 Fabrizio Antonangeli1*
Fabrizio Antonangeli1* Ambra Natalini1
Ambra Natalini1 Marina Chiara Garassino2
Marina Chiara Garassino2 Antonio Sica3,4
Antonio Sica3,4 Angela Santoni5
Angela Santoni5 Francesca Di Rosa1
Francesca Di Rosa1- 1Institute of Molecular Biology and Pathology, National Research Council (CNR), Rome, Italy
- 2Medical Oncology Department, Istituto Nazionale dei Tumori, Istituto di Ricovero e Cura a Carattere Scientifico, Milan, Italy
- 3Department of Pharmaceutical Sciences, University of Eastern Piedmont, A. Avogadro, Novara, Italy
- 4Humanitas Clinical and Research Center, Istituto di Ricovero e Cura a Carattere Scientifico, Milan, Italy
- 5Department of Molecular Medicine, Laboratory Affiliated to Istituto Pasteur Italia, Sapienza University of Rome, Rome, Italy
Immune checkpoints are inhibitory receptor/ligand pairs regulating immunity that are exploited as key targets of anti-cancer therapy. Although the PD-1/PD-L1 pair is one of the most studied immune checkpoints, several aspects of its biology remain to be clarified. It has been established that PD-1 is an inhibitory receptor up-regulated by activated T, B, and NK lymphocytes and that its ligand PD-L1 mediates a negative feedback of lymphocyte activation, contributing to the restoration of the steady state condition after acute immune responses. This loop might become detrimental in the presence of either a chronic infection or a growing tumor. PD-L1 expression in tumors is currently used as a biomarker to orient therapeutic decisions; nevertheless, our knowledge about the regulation of PD-L1 expression is limited. The present review discusses how NF-κB, a master transcription factor of inflammation and immunity, is emerging as a key positive regulator of PD-L1 expression in cancer. NF-κB directly induces PD-L1 gene transcription by binding to its promoter, and it can also regulate PD-L1 post-transcriptionally through indirect pathways. These processes, which under conditions of cellular stress and acute inflammation drive tissue homeostasis and promote tissue healing, are largely dysregulated in tumors. Up-regulation of PD-L1 in cancer cells is controlled via NF-κB downstream of several signals, including oncogene- and stress-induced pathways, inflammatory cytokines, and chemotherapeutic drugs. Notably, a shared signaling pathway in epithelial cancers induces both PD-L1 expression and epithelial–mesenchymal transition, suggesting that PD-L1 is part of the tissue remodeling program. Furthermore, PD-L1 expression by tumor infiltrating myeloid cells can contribute to the immune suppressive features of the tumor environment. A better understanding of the interplay between NF-κB signaling and PD-L1 expression is highly relevant to cancer biology and therapy.
Introduction
The immune system relies on a complex balance between activating and inhibitory mechanisms to counteract infections and other threats while avoiding excessive tissue damage. Among the inhibitory molecules, a distinct set of inhibitory receptors and their ligands, collectively called “immune checkpoints,” has recently attracted a lot of attention for its relevance in cancer therapy, chronic infections, and autoimmune diseases (1). Programmed cell death protein-1 (PD-1) is a member of the CD28 family expressed by activated lymphocytes. PD-1 triggers immunosuppressive signals upon engagement by its ligands, i.e., PD-L1 (CD274 or B7-H1) and PD-L2 (CD273), which are members of the B7 family. While PD-L2 expression is largely restricted to antigen-presenting cells (APCs) and B1 lymphocytes, PD-L1 is expressed by APCs (mostly macrophages and dendritic cells), activated/exhausted T and B lymphocytes, and regulatory T cells (Treg), among others (2, 3). PD-L1 is also expressed by the cardiac endothelium, placenta, and pancreatic islets, with a possible role in maintaining immunological tolerance in these districts (4). Cancer cells can express PD-L1 and exploit the PD-L1-driven inhibitory pathway to their benefit as a key mechanism of immune evasion (5).
Return to the steady state at the end of immune response is tightly regulated (6, 7), and it is widely recognized that the PD-1/PD-L1 axis plays a central role in physiological immune homeostasis, contributing to the prevention of lymphocyte over-activation and immunopathology (1, 8). A variety of mechanisms have been involved in PD-1-mediated suppression of activated T lymphocytes, including exhaustion, inflammatory cytokine secretion inhibition, anergy, and apoptosis (8). PD-1 expression by antigen-responding T and B cells is tightly regulated, which allows for a stringent control of lymphocyte response (9, 10). Accordingly, PD-1 deficiency is associated with the development of autoimmune diseases (11, 12). PD-1 can be expressed also by γδ T cells, natural killer (NK) cells, and innate lymphoid cells (ILCs), which are circulating and tissue-resident lymphocytes involved in tissue repair and early responses against pathogens and cellular stress (13–16).
Regulation of PD-L1 expression and function takes place at different levels, as extensively reviewed by Sun and colleagues (17). Several mediators of inflammation are PD-L1 inducers, including TNFα, IFN-γ, IL-10, IL-17, and C5a (18–21). The JAK/STAT, RAS/MAPK, and PTEN-PI3K/AKT pathways are involved in the control of PD-L1 gene expression via different downstream transcription factors, such as STAT1, STAT3, IRF1, IRF3, HIF-1α, MYC, JUN, BRD4, and NF-κB (22). The corresponding DNA-binding elements, except for IRF3, have been described on the PD-L1 promoter (23–32). Additional mechanisms of regulation include microRNA-mediated post-transcriptional inhibition (e.g., miR-513, miR-34a, miR-200, and miR-570) and the presence of a soluble form of PD-L1 (sPD-L1) in the blood, which possibly competes with the membrane-bound PD-L1 for binding to PD-1 (17, 33). Though only partially investigated, reverse signaling of PD-L1 has been reported in tumor cells and macrophages, resulting in pro-survival and inhibitory signals, respectively (34, 35). In addition to the PD-1/PD-L1 pair, a further interaction between PD-L1 and B7.1 (CD80) has been implicated in the inhibition of T-cell proliferation and cytokine production (36).
IFN-γ is one of the most studied PD-L1 inducers in tumors but PD-L1 expression does not necessarily mirror the IFN-γ signature (37, 38). NF-κB, a central player of inflammation and immunity, is emerging as a key positive regulator of PD-L1 expression. Notably, two recent studies, by using a CRISPR-Cas9-based wide screening approach, have identified NF-κB as a major determinant of cancer cell resistance against immune attack (39, 40). Considering the pivotal role played by PD-L1 for tumor cell immune evasion, the disclosure of the relationship between NF-κB signaling and PD-L1 expression is of great relevance (41). The present review gives an overview of the experimental works linking NF-κB to the regulation of PD-L1 expression in tumors. Furthermore, the implications of NF-κB-mediated control of PD-L1 expression for tissue homeostasis, cancer biology, and immune-therapy are discussed.
NF-κB Among Inflammation, Immunity, and Cancer
NF-κB [nuclear factor kappa-light-chain-enhancer of activated B cells, discovered by Sen and Baltimore in 1986 (42)] is a transcription factor supporting host responses to cellular stress and immune responses to pathogens and other challenges. NF-κB can be composed of different dimers of the NF-κB family, activated downstream of multiple signaling pathways [for a comprehensive review on NF-κB see (43)]. Briefly, five proteins belong to the NF-κB family: p50, p52, p65 (RelA), RelB, and c-Rel; they are encoded by NFKB1, NFKB2, RELA, RELB, and REL genes, respectively. NFKB1 and NFKB2 codify for the p105 and p100 precursors, which are then processed to the active forms p50 and p52, respectively. The canonical (or classical) pathway leads to the activation of the p50/p65 (RelA) or p50/c-Rel heterodimers, while the non-canonical (or alternative) pathway leads to the activation of the p52/RelB heterodimer. The different heterodimers play distinct biological roles, controlling lymphoid organ development, immune activation, and cell survival (44–46). In healthy cells, NF-κB complexes are retained in the cytoplasm by inhibitory proteins belonging to the IκB family. Activating signals of the canonical pathway, which include TNFα, IL-1, and Lypopolysaccharide (LPS), cause IκB phosphorylation via IκB kinase (IKK). IKKα and IKKβ are the catalytic subunits of the multimeric IKK, which also includes the scaffold protein IKKγ (also named NEMO). Upon phosphorylation, IκB is ubiquitinated and targeted to degradation by the proteasome. This allows NF-κB's translocation to the nucleus where it regulates gene transcription by binding to the promoters of its target genes. Direct phosphorylation of p65 further enhances NF-κB nuclear translocation. In the non-canonical pathway, the inactive precursor of p52/RelB heterodimer is matured into its active form by IKKα phosphorylation and proteasomal processing upon NF-κB-induced kinase (NIK) activation by signals such as CD40L and lymphotoxin (47). A third atypical IKK-independent pathway is mainly triggered by hypoxia and UV radiation and leads to p50/p65 activation (48). NF-κB activation is regulated by several negative loops, including phosphorylation/de-phosphorylation and ubiquitination events. The NF-κB inhibitor IκBα is itself a transcriptional target of NF-κB. Different microRNAs (e.g., miR-146a, miR-302b) can contrast mRNA translation of proteins involved in the NF-κB cascade (49, 50).
Within the context of inflammation and immunity, NF-κB is activated downstream of the toll-like receptor (TLR)-MyD88 pathway that senses both pathogen-associated molecular patterns (PAMPs), such as LPS and other microbial products, and damage-associated molecular patterns (DAMPs), which are released by either stressed or dying host cells (51). NF-κB positively regulates the expression of inflammatory cytokines (TNFα, IL-1, IL-6), chemokines (CCL2, CCL5, CXCL8), adhesion molecules (VCAM1, ICAM1), angiogenic (VEGF), and anti-apoptotic factors (BCL-2, BCL-XL, FLIP), enzymes required for prostaglandin and NO synthesis (COX2, iNOS) (52). Furthermore, NF-κB is activated downstream of both the T-cell receptor (TCR) and B-cell receptor (BCR), sustaining the adaptive immune response (e.g., by controlling IL-2 expression) (53). NF-κB is also involved in NK-cell activation regulating IFN-γ production (54).
Within the context of cancer, NF-κB activation can support the neoplastic process (55, 56). One of the earliest pieces of evidence dates back to the discovery of the retroviral oncogene v-Rel (57). Furthermore, NF-κB can induce the transcription of the mitogenic factors MYC and Cyclin-D (58, 59). Finally, the AKT-mediated NF-κB activation, which frequently occurs in tumors, promotes cell survival and contributes to chemotherapy resistance of cancer cells (60). Remarkably, NF-κB activation in cells of the tumor immune infiltrate can have both anti-tumoral and pro-tumoral consequences depending on the immune cell type. On one hand, T cells, NK cells, and NKT cells require NF-κB for their anti-tumoral effector activity (61–63), but on the other hand, NF-κB sustains Treg and myeloid-derived suppressor cell (MDSC) activity, resulting in pro-tumoral effects (64–66).
NF-κB-Mediated Expression of PD-L1 by Cancer Cells
PD-L1 expression is often observed in tumors and correlates with aggressive behavior and poor prognosis. Whether PD-L1 is expressed by cancers cells, especially at the later stages of the disease, as negative feedback of a chronic inflammation process intertwined with cancer progression or as a consequence of cell selection by the immune system is still unclear. Certainly, PD-L1 expression confers a selective advantage to cancer cells, e.g., by enabling them to avoid host immune response by activated CD8 T lymphocytes and NK cells.
Considering the key role played by NF-κB in inflammation, immunity, and cancer, perhaps it is not surprising that NF-κB regulates PD-L1 expression in tumors, either directly at the transcriptional level or via indirect mechanisms. Different binding sequences for NF-κB have been described on the promoter of the PD-L1 gene (Table 1). The canonical consensus for NF-κB DNA binding, named κB, consists of a nearly palindromic sequence, 5′-GGGRNWYYCC-3′ (where R: purine, Y: pyrimidine, W: adenine or thymine, and N: any base), which recently has been broadened to 5′-GGGNNNNNCC-3′ (32, 71, 72).
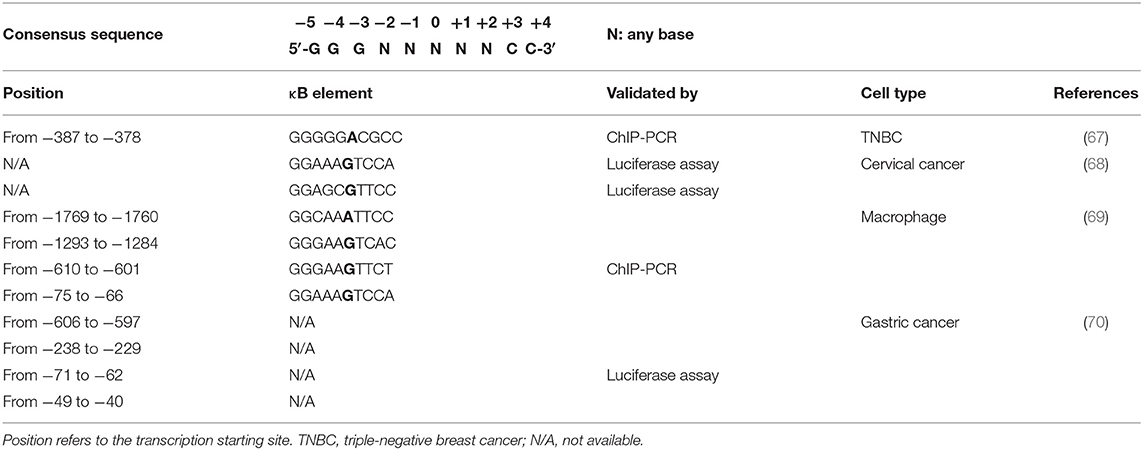
Table 1. Predicted binding sites for NF-κB on the PD-L1 gene promoter.
PD-L1 expression by cancer cells can be related to endogenous oncogenic pathways or oncogenic virus infection. In addition, PD-L1 expression by either cancer cells or tumor infiltrating cells can be driven by different kinds of exogenous stimuli, including cellular stress, e.g. stress induced by UV exposure or chemotherapy, as well as pro-inflammatory cytokines (such as TNFα and IFN-γ) in the tumor bed (Figure 1 and paragraphs below).
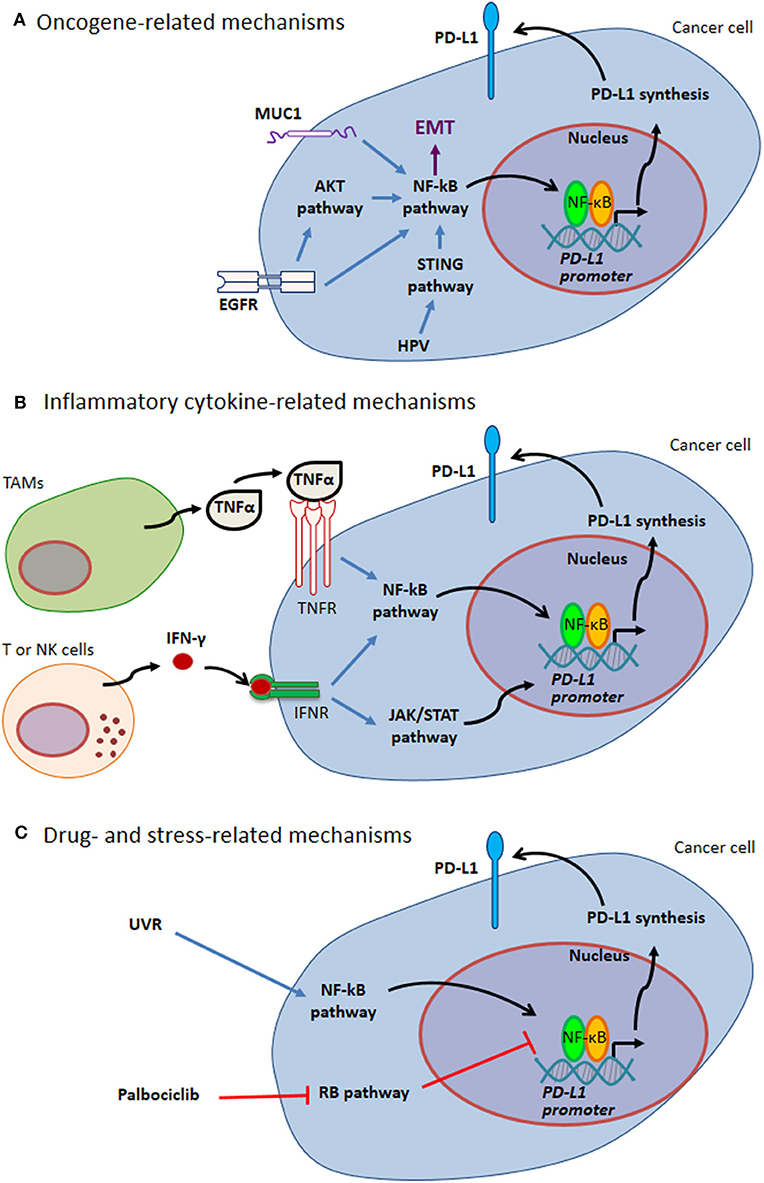
Figure 1. Mechanisms of PD-L1 expression through NF-κB. (A) Oncogene-related mechanisms. MUC1 and EGFR up-regulate PD-L1 expression by activating NF-κB pathway. These pathways are intertwined with EMT. HPV modulates PD-L1 expression triggering STING that in turn activates NF-κB. (B) Inflammatory cytokine-related mechanisms. Tumor-infiltrating immune cells can produce several cytokines regulating PD-L1 expression. Two well-known cytokines acting via NF-κB pathway are TNFα produced by TAMs and IFN-γ produced by tumor infiltrating T and NK cells. (C) Drug- and stress-related mechanisms. Different drugs act on NF-κB transcriptional activity (e.g., Palblociclib). Stress response to UVR activates NF-κB, thus mediating PD-L1 up-regulation. Blue arrows indicate activation of NF-kB pathway; black arrows indicate NF-κB-mediated PD-L1 up-regulation; red T-arrows indicate negative regulation. EGFR, Epidermal Growth Factor Receptor; EMT, Epithelial-Mesenchymal Transition; HPV, Human Papilloma Virus; IFN, Interferon; JAK, Janus Kinase; MUC1, Mucin 1; NK, Natural Killer; PD-L1, Programmed Cell Death Protein 1 Ligand; RB, Retinoblastoma; STAT, Signal Transducer and Activator of Transcription; STING, Stimulator of Interferon Genes; TAMs, Tumor Associated Macrophages; TNF, Tumor Necrosis Factor; UVR, ultraviolet radiation.
Oncogene-Related Mechanisms
A direct link between the epidermal growth factor receptor (EGFR) and PD-L1 expression via NF-κB has been described in non-small-cell lung cancer (NSCLC), the most common lung cancer (73, 74). EGFR mutations associated with constitutive tyrosine kinase-mediated phosphorylation lead to NF-κB activation along with PD-L1 overexpression. Accordingly, the tyrosine kinase inhibitor gefitinib, approved for NSCLC treatment, reduces PD-L1 levels by inhibiting the NF-κB pathway. Notably, even wild-type EGFR induces PD-L1 up-regulation after stimulation with EGF (73). Mechanistically, EGFR triggers ERK, AKT, and IκBα phosphorylation, resulting in HIF-1α and PD-L1 accumulation (74). Activation of AKT is reported to increase HIF-1α protein translation, and it is known that the PD-L1 promoter contains an HIF-1α response element (27, 75, 76). As a consequence, EGFR activation in NSCLC promotes PD-L1 expression directly by phosphorylating IκBα and indirectly via HIF-1α. Furthermore, HIF-1α can support NF-κB pathway activation by driving IKKβ gene transcription through a hypoxia response element present in the promoter and, at the same time, by directly inducing p65 (77, 78). Conversely, it has been reported that NF-κB can induce HIF-1α by binding directly to the HIF-1α promoter (79, 80). Thus, both HIF-1α and NF-κB pathways sustain PD-L1 expression and reinforce each other.
Overexpression of the oncoprotein Mucin 1 (MUC1) is correlated to NF-κB-mediated PD-L1 expression in both NSCLC and triple-negative breast cancer (TNBC) (81, 82). MUC1, through its MUC1-C subunit, activates the PI3K/AKT, MAPK, β-catenin/MYC, and NF-κB pathways (83). NF-κB activation by MUC1 occurs, at least, through three different mechanisms: (i) binding to p65, thus forming a p65/MUC1 transcriptionally active complex (84); (ii) binding to IKKβ-IKKγ complexes, which triggers IκBα phosphorylation (85); and (iii) associating with TGF-β-activated kinase-1 (TAK1), which phosphorylates IKKβ on Ser181 (86). Once activated, NF-κB enhances the expression of MUC1, thus creating a self-sustaining loop. At the same time, MUC1, via the YAP/β-catenin pathway, mediates the induction of the WNT target gene MYC (87). PD-L1 promoter contains both an E-box sequence (CAGCTT) for MYC binding at positions from −164 to −159 nt and a p65-binding site (GGGGGACGCC) at positions from −387 to −378 nt upstream of the transcription-starting site (67) (Table 1). Hence, MUC1 can induce the expression of PD-L1 triggering the occupancy of its promoter by both p65 and MYC.
It is remarkable that the signaling of PD-L1 induction shares important elements with that occurring during epithelial-mesenchymal transition (EMT), a tissue remodeling process typical of advanced epithelial cancers (88). PD-L1 expression is observed during EMT in NSCLC (89). Hypoxia and chronic inflammation are the main drivers of EMT, which is largely mediated by TGF-β1, TNFα, HGF, EGF, and PDGF (90, 91). Interestingly, PD-L1 expression is not regulated by the EMT-specific transcription factor SNAIL, but it involves a non-canonical NF-κB signaling. In particular, TNFα activates IKKε, which leads to p65 recruitment on the PD-L1 promoter. Concomitantly, TGF-β1 reduces DNA methyl transferase-1 (DNMT1) recruitment on the PD-L1 promoter, causing its de-methylation and thus enhancing its transcription. Notably, TNFα and TGF-β1 withdrawal reverts the EMT phenotype and, at the same time, abolishes PD-L1 expression (89). The interconnection between EMT and PD-L1 is evident in the MUC1 signaling with NF-κB as the matchmaker. NF-κB not only drives PD-L1 expression, but also the expression of ZEB1, a transcription factor that is able to mediate EMT (92). ZEB1, in turn, suppresses the transcription of miR-200, an inducer of epithelial differentiation, thus enhancing EMT and PD-L1 expression (93).
Taken together, these findings indicate that PD-L1 expression is a feature of EMT in cancer. Considering that (i) EMT occurs physiologically during embryonic development and (ii) PD-L1 expression can be enhanced by OCT4 and SOX2, two master “stemness” transcription factors driving the expression of genes necessary for the stem cell phenotype (94), it is tempting to speculate that NF-κB drives PD-L1 expression in the context of a general mechanism that has evolved to protect mesenchymal cells and stem cells from immune attack during physiological development.
It is now emerging that PD-L1 not only mediates negative feedback in the context of immune response, but it is also a component of a homeostatic response of epithelial cells to stress (95). In this case, PD-L1 expression can be linked to cellular conditions such as proliferation, adhesion, and migration (22). Supporting this consideration, MUC1, whose role in mammalians is to protect epithelial layers by forming a mucous barrier, senses cell stress and transduces signals that are able to activate the NF-κB pathway, which leads to PD-L1 expression (96). It is therefore convincing that NF-κB-mediated expression of PD-L1 takes part in cancer biology by echoing a process evolved to restore tissue integrity during the epithelial stress response.
Oncogenic viruses are also PD-L1 inducers. For example, infection by papilloma virus promotes PD-L1 expression in cervical cancer cells. Interferon-inducible 16 (IFI16) acts as viral DNA sensor and activates stimulator of IFN genes (STING), leading to TANK-binding kinase-1 (TBK1) activation that, in turn, initiates a cascade signaling that is able to recruit p-p65 on the PD-L1 promoter. The STING/TBK1 pathway activates both IRF3 and NF-κB, with NF-κB mediating a major contribution to PD-L1 gene transcription (68). PD-L1 expression is also observed in NK/T cell lymphoma infected by Epstein-Barr virus. Both MAPK and NF-κB pathways are involved in PD-L1 induction in these cells (97).
Inflammatory Cytokine-Related Mechanisms
NF-κB regulates PD-L1 expression downstream of inflammatory cytokine-induced pathways in the tumor microenvironment, contributing to linking together two immune-related hallmarks of cancer, i.e., tumor-promoting chronic inflammation and immune escape (98). IFN-γ and TNFα will be considered here in more detail.
IFN-γ is one of the most studied PD-L1-inducer inflammatory cytokines, which is produced by highly activated T and NK cells infiltrating the tumor (38, 99). Although it is well-established that IFN-γ signals via JAK/STAT (100), there is evidence that IFN-γ can also activate the NF-κB pathway, which in turn mediates PD-L1 up-regulation. For example, in melanoma cells, IFN-γ inducible expression of PD-L1 is linked to the activity of p50 and p65, and not to the activation of the interferon-related STAT factors (101). Moreover, inhibitors of the bromodomain and extra terminal (BET) proteins, a class of epigenetic regulators with immunomodulatory activity (102), reduce PD-L1 expression by inhibiting p50 transcription (103). Transcriptional up-regulation of PD-L1 gene by NF-κB occurs also in clonal blasts from myelodysplastic diseases treated with IFN-γ and TNFα (104). Notably, also type I IFN, which can be produced by many types of cell stimulated by either PAMPs or DAMPs, is a PD-L1 inducer (105).
TNFα, which can be released by activated tumor associated macrophages (TAMs), is a major driver of inflammation and one of the main inducers of NF-κB in tumor microenvironment. It has been mentioned above that TNFα drives EMT and regulates PD-L1 expression (see previous paragraph). Apart from EMT, TNFα, IL-17, or a combination of both, can induce PD-L1 up-regulation via NF-κB (20). Moreover, Lim and colleagues have shown that TNFα-activated NF-κB can regulate PD-L1 post-transcriptionally through an indirect way (106). TNFα binds TNFR on cancer cells and induces, among other pathways, a signaling cascade that promotes p65 nuclear translocation via IKKβ. In the nucleus, p65 trans-activates the COPS5 gene by binding to its promoter. COPS5 codifies for COP9 signalosome 5 (CSN5), which is the catalytic subunit of a large multiprotein complex with deubiquitination activity (107). CSN5 is able to interact and deubiquitinate PD-L1 protein, increasing its stability and consequently its surface expression. The biological relevance of this regulatory mechanism is confirmed by the positive correlation observed in breast cancer specimens between p-p65, CSN5, and PD-L1 expression as well as the inverse correlation with granzyme B, a cytotoxic lymphocyte effector molecule (106). Notably, TNFα, through the activation of the NF-κB pathway, also up-regulates CSN2, which, by blocking the ubiquitination of the transcription factor SNAIL, promotes tumor invasiveness (108). Therefore, the inflammatory cytokine TNFα coordinates both EMT and tumor immune evasion by using NF-κB signaling (see also the previous paragraph). This pathway is negatively regulated by curcumin, a natural anti-inflammatory compound that is known to inhibit NF-κB signaling as well as CSN5 activity (109, 110). Accordingly, it has been shown that curcumin reduces TNFα-mediated PD-L1 stabilization (106).
Drug- and Stress-Related Mechanisms
Drugs currently used in the clinic or in pre-clinical studies can influence PD-L1 expression via NF-κB, also influencing epigenetic regulation. Epigenetic events, changing the chromatin structure via methylation or acetylation/deacetylation, modulate gene expression. For example, taxolo (named also paclitaxel) and gemcitabine induce transient expression of PD-L1 mRNA in ovarian cancer by up-regulating p65, even in the absence of IFN-γ signaling (111). NF-κB nuclear activity is regulated by reversible acetylation/deacetylation of p65 operated by HDAC3 (112). As a result, two histone deacetylase (HDAC) inhibitors, namely resminostat and entinostat, affect NF-κB mediated PD-L1 expression (113).
Palbociclib, a recently developed CDK4/6 inhibitor, promotes PD-L1 protein stabilization and increases PD-L1 gene transcription (114, 115). The latest effect is due to an indirect mechanism involving the retinoblastoma protein (RB), which acts as a negative regulator of NF-κB functions, as follows. Hyper-phosphorylated RB (phosphorylation at S249 and T252) specifically interacts via its N-terminal portion with p65 in the nucleus and, in this manner, blocks NF-κB transcriptional activity, including the NF-κB-dependent transcription of PD-L1. Inhibition of the RB pathway by palbociclib induces the hypo-phosphorylated status of RB, thus enhancing NF-κB-mediated PD-L1 transcription (115).
Solar ultraviolet radiation (UVR) is a common environmental stress for the skin and is largely involved in the carcinogenesis of skin cancers. Besides the well-known mutagenic effects, UVR can establish an immunosuppressive environment by different mechanisms, including CTLA-4 and PD-L1 up-regulation (116). PD-L1 transcriptional up-regulation in melanocytes and melanoma cells has been linked to the transcriptional activity of either NRF2, a regulator of antioxidant proteins, or NF-κB (117, 118). Regarding NF-κB, UVR B exposure causes in keratinocytes and melanocytes the subcellular translocation from the nucleus and release outside the cell of the high mobility group box 1 protein (HMGB1), an early stress response DAMP. HMGB1 acts in an autocrine and/or paracrine fashion and binds the receptor for advanced glycation endproducts (RAGE) leading to downstream kinase TBK1 activation. TBK1, and not TAK1, is involved in starting the NF-κB cascade after UVR B exposure even though TAK1 has been shown to play a role in the DNA-damage induced NF-κB signaling (119). TBK1 phosphorylates its canonical target IRF3 and IKKβ, which, by phosphorylating IκBα, removes its inhibition on p65. PD-L1 promoter contains two putative IFN-stimulated response elements (ISRE) along with NF-κB binding sites. Once activated, p65 forms the canonical p50/p65 heterodimer but also interacts with IRF3 itself forming a complex that is recruited to PD-L1 promoter at the NF-κB binding sites, thus starting PD-L1 gene transcription. In agreement with PD-L1 up-regulation, and possibly mediated also by additional mechanisms, melanoma cells show a reduced susceptibility to CTL-dependent cytotoxicity after UVR B treatment (118).
NF-κB-Mediated Expression of PD-L1 by Tumor Infiltrating Macrophages
Chronic inflammation can pave the way to tumor onset and cancers are often embedded in an inflammatory microenvironment that enhance tumor progression (120). In this context, investigating the regulation of PD-L1 expression by tumor infiltrating cells can shed light on the link between chronic inflammation, tumor progression, and immune escape. TAMs are key cellular players of the tumor infiltrate that can regulate the inflammatory process and, at the same time, act as antigen-presenting cells for CD4 T lymphocytes. Macrophages have heterogeneous phenotypes, ranging from classical M1 to alternative M2 cells, which represent extremes in a continuous spectrum of activation states, with M1 cells having tumoricidal activity and M2 cells favoring tumor progression (121, 122). In addition to the identification of PD-L1+ TAMs (123), PD-L1 has been found highly expressed in MDSCs, a population of tumor-infiltrating myeloid cells implicated in inhibition of other cells of the immune system (124, 125), and a MDSC molecular program linked to NF-κB activation has been related to PD-L1 expression (126).
NF-κB can directly regulates PD-L1 expression in macrophages and other myeloid cells stimulated by inflammatory cytokines (IL-12, IFN-γ), PAMPs, and DAMPs (69, 107, 127, 128). LPS, a prototypical PAMP, can stimulate PD-L1 expression by macrophages via TLR signaling. Indeed, within 1 h after LPS sensing, p65 translocates to the nucleus where it binds to the PD-L1 promoter, inducing PD-L1 gene transcription independently of AP-1 and IRF3, two canonical transcription factors activated by LPS-TLR4 signaling (69). The mechanism whereby LPS promotes PD-L1 gene expression through the transcriptional activity of NF-κB links inflammation to its control, and it is not restricted to macrophages but also occurs in tumors, as demonstrated in gastric cancer cells (70). Furthermore, melanoma-derived extracellular vesicles carrying Heat Shock Protein (HSP)-86, a typical DAMP, can stimulate PD-L1 expression by myeloid cells via TLR4 signaling. Interestingly, a strong NF-κB activation is observed in the immortalized myeloid suppressor cell line MSC-2 stimulated with the extracellular vesicles, and PD-L1 up-regulation is reduced in a dose-dependent manner by the NF-κB inhibitor Bay11-8082 (128).
Despite a direct role for NF-κB in the expression of PD-L1 by macrophages has been demonstrated in response to inflammatory cytokines, NF-κB activation in TAMs is the result of the combined action of both microenvironmental signals and microphysiological conditions (i.e., hypoxia, glucose levels, and pH), and accumulating evidence indicates that different activation states of NF-κB regulate functions and phenotypic heterogeneity of TAMs (121). Moreover, along with tumor-promoting functions, TAMs and monocytic MDSCs share similar molecular traits, such as nuclear accumulation of p50 NF-κB inhibitory homodimer, which drives M2 macrophage polarization and suppressive activity (66, 129, 130). The nuclear accumulation of p50 homodimer hinders the expression of inflammatory cytokines (TNFα, IL-1, IL-6) while increasing anti-inflammatory cytokines (IL-10, TGFβ) and chemokines (CCL17, CCL2), being therefore essential for the resolution of the inflammatory response (131). It thus appears that tumors co-opt transcriptional mechanisms guiding the resolution of inflammation, to promote cancer development. Inhibition of classical NF-κB activation in TAMs has been also observed in response to the M2 polarizing signal TGFβ, through the induced expression of kinase IRAK-M, an inactive serine/threonine kinase that acts as a negative regulator of TLR signaling (132). Of relevance, a recent report has associated M2-like macrophage infiltration with PD-L1 expression in gastric adenocarcinoma (133). Hence, as a key transcriptional component setting the onset and the resolution phase of inflammation, the different forms of NF-κB activation appear as the main regulators of TAMs functional heterogeneity, including their suppressive activity mediated by PD-L1.
Concluding Remarks: Translational Implications
NF-κB, being a master regulator of inflammation, represents a link between immune response and cell growth (134). According to this view, inhibiting NF-κB signaling might counteract inflammation, tumor growth, and possibly reduce PD-L1 expression. NF-κB pathway is the primary or secondary target of different currently used drugs for the treatment of multiple myeloma (135). IKK inhibitors are commercially available and have been tested in preclinical studies for the management of different tumoral and inflammatory pathologies [for comprehensive reviews on the therapeutic implications of IKK targeting see (136, 137)]. Nevertheless, several concerns exist regarding the administration of NF-κB inhibitors due to their pleiotropic effects. To address this issue, current approaches comprise intermittent administration and use as adjuvant therapy. Alternative approaches that can be considered to reduce unwanted effects include either targeting components of the NF-κB pathway other than IKK or modulating NF-κB-dependent downstream effectors. In brief, NF-κB inhibition could be especially relevant in the context of cancer immunotherapies aiming to prevent PD-L1 overexpression and to modulate TAM survival/polarization.
Conversely, immunotherapy based on immune checkpoint inhibition (ICI) has revolutionized cancer treatment and PD-1/PD-L1 axis is the target of different monoclonal antibodies approved for human use (Pembrolizumab and Nivolumab are approved anti-PD-1; Atezolizumab, Avelumab, Durvalumab are approved anti-PD-L1). It is still unclear if they have different activity and/or toxicity. The majority of these compounds are engineered in order to prevent antibody-dependent cell cytotoxicity (ADCC), but Avelumab, an anti-PD-L1 IgG1 isotype, is able to perform both immune checkpoint inhibition and ADCC so that TAMs, MDSCs, and Treg, which can express high levels of PD-L1 in the tumor infiltrate, can be targeted as well as cancer cells (35, 138, 139). Immune-related adverse effects (irAEs) that resemble autoimmune responses occur during ICI therapy. Indeed, breakdown of the homeostatic PD-1/PD-L1 axis can provoke colitis, hepatitis, endocrinopathies, kidney injury, and skin problems (4, 140).
PD-L1 expression, as evaluated by immunohistochemistry, is routinely used as a biomarker for patient eligibility to anti-PD-1/PD-L1 therapy, and it is the only reliable molecular biomarker nowadays. Nevertheless, responsiveness to therapy does not mirror PD-L1 expression, and unfortunately a few PD-L1+ patients undergo hyper-progressive disease after ICI therapy (141–143). These discrepancies can be ascribed to several issues, including technical limitations of PD-L1 expression analysis, intra-tumoral heterogeneity, tumor mutational burden, inefficient priming of anti-tumoral T cells, and inadequate T-cell responses due to either tumor-intrinsic (compensatory immune checkpoints) or tumor-extrinsic (immune suppressive milieu) factors (144). It remains to be determined whether there are differences among individual drugs targeting PD-1/PD-L1 axis that are relevant for their clinical use and whether some patients would better benefit of either anti-PD-1 or anti-PD-L1 treatment. Although blocking either PD-L1 or PD-1 should similarly inhibit their molecular interaction, it is conceivable that, at least in some anti-PD-L1-treated patients, unblocked PD-L2 activity on PD-1 receptor can still inhibit anti-tumor response. Furthermore, considering that anti-CTLA-4 therapeutic efficacy can heavily depend on depletion of intra-tumoral CTLA-4+ Treg by ADCC (145), it is possible that anti-PD-1/PD-L1 therapy efficacy could similarly depend on the depletion of either PD-1+ and/or PD-L1+ tumor-infiltrating Treg, at least in those patients in which these types of inhibitory cells dominate suppression of anti-tumor response. Accordingly, patient-tailored anti-PD-1/PD-L1 therapy is a subject of intense investigation [for example, see (146)].
It is conceivable that it is not PD-L1 expression per se, but rather the tumor microenvironment that induces PD-L1, that accounts for therapy success. A more comprehensive characterization of the tumor environment in terms of cytokine milieu, type of lymphocyte infiltration, macrophage phenotype would lead to improved approaches, most likely involving combined therapies (147, 148).
In this regard, the NF-κB state of activation, rather than PD-L1 alone, could have a prognostic value, as recently suggested by a detailed investigation of different types of human cancers, which reports how the local immune landscape drives clinical outcome (38). In patients with cancer, a positive or negative prognostic value has been correlated to the activation state of STAT1 and NF-κB pathways, respectively, despite the signaling of either being enough to lead to PD-L1 expression (38). It is tempting to speculate that the STAT factors, evolved in response to viral infections as components of the IFN-activated pathways that limit viremia, have anti-cancer features mostly by mediating pro-apoptotic effects. In contrast, NF-κB, evolved to regulate inflammation and tissue healing, and thus supporting cell survival and proliferation, drives pro-survival functions in cancer settings.
PD-L1 expression controls the strength of the immune response and acts as a rheostat of inflammation. Unfortunately, this mechanism is exploited by cancer cells to perform immune evasion. PD-L1 regulation during tumor progression evokes its physiological modulation, as discussed in the present review. In this scenario, uncovering the NF-κB-mediated regulation of PD-L1 in tumors can pave the way towards tailored therapeutic approaches targeting the PD-1/PD-L1 axis.
Author Contributions
FA and FD searched for literature articles, conceived, and wrote the manuscript. AN, MG, ASi, and ASa revised and critically contributed to the manuscript drafting. FA, AN, MG, ASi, ASa, and FD approved the final version of the manuscript.
Funding
Work supported by the Associazione Italiana Ricerca sul Cancro (AIRC) IG number 19885; AIRC 5 x 1000 number 22757; Fondazione Cariplo, and Ministero Università Ricerca (MIUR) (project: 2017BA9LM5_001); Associazione Augusto per la Vita, and Associazione Medicine Rocks (ASi's laboratory); AIRC 5 x 1000 number 21147 (ASa's laboratory); MIUR project: 2017K55HLC_006 (FD's laboratory). MG received institutional clinical research funding from Eli Lilly, Otsuka Pharma, Astra Zeneca, Novartis, BMS, Roche, Pfizer, Celgene, Incyte, Tiziana Sciences, Clovis, Merck Serono, Bayer, MSD, GlaxoSmithKline S.p.A., Spectrum Pharmaceuticals, Blueprint Medicine, Merck KGaA, BAYER, IPSEN, MedImmune, EXELISIS. The funder bodies were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
MG reports personal fees as consultant/member of advisory board from Eli Lilly, Boehringer Ingelheim, Otsuka Pharma, Astra Zeneca, Novartis, BMS, Roche, Pfizer, Celgene, Incyte, Inivata, Takeda, Bayer, MSD, GlaxoSmithKline S.p.A., Sanofi-Aventis, Spectrum Pharmaceuticals, Blueprint Medicine, Seattle Genetics, Daiichi Sankyo, Jannesen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity. (2016) 44:955–72. doi: 10.1016/j.immuni.2016.05.002
2. Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur J Immunol. (2007) 37:2405–10. doi: 10.1002/eji.200737461
3. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. (2020) 21:442–54. doi: 10.1038/s41590-020-0620-x
4. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of PD-1/PD-L pathway beyond cancer. Front Immunol. (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
5. Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. (2017) 5:106–17. doi: 10.1158/2326-6066.CIR-16-0391
6. Di Rosa F, Francesconi A, Di Virgilio A, Finocchi L, Santilio I, Barnaba V. Lack of Th2 cytokine increase during spontaneous remission of experimental allergic encephalomyelitis. Eur J Immunol. (1998) 28:3893–903. doi: 10.1002/(SICI)1521-4141(199812)28:12%3C3893::AID-IMMU3893%3E3.0.CO;2-%23
7. Di Rosa F, Serafini B, Scognamiglio P, Di Virgilio A, Finocchi L, Aloisi F, et al. Short-lived immunization site inflammation in self-limited active experimental allergic encephalomyelitis. Int Immunol. (2000) 12:711–9. doi: 10.1093/intimm/12.5.711
8. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
9. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. (1996) 8:765–72. doi: 10.1093/intimm/8.5.765
10. Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. (2014) 40:235–47. doi: 10.1016/j.immuni.2013.11.017
11. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
12. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. (2001) 291:319–22. doi: 10.1126/science.291.5502.319
13. Antonangeli F, Soriani A, Cerboni C, Sciume G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol. (2017) 8:1583. doi: 10.3389/fimmu.2017.01583
14. Antonangeli F, Zingoni A, Soriani A, Santoni A. Senescent cells: living or dying is a matter of NK cells. J Leukoc Biol. (2019) 105:1275–83. doi: 10.1002/JLB.MR0718-299R
15. Mariotti FR, Quatrini L, Munari E, Vacca P, Moretta L. Innate lymphoid cells: expression of PD-1 and other checkpoints in normal and pathological conditions. Front Immunol. (2019) 10:910. doi: 10.3389/fimmu.2019.00910
16. Mariotti FR, Quatrini L, Munari E, Vacca P, Tumino N, Pietra G, et al. Inhibitory checkpoints in human natural killer cells: IUPHAR review 28. Br J Pharmacol. (2020) 177:2889–903. doi: 10.1111/bph.15081
17. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
18. An LL, Gorman JV, Stephens G, Swerdlow B, Warrener P, Bonnell J, et al. Complement C5a induces PD-L1 expression and acts in synergy with LPS through Erk1/2 and JNK signaling pathways. Sci Rep. (2016) 6:33346. doi: 10.1038/srep33346
19. Wang X, Ni S, Chen Q, Ma L, Jiao Z, Wang C, et al. Bladder cancer cells induce immunosuppression of T cells by supporting PD-L1 expression in tumour macrophages partially through interleukin 10. Cell Biol Int. (2017) 41:177–86. doi: 10.1002/cbin.10716
20. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
21. Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, et al. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. (2017) 22:1026–33. doi: 10.1007/s10147-017-1161-7
22. Ritprajak P, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol. (2015) 51:221–8. doi: 10.1016/j.oraloncology.2014.11.014
23. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. (2006) 580:755–62. doi: 10.1016/j.febslet.2005.12.093
24. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. (2007) 110:296–304. doi: 10.1182/blood-2006-10-051482
25. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. (2007) 13:84–8. doi: 10.1038/nm1517
26. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA. (2008) 105:20852–7. doi: 10.1073/pnas.0810958105
27. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. (2014) 74:665–74. doi: 10.1158/0008-5472.CAN-13-0992
28. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. (2016) 352:227–31. doi: 10.1126/science.aac9935
29. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. (2016) 16:2829–37. doi: 10.1016/j.celrep.2016.08.032
30. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
31. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. (2017) 109:djw199. doi: 10.1093/jnci/djw199
32. Mulero MC, Wang VY, Huxford T, Ghosh G. Genome reading by the NF-kappaB transcription factors. Nucleic Acids Res. (2019) 47:9967–89. doi: 10.1093/nar/gkz739
33. Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, et al. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine. (2011) 56:231–8. doi: 10.1016/j.cyto.2011.06.004
34. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. (2008) 111:3635–43. doi: 10.1182/blood-2007-11-123141
35. Lecis D, Sangaletti S, Colombo MP, Chiodoni C. Immune checkpoint ligand reverse signaling: looking back to go forward in cancer therapy. Cancers. (2019) 11:624. doi: 10.3390/cancers11050624
36. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
37. Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual faces of IFNgamma in cancer progression: a role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin Cancer Res. (2016) 22:2329–34. doi: 10.1158/1078-0432.CCR-16-0224
38. Wei Y, Zhao Q, Gao Z, Lao XM, Lin WM, Chen DP, et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J Clin Invest. (2019) 129:3347–60. doi: 10.1172/JCI127726
39. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. (2017) 547:413–8. doi: 10.1038/nature23270
40. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. (2018) 359:770–5. doi: 10.1126/science.aao1710
41. Betzler AC, Theodoraki MN, Schuler PJ, Doscher J, Laban S, Hoffmann TK, et al. NF-kappaB and its role in checkpoint control. Int J Mol Sci. (2020) 21:3949. doi: 10.3390/ijms21113949
42. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. (1986) 46:705–16. doi: 10.1016/0092-8674(86)90346-6
43. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
44. Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res. (2011) 21:223–44. doi: 10.1038/cr.2011.13
45. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
46. Rossi MN, Pascarella A, Licursi V, Caiello I, Taranta A, Rega LR, et al. NLRP2 regulates proinflammatory and antiapoptotic responses in proximal tubular epithelial cells. Front Cell Dev Biol. (2019) 7:252. doi: 10.3389/fcell.2019.00252
47. Hostager BS, Bishop GA. CD40-mediated activation of the NF-kappaB2 pathway. Front Immunol. (2013) 4:376. doi: 10.3389/fimmu.2013.00376
48. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. (2005) 30:43–52. doi: 10.1016/j.tibs.2004.11.009
49. Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol. (2011) 12:709–14. doi: 10.1038/ni.2055
50. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol. (2017) 18:861–9. doi: 10.1038/ni.3772
51. Karin M, Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol. (2000) 12:85–98. doi: 10.1006/smim.2000.0210
52. Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
53. Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol. (2010) 2:a000182. doi: 10.1101/cshperspect.a000182
54. Tato CM, Mason N, Artis D, Shapira S, Caamano JC, Bream JH, et al. Opposing roles of NF-kappaB family members in the regulation of NK cell proliferation and production of IFN-gamma. Int Immunol. (2006) 18:505–13. doi: 10.1093/intimm/dxh391
55. DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. (2012) 246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x
56. Pires BRB, Silva R, Ferreira GM, Abdelhay E. NF-kappaB: two sides of the same coin. Genes. (2018) 9:24. doi: 10.3390/genes9010024
57. Gilmore TD. Multiple mutations contribute to the oncogenicity of the retroviral oncoprotein v-Rel. Oncogene. (1999) 18:6925–37. doi: 10.1038/sj.onc.1203222
58. Renan MJ. Does NF-kappa B relieve the transcription block in c-myc? Cancer Lett. (1989) 47:1–9. doi: 10.1016/0304-3835(89)90170-5
59. Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. (2001) 12:73–90. doi: 10.1016/S1359-6101(00)00018-6
60. Fahy BN, Schlieman MG, Virudachalam S, Bold RJ. Inhibition of AKT abrogates chemotherapy-induced NF-kappaB survival mechanisms: implications for therapy in pancreatic cancer. J Am Coll Surg. (2004) 198:591–9. doi: 10.1016/j.jamcollsurg.2003.12.005
61. Zhou J, Zhang J, Lichtenheld MG, Meadows GG. A role for NF-kappa B activation in perforin expression of NK cells upon IL-2 receptor signaling. J Immunol. (2002) 169:1319–25. doi: 10.4049/jimmunol.169.3.1319
62. Vallabhapurapu S, Powolny-Budnicka I, Riemann M, Schmid RM, Paxian S, Pfeffer K, et al. Rel/NF-kappaB family member RelA regulates NK1.1- to NK1.1+ transition as well as IL-15-induced expansion of NKT cells. Eur J Immunol. (2008) 38:3508–19. doi: 10.1002/eji.200737830
63. Clavijo PE, Frauwirth KA. Anergic CD8+ T lymphocytes have impaired NF-kappaB activation with defects in p65 phosphorylation and acetylation. J Immunol. (2012) 188:1213–21. doi: 10.4049/jimmunol.1100793
64. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. (2014) 193:2574–86. doi: 10.4049/jimmunol.1400833
65. Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. NF-kappaB c-rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell. (2017) 170:1096–108; e1013. doi: 10.1016/j.cell.2017.08.004
66. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, et al. Tumor-derived prostaglandin E2 promotes p50 NF-kappaB-dependent differentiation of monocytic MDSCs. Cancer Res. (2020) 80:2874–88. doi: 10.1158/0008-5472.CAN-19-2843
67. Maeda T, Hiraki M, Jin C, Rajabi H, Tagde A, Alam M, et al. MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res. (2018) 78:205–15. doi: 10.1158/0008-5472.CAN-17-1636
68. Cai H, Yan L, Liu N, Xu M, Cai H. IFI16 promotes cervical cancer progression by upregulating PD-L1 in immunomicroenvironment through STING-TBK1-NF-kB pathway. Biomed Pharmacother. (2020) 123:109790. doi: 10.1016/j.biopha.2020.110077
69. Huang G, Wen Q, Zhao Y, Gao Q, Bai Y. NF-kappaB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment. PLoS ONE. (2013) 8:e61602. doi: 10.1371/journal.pone.0061602
70. Li H, Xia JQ, Zhu FS, Xi ZH, Pan CY, Gu LM, et al. LPS promotes the expression of PD-L1 in gastric cancer cells through NF-kappaB activation. J Cell Biochem. (2018) 119:9997–10004. doi: 10.1002/jcb.27329
71. Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-kappaB transcription factors: structural views. Oncogene. (1999) 18:6845–52. doi: 10.1038/sj.onc.1203224
72. Wong D, Teixeira A, Oikonomopoulos S, Humburg P, Lone IN, Saliba D, et al. Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. (2011) 12:R70. doi: 10.1186/gb-2011-12-7-r70
73. Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-kappaB. Biochem Biophys Res Commun. (2015) 463:95–101. doi: 10.1016/j.bbrc.2015.05.030
74. Guo R, Li Y, Wang Z, Bai H, Duan J, Wang S, et al. Hypoxia-inducible factor-1alpha and nuclear factor-kappaB play important roles in regulating programmed cell death ligand 1 expression by epidermal growth factor receptor mutants in non-small-cell lung cancer cells. Cancer Sci. (2019) 110:1665–75. doi: 10.1111/cas.13989
75. Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. (2001) 12:363–9.
76. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
77. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
78. Mak P, Li J, Samanta S, Mercurio AM. ERbeta regulation of NF-kB activation in prostate cancer is mediated by HIF-1. Oncotarget. (2015) 6:40247–54. doi: 10.18632/oncotarget.5377
79. Jiang Y, Zhu Y, Wang X, Gong J, Hu C, Guo B, et al. Temporal regulation of HIF-1 and NF-kappaB in hypoxic hepatocarcinoma cells. Oncotarget. (2015) 6:9409–19. doi: 10.18632/oncotarget.3352
80. Yao G, Zhang Q, Doeppner TR, Niu F, Li Q, Yang Y, et al. LDL suppresses angiogenesis through disruption of the HIF pathway via NF-kappaB inhibition which is reversed by the proteasome inhibitor BSc2118. Oncotarget. (2015) 6:30251–62. doi: 10.18632/oncotarget.4943
81. Soliman H, Khalil F, Antonia S. PD-L1 expression is increased in a subset of basal type breast cancer cells. PLoS ONE. (2014) 9:e88557. doi: 10.1371/journal.pone.0088557
82. Bouillez A, Rajabi H, Jin C, Samur M, Tagde A, Alam M, et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene. (2017) 36:4037–46. doi: 10.1038/onc.2017.47
83. Kufe DW. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. (2013) 32:1073–81. doi: 10.1038/onc.2012.158
84. Ahmad R, Raina D, Joshi MD, Kawano T, Ren J, Kharbanda S, et al. MUC1-C oncoprotein functions as a direct activator of the nuclear factor-kappaB p65 transcription factor. Cancer Res. (2009) 69:7013–21. doi: 10.1158/0008-5472.CAN-09-0523
85. Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S, et al. MUC1 oncoprotein activates the IkappaB kinase beta complex and constitutive NF-kappaB signalling. Nat Cell Biol. (2007) 9:1419–27. doi: 10.1038/ncb1661
86. Takahashi H, Jin C, Rajabi H, Pitroda S, Alam M, Ahmad R, et al. MUC1-C activates the TAK1 inflammatory pathway in colon cancer. Oncogene. (2015) 34:5187–97. doi: 10.1038/onc.2014.442
87. Alam M, Bouillez A, Tagde A, Ahmad R, Rajabi H, Maeda T, et al. MUC1-C represses the crumbs complex polarity factor CRB3 and downregulates the hippo pathway. Mol Cancer Res. (2016) 14:1266–76. doi: 10.1158/1541-7786.MCR-16-0233
88. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
89. Asgarova A, Asgarov K, Godet Y, Peixoto P, Nadaradjane A, Boyer-Guittaut M, et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology. (2018) 7:e1423170. doi: 10.1080/2162402X.2017.1423170
90. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. (2006) 7:131–42. doi: 10.1038/nrm1835
91. Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. (2009) 1:303–14. doi: 10.1002/emmm.200900043
92. Rajabi H, Alam M, Takahashi H, Kharbanda A, Guha M, Ahmad R, et al. MUC1-C oncoprotein activates the ZEB1/miR-200c regulatory loop and epithelial-mesenchymal transition. Oncogene. (2014) 33:1680–9. doi: 10.1038/onc.2013.114
93. Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. (2017) 6:e1263412. doi: 10.1080/2162402X.2016.1263412
94. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front Oncol. (2018) 8:386. doi: 10.3389/fonc.2018.00386
95. Cao Y, Zhang L, Ritprajak P, Tsushima F, Youngnak-Piboonratanakit P, Kamimura Y, et al. Immunoregulatory molecule B7-H1 (CD274) contributes to skin carcinogenesis. Cancer Res. (2011) 71:4737–41. doi: 10.1158/0008-5472.CAN-11-0527
96. Rajabi H, Kufe D. MUC1-C oncoprotein integrates a program of EMT, epigenetic reprogramming and immune evasion in human carcinomas. Biochim Biophys Acta Rev Cancer. (2017) 1868:117–22. doi: 10.1016/j.bbcan.2017.03.003
97. Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol. (2016) 9:109. doi: 10.1186/s13045-016-0341-7
98. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
99. Gao Y, Yang J, Cai Y, Fu S, Zhang N, Fu X, et al. IFN-gamma-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int J Cancer. (2018) 143:931–43. doi: 10.1002/ijc.31357
100. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
101. Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS ONE. (2015) 10:e0123410. doi: 10.1371/journal.pone.0123410
102. Abruzzese MP, Bilotta MT, Fionda C, Zingoni A, Soriani A, Petrucci MT, et al. The homeobox transcription factor MEIS2 is a regulator of cancer cell survival and IMiDs activity in Multiple Myeloma: modulation by Bromodomain and Extra-Terminal (BET) protein inhibitors. Cell Death Dis. (2019) 10:324. doi: 10.1038/s41419-019-1562-9
103. Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. (2014) 27:1126–37. doi: 10.1111/pcmr.12282
104. Kondo A, Yamashita T, Tamura H, Zhao W, Tsuji T, Shimizu M, et al. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood. (2010) 116:1124–31. doi: 10.1182/blood-2009-12-255125
105. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
106. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. (2016) 30:925–39. doi: 10.1016/j.ccell.2016.10.010
107. Liu Y, Shah SV, Xiang X, Wang J, Deng ZB, Liu C, et al. COP9-associated CSN5 regulates exosomal protein deubiquitination and sorting. Am J Pathol. (2009) 174:1415–25. doi: 10.2353/ajpath.2009.080861
108. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. (2009) 15:416–28. doi: 10.1016/j.ccr.2009.03.016
109. Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. (2003) 23:363–98.
110. Uhle S, Medalia O, Waldron R, Dumdey R, Henklein P, Bech-Otschir D, et al. Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J. (2003) 22:1302–12. doi: 10.1093/emboj/cdg127
111. Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-kappaB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. (2015) 75:5034–45. doi: 10.1158/0008-5472.CAN-14-3098
112. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. (2001) 293:1653–7. doi: 10.1126/science.1062374
113. Lucas J, Hsieh TC, Halicka HD, Darzynkiewicz Z, Wu JM. Upregulation of PDL1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occurs via HDAC3/p300mediated NFkappaB signaling. Int J Oncol. (2018) 53:1469–80. doi: 10.3892/ijo.2018.4512
114. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. (2018) 553:91–5. doi: 10.1038/nature25015
115. Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-kappaB activation and PD-L1 expression. Mol Cell. (2019) 73:22–35; e26. doi: 10.1016/j.molcel.2018.10.034
116. Kripke ML. Reflections on the field of photoimmunology. J Invest Dermatol. (2013) 133:27–30. doi: 10.1038/jid.2012.234
117. Zhu B, Tang L, Chen S, Yin C, Peng S, Li X, et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene. (2018) 37:4941–54. doi: 10.1038/s41388-018-0314-0
118. Wang W, Chapman NM, Zhang B, Li M, Fan M, Laribee RN, et al. Upregulation of PD-L1 via HMGB1-activated IRF3 and NF-kappaB contributes to UV radiation-induced immune suppression. Cancer Res. (2019) 79:2909–22. doi: 10.1158/0008-5472.CAN-18-3134
119. Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. (2010) 40:75–86. doi: 10.1016/j.molcel.2010.09.010
120. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
121. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
122. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
123. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. (2006) 203:871–81. doi: 10.1084/jem.20050930
124. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
125. Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. (2016) 5:e1247135. doi: 10.1080/2162402X.2016.1247135
126. Fiore A, Ugel S, De Sanctis F, Sandri S, Fracasso G, Trovato R, et al. Induction of immunosuppressive functions and NF-kappaB by FLIP in monocytes. Nat Commun. (2018) 9:5193. doi: 10.1038/s41467-018-07654-4
127. Xiong HY, Ma TT, Wu BT, Lin Y, Tu ZG. IL-12 regulates B7-H1 expression in ovarian cancer-associated macrophages by effects on NF-kappaB signalling. Asian Pac J Cancer Prev. (2014) 15:5767–72. doi: 10.7314/APJCP.2014.15.14.5767
128. Fleming V, Hu X, Weller C, Weber R, Groth C, Riester Z, et al. Melanoma extracellular vesicles generate immunosuppressive myeloid cells by upregulating PD-L1 via TLR4 signaling. Cancer Res. (2019) 79:4715–28. doi: 10.1158/0008-5472.CAN-19-0053
129. Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. (2006) 66:11432–40. doi: 10.1158/0008-5472.CAN-06-1867
130. Consonni FM, Porta C, Marino A, Pandolfo C, Mola S, Bleve A, et al. Myeloid-derived suppressor cells: ductile targets in disease. Front Immunol. (2019) 10:949. doi: 10.3389/fimmu.2019.00949
131. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci USA. (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
132. Standiford TJ, Kuick R, Bhan U, Chen J, Newstead M, Keshamouni VG. TGF-beta-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene. (2011) 30:2475–84. doi: 10.1038/onc.2010.619
133. Harada K, Dong X, Estrella JS, Correa AM, Xu Y, Hofstetter WL, et al. Tumor-associated macrophage infiltration is highly associated with PD-L1 expression in gastric adenocarcinoma. Gastric Cancer. (2018) 21:31–40. doi: 10.1007/s10120-017-0760-3
134. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. (2011) 12:715–23. doi: 10.1038/ni.2060
135. Vrabel D, Pour L, Sevcikova S. The impact of NF-kappaB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. (2019) 34:56–66. doi: 10.1016/j.blre.2018.11.003
136. Paul A, Edwards J, Pepper C, Mackay S. Inhibitory-kappaB kinase (IKK) alpha and nuclear factor-kappaB (NFkappaB)-inducing kinase (NIK) as anti-cancer drug targets. Cells. (2018) 7:176. doi: 10.3390/cells7100176
137. Prescott JA, Cook SJ. Targeting IKKbeta in cancer: challenges and opportunities for the therapeutic utilisation of IKKbeta inhibitors. Cells. (2018) 7:115. doi: 10.3390/cells7090115
138. Hamilton G, Rath B. Avelumab: combining immune checkpoint inhibition and antibody-dependent cytotoxicity. Expert Opin Biol Ther. (2017) 17:515–23. doi: 10.1080/14712598.2017.1294156
139. Zhang N, Tu J, Wang X, Chu Q. Programmed cell death-1/programmed cell death ligand-1 checkpoint inhibitors: differences in mechanism of action. Immunotherapy. (2019) 11:429–41. doi: 10.2217/imt-2018-0110
140. van der Vlist M, Kuball J, Radstake TR, Meyaard L. Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nat Rev Rheumatol. (2016) 12:593–604. doi: 10.1038/nrrheum.2016.131
141. Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin Cancer Res. (2019) 25:989–99. doi: 10.1158/1078-0432.CCR-18-1390
142. Proto C, Ferrara R, Signorelli D, Lo Russo G, Galli G, Imbimbo M, et al. Choosing wisely first line immunotherapy in non-small cell lung cancer (NSCLC): what to add and what to leave out. Cancer Treat Rev. (2019) 75:39–51. doi: 10.1016/j.ctrv.2019.03.004
143. Kocikowski M, Dziubek K, Parys M. Hyperprogression under immune checkpoint-based immunotherapy-current understanding, the role of PD-1/PD-L1 tumour-intrinsic signalling, future directions and a potential large animal model. Cancers. (2020) 12:804. doi: 10.3390/cancers12040804
144. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118:9–16. doi: 10.1038/bjc.2017.434
145. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33:649–63; e644. doi: 10.1016/j.ccell.2018.02.010
146. Duan J, Cui L, Zhao X, Bai H, Cai S, Wang G, et al. Use of immunotherapy with programmed cell death 1 vs programmed cell death ligand 1 inhibitors in patients with cancer: a systematic review and meta-analysis. JAMA Oncol. (2019) 6:375–84. doi: 10.1001/jamaoncol.2019.5367
147. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
Keywords: tumor associated macrophages, T cells, immune checkpoint inhibitors, tumor immunity, non-small-cell-lung cancer, tissue homeostasis, epithelial-mesenchymal transition, inflammation
Citation: Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A and Di Rosa F (2020) Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 11:584626. doi: 10.3389/fimmu.2020.584626
Received: 17 July 2020; Accepted: 25 August 2020;
Published: 25 November 2020.
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Viktor Umansky, German Cancer Research Center (DKFZ), GermanyFernando O. Martinez, University of Surrey, United Kingdom
Copyright © 2020 Antonangeli, Natalini, Garassino, Sica, Santoni and Di Rosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrizio Antonangeli, fabrizio.antonangeli@cnr.it