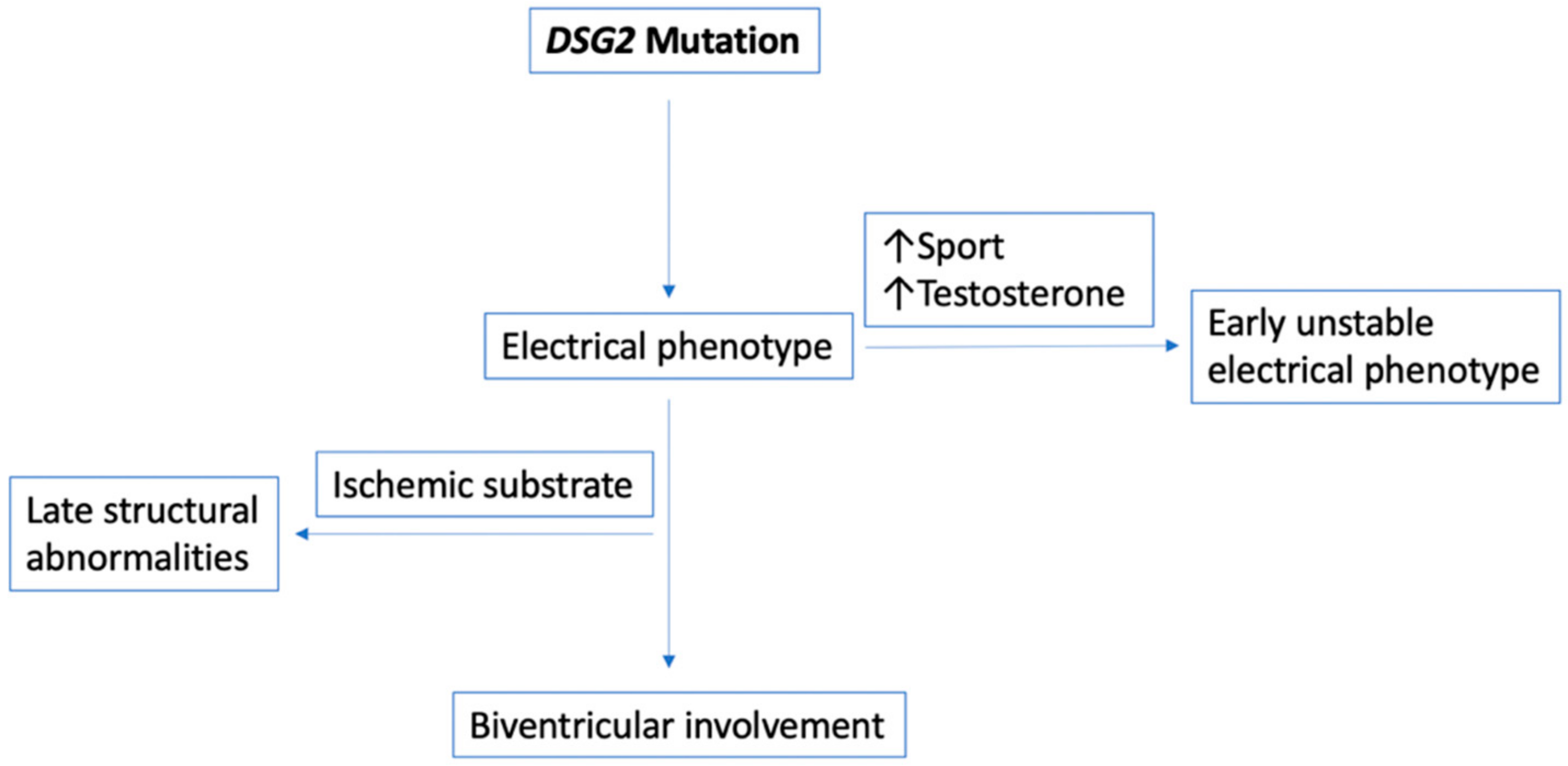
4.3. Early and Unstable Electrical Phenotype vs. Late and Stable Phenotype
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sen-Chowdhry, S.; Morgan, R.D.; Chambers, J.C.; McKenna, W.J. Arrhythmogenic cardiomyopathy: Etiology, diagnosis, and treatment. Ann. Rev. Med. 2010, 61, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ. Cardiovasc Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, E.; Jongbloed, J.D.H.; Pilichou, K.; Thiene, G.; Basso, C.; Bikker, H.; Charbon, B.; Swertz, M.; van Tintelen, J.P.; van der Zwaag, P.A. The ARVD/C genetic variants database: 2014 update. Hum. Mutat. 2015, 36, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Medeiros-Domingo, A.; Gaertner-Rommel, A.; Kast, J.I.; Enseleit, F.; Bode, P.; Klingel, K.; Kandolf, R.; Renois, F.; Andreoletti, L.; et al. Myocardial expression profiles of candidate molecules in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia compared to those with dilated cardiomyopathy and healthy controls. Heart Rhythm. 2016, 13, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Delmar, M.; McKenna, W.J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.P.; Wilde, A.A.M.; van der Smagt, J.; Boven, L.G.; Mannens, M.M.A.M.; van Langen, I.M.; et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Peng, H.; Zheng, C.; Zhang, H.; Yan, C.; Ma, H.; Dai, X.; Li, X. Two pedigrees with arrhythmogenic right ventricular cardiomyopathy linked with R49H and F531C mutation in DSG2. Hum Genome Var. 2019, 6, 38. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm. Electrophysiol. Rev. 2016, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Dalal, D.; Tandri, H.; Judge, D.P.; Amat, N.; Macedo, R.; Jain, R.; Tichnell, C.; Daly, A.; James, C.; Russell, S.D.; et al. Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2009, 53, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorzi, A.; Cipriani, A.; Mattesi, G.; Vio, R.; Bettella, N.; Corrado, D. Arrhythmogenic cardiomyopathy and sports activity. J. Cardiovasc. Trans. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy—Associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, M.; Gasperetti, A.; Gaetano, F.; Busana, M.; Sommariva, E.; Catto, V.; Sicuso, R.; Rizzo, S.; Conte, E.; Mushtaq, S.; et al. Long-term follow-up analysis of a highly characterized arrhythmogenic cardiomyopathy cohort with classical and non-classical phenotypes—A real-world assessment of a novel prediction model: Does the subtype really matter. EP Eur. 2020, 22, 797–805. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.M.; James, C.A.; Rastegar, N.; Bhonsale, A.; Murray, B.; Tichnell, C.; Judge, D.P.; Bluemke, D.A.; Zimmerman, S.L.; Kamel, I.R.; et al. Yield of serial evaluation in at-risk family members of patients with ARVD/C. J. Am. Coll. Cardiol. 2014, 64, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Kléber, A.G.; MacRae, C.A.; Saffitz, J.E. Arrhythmogenic cardiomyopathy—New insights into disease mechanisms and drug discovery. Prog. Pediatric Cardiol. 2014, 37, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Kirchhof, P.; Fabritz, L.; Zwiener, M.; Witt, H.; Schäfers, M.; Zellerhoff, S.; Paul, M.; Athai, T.; Hiller, K.-H.; Baba, H.A.; et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 2006, 114, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Gasperetti, A.; Dello Russo, A.; Busana, M.; Dessanai, M.; Pizzamiglio, F.; Saguner, A.M.; te Riele, A.S.J.M.; Sommariva, E.; Vettor, G.; Bosman, L.; et al. Novel risk calculator performance in athletes with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2020, 17, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Roli, L.; De Vincentis, S.; Rocchi, M.B.L.; Trenti, T.; De Santis, M.C.; Savino, G. Testosterone, cortisol, hGH, and IGF-1 levels in an Italian female elite volleyball team. Health Sci. Rep. 2018, 1, e32. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Lüscher, T.F.; Brunckhorst, C.; Chen, H.S.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chen, L.; Zhang, N.; Chen, X.; Zhao, Q.; Chen, K.; Li, X.; Ruschitzka, F.; Duru, F.; Song, J. Plasma testosterone and arrhythmic events in male patients with arrhythmogenic right ventricular cardiomyopathy. Esc. Heart Fail. 2020, 7, 1547–1559. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Task Force Criteria | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structural | Histological | Repolarization | Conduction | Arrhythmias | Familial | TFC Score | ||||||||
| Major | Minor | Major | Minor | Major | Minor | Major | Minor | Major | Minor | Major | Minor | |||
| Family 1 | Proband | X | N/A | N/A | X | X | X * | X | 9 | |||||
| Niece | X | N/A | N/A | X | X * | X | 7 | |||||||
| Family 2 | Proband | X | N/A | N/A | X | X | X | 7 | ||||||
| Mother | N/A | N/A | X | 2 | ||||||||||
| Brother | N/A | N/A | X | 2 | ||||||||||
| Family 3 | Proband | X | N/A | N/A | X | X | X | 7 | ||||||
| Father | X | N/A | N/A | X | X | 5 | ||||||||
| Family 4 | Proband | X | N/A | N/A | X | X | X | 8 | ||||||
| Brother | X | N/A | N/A | X | X | 5 | ||||||||
| Patients Clinical Characteristics | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at Diagnosis | Gender | Endurance Athlete | TTE (RV) | MRI | ECG | |||||||
| PLAX (cm/m2) | PSAX (cm/m2) | FAC (%) | RVEDVi (ml/m2) | RVEF (%) | LGE | T-Wave Inversions (Leads) | PVC Burden (%) | |||||
| Family 1 | Proband | 26 | M | Yes | 2.9 * | 2.6 * | 26 * | N/A | N/A | N/A | III, aVR, V1-V4 * | 2.5 * |
| Niece | 34 | F | No | 2.3 * | 2.1 * | 30 * | 147 * | 38 * | Yes * | II, III, aVF, V1-V6 * | 1.5 * | |
| Family 2 | Proband | 19 | M | Yes | 2.1 * | 2.1 * | 24 * | 176 * | 40 * | Yes * | II, III, aVF, V1-V3 * | 3 * |
| Mother | 58 | F | No | N/A | N/A | 45 | 71 | 60 | No | aVR, aVL | 0 | |
| Brother | 30 | M | No | N/A | N/A | 52 | 93 | 60 | No | aVR, V1 | 0 | |
| Family 3 | Proband | 18 | F | Yes | 1.8 * | 1.87 * | 44 | 97 | 57 | No | aVR, V1-V3 * | 3 * |
| Father | 52 | M | No | 2 * | 1.8 * | 35 | 79 | 62 | No | aVR | 13.8 * | |
| Family 4 | Proband | 21 | F | Yes | N/A | N/A | 26 * | N/A | N/A | N/A | II, III, aVF, V1-V6 * | 4 * |
| Brother | 53 | M | No | N/A | N/A | 28 * | 81 | 61 | Yes * | II, III, aVF, V5-V6 * | N/A | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, S.; Gasperetti, A.; Medeiros-Domingo, A.; Akdis, D.; Brunckhorst, C.; Saguner, A.M.; Duru, F. Familial Arrhythmogenic Cardiomyopathy: Clinical Determinants of Phenotype Discordance and the Impact of Endurance Sports. J. Clin. Med. 2020, 9, 3781. https://doi.org/10.3390/jcm9113781
Costa S, Gasperetti A, Medeiros-Domingo A, Akdis D, Brunckhorst C, Saguner AM, Duru F. Familial Arrhythmogenic Cardiomyopathy: Clinical Determinants of Phenotype Discordance and the Impact of Endurance Sports. Journal of Clinical Medicine. 2020; 9(11):3781. https://doi.org/10.3390/jcm9113781
Chicago/Turabian StyleCosta, Sarah, Alessio Gasperetti, Argelia Medeiros-Domingo, Deniz Akdis, Corinna Brunckhorst, Ardan M. Saguner, and Firat Duru. 2020. "Familial Arrhythmogenic Cardiomyopathy: Clinical Determinants of Phenotype Discordance and the Impact of Endurance Sports" Journal of Clinical Medicine 9, no. 11: 3781. https://doi.org/10.3390/jcm9113781