 Elizabeth R. Aguilera
Elizabeth R. Aguilera Laurel L. Lenz
Laurel L. Lenz- Department of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO, United States
Bacterial and viral pathogens are predominant causes of pulmonary infections and complications. Morbidity and mortality from these infections is increased in populations that include the elderly, infants, and individuals with genetic disorders such as Down syndrome. Immune senescence, concurrent infections, and other immune alterations occur in these susceptible populations, but the underlying mechanisms that dictate increased susceptibility to lung infections are not fully defined. Here, we review unique features of the lung as a mucosal epithelial tissue and aspects of inflammatory and immune responses in model pulmonary infections and co-infections by influenza virus and Streptococcus pneumoniae. In these models, lung inflammatory responses are a double-edged sword: recruitment of immune effectors is essential to eliminate bacteria and virus-infected cells, but inflammatory cytokines drive changes in the lung conducive to increased pathogen replication. Excessive accumulation of inflammatory cells also hinders lung function, possibly causing death of the host. Some animal studies have found that targeting host modulators of lung inflammatory responses has therapeutic or prophylactic effects in these infection and co-infection models. However, conflicting results from other studies suggest microbiota, sequence of colonization, or other unappreciated aspects of lung biology also play important roles in the outcome of infections. Regardless, a predisposition to excessive or aberrant inflammatory responses occurs in susceptible human populations. Hence, in appropriate contexts, modulation of inflammatory responses may prove effective for reducing the frequency or severity of pulmonary infections. However, there remain limitations in our understanding of how this might best be achieved—particularly in diverse human populations.
Introduction
Pulmonary disease constitutes four of the ten leading causes of death in the human population [chronic obstructive pulmonary disease (COPD), lung cancers, pneumonias, and tuberculosis]1. Each of these conditions is also associated with inflammatory reactions. Therefore, a better understanding of lung biology and the control of inflammation in the lungs during infection has potential to substantially impact human health.
The lungs are a vital organ that facilitate efficient transfer of oxygen and carbon dioxide. Their large surface area is comprised of small terminal air sacs called alveoli. In the alveoli, a single layer of epithelial cells separates inhaled air from underlying small capillaries. Maintenance of the alveolar structure and function is thus crucial for proper functioning of the lungs. Breathing exposes the upper respiratory tract and lung alveolar surface to microbes and other environmental substances. At an average of 15 breaths each minute, more than 10,000 L of air passes over airway mucosal surfaces in the course of a day (1). Each liter of air contains hundreds of thousands or even millions of microbes, thus nasal tissues and the lung alveoli may contact upwards of 109 inhaled microbes each day (2). Commensal microbes also inhabit these tissues (3). To protect the lungs from overgrowth or invasion by microbes, the upper respiratory tract is coated with a mucus layer containing antimicrobial peptides and proteins. Mucus traps many inhaled microbes, which are then cleared from the respiratory tract through the activity of ciliated cells (4). The lung luminal (environmental) alveolar surface is similarly coated with a thin layer of liquid surfactant with dissolved proteins and lipids (3, 4). This surfactant adsorbs at the air/water interface to reduce surface tension, maintain lung elasticity, and capture particles from the air. Beneath the surfactant, alveolar macrophages (AMs) patrol the apical surface of epithelial cells to engulf and remove inhaled microbes (1). Though generally effective, certain pathogens can overcome these upper and lower airway defenses. Infection by such pathogens elicits innate immune cell activation and the initiation of inflammatory responses. In this review, we focus on these innate immune players in the context of lung infections.
Bacterial and viral pathogens are common causes of human pulmonary infections and will be the focus of this review. Bacteria that commonly cause human lung infections include Haemophilus influenzae, Staphylococcus aureus, and Streptococcus pneumoniae (5). Viruses that commonly cause human lung infections include Respiratory syncytial virus (RSV) and influenza viruses (5, 6). Co-infections with these bacteria and viruses is also common and is generally associated with more severe disease and a higher incidence of mortality (5). The current review provides an update and expands on elements previously reviewed by others [e.g., (7)]. Fungal pathogens also cause lung infections and co-infections with bacteria or viruses—particularly in immunocompromised individuals and individuals with polymorphisms in innate immune detection systems (8, 9). However, due to space limitations, fungal infections will not be further discussed in this review.
In this review, we provide an overview of the events that occur when innate lung defenses are overwhelmed by viral and/or bacterial pathogens. Our focus is on innate immune players in animal models of influenza A virus (IAV) and/or S. pneumoniae infection, though some relevant human subject studies are also mentioned. The available data support the hypothesis that the nature and magnitude of the inflammatory response contributes to host susceptibility and thus can drive overwhelmingly severe lung infection.
Inflammatory Responses to Pulmonary Infection
The healthy lung houses both epithelial and resident immune cell populations. Resident immune cells typically found in the healthy lung include neutrophils, monocytes, macrophages, dendritic cells, natural killer (NK), and other innate lymphocyte (ILC) populations, as well as B and T cells (10). Of these cell populations, resident AMs are most abundant (Figure 1A). When an invasive pathogen overwhelms AMs and has established an active infection, pathogen-associated molecular pattern (PAMPs) and damage-associated molecular patterns (DAMPs) can engage pattern recognition receptors (PRRs) on these cell populations (10). Ligation of PRRs leads to activation of cellular signaling pathways and the production of soluble interferons (IFNs) which drive expression of IFN-stimulated genes (ISGs) that act in a cell-intrinsic manner to prevent or limit replication of invading pathogens (10). Simultaneously, PRR ligation induces the expression and production of cytokines and chemokines which regulate the activation and recruitment of additional immune and inflammatory cell populations in the lung (10). Recognition of PAMPs by specific PRRs [e.g., toll-like receptors (TLRs) 2 and 4] thus has substantial impact on disease susceptibility and pathogen transmission. In the initial stages of infections, recruited neutrophils, monocytes, and resident AMs are considered the primary effectors of pathogen clearance (Figure 1). The influx of inflammatory myeloid and other immune cells is necessary to contain and kill invasive microbes. However, the recruitment and activities of these cells can also impair gas exchange and cause damage to the lung epithelium. Thus, fine-tuning of these responses is essential for efficient pathogen clearance and to reduce host damage associated with severe lung infections (10). When accumulation of inflammatory cells and fluid in the lung alveoli disrupts their ability to mediate gas exchange, the clinical condition known as pneumonia ensues. Pneumonias occur with increased frequency in infants, the elderly, and/or individuals who are immunocompromised or have specific genetic conditions that include Down syndrome2 (11). This suggests these groups have an inherent impaired ability to combat lung pathogens and/or to constrain the inflammation associated with these infections.
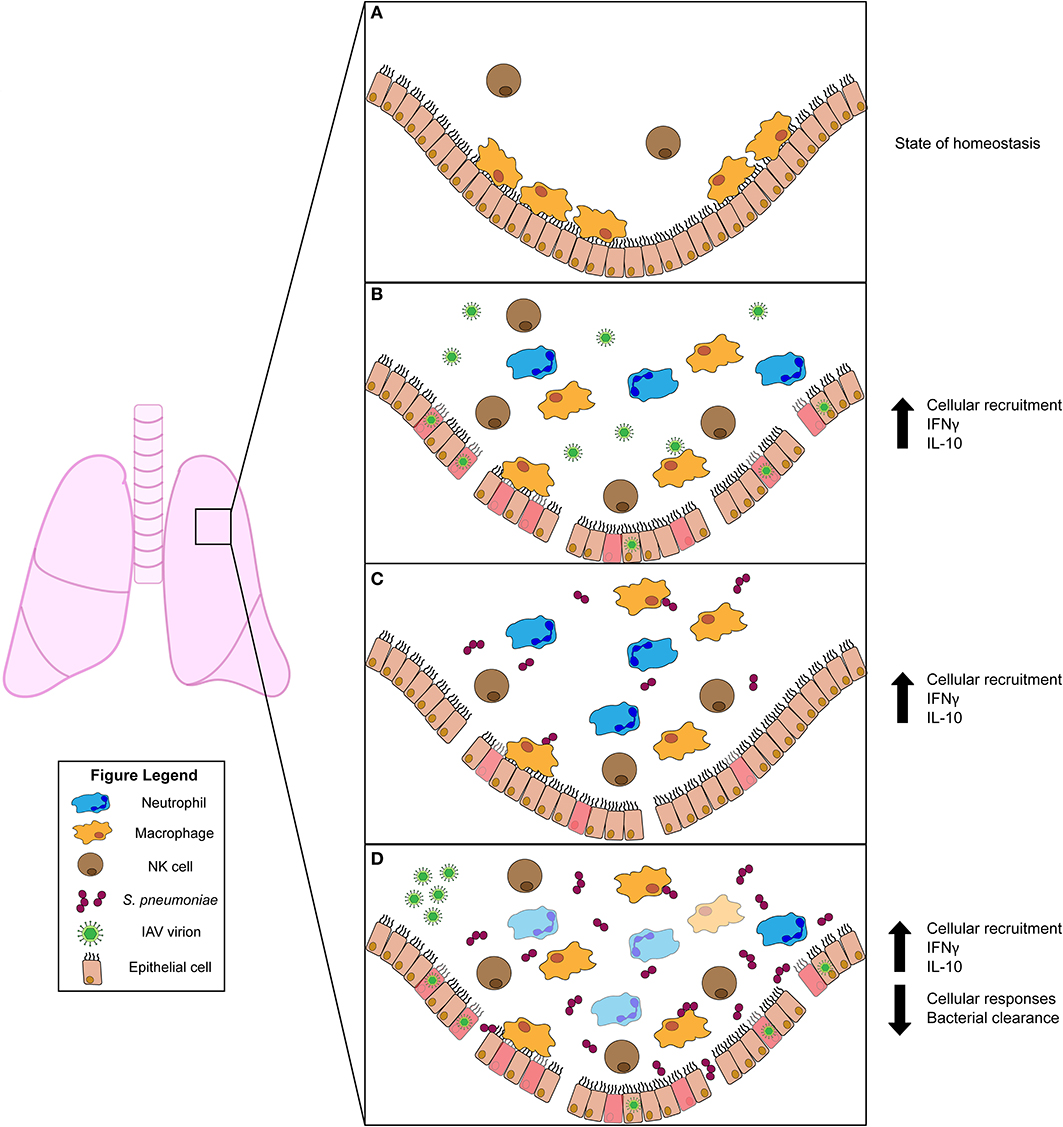
Figure 1. Innate immune responses to viral and/or bacterial infection in the lung. (A) Uninfected lung in homeostatic state harbors resident alveolar macrophages (AMs) and natural killer (NK) cells. (B) Influenza A virus (IAV) infection activates AMs and NK cells. Infected cells produce chemokines that recruit inflammatory phagocytic cells (such as neutrophils) to aid in clearance. The pro-inflammatory cytokine IFNγ is induced during IAV infection but has conflicting roles in animal studies. The anti-inflammatory cytokine IL-10 is produced and plays a role in regulating lung inflammation. (C) Streptococcus pneumoniae infection also stimulates activation of AM and NK cells. NK cells produce IFNγ, which plays a protective role for the host. IL-10 is also induced and modulates the influx of neutrophils into the lung. (D) Primary IAV infection predisposes the host to secondary S. pneumoniae infection. This co-infection drives increased bacterial burdens in the lung. Increased burdens correlate with diminished bacterial clearance by AMs, which is attributed to stimulation of AMs by IFNγ. Similar to S. pneumoniae single infection, IL-10 is also induced during co-infection. This suppresses excessive neutrophil accumulation and subsequent lung damage.
Influenza Virus Infection in the Lung
One of the most common viral pathogens associated with lung disease in humans is influenza virus. Influenza virus is a segmented RNA virus belonging to the Orthomyxoviridae family (12). Influenza, like other RNA viruses have high genetic variability due to poor proofreading activity during replication (12). In addition, co-infection by different influenza viruses increases genetic diversity through reassortment of viral genome segments (12). These, and other factors result in antigenic and pathogenic diversity which limits effectiveness of vaccination. Consequently, influenza viruses continue to pose a serious health risk to the human population with over 3,000,000 cases and up to 650,000 deaths per year globally (average of 25,000–36,000 in the US)3 (6, 13).
Upon surpassing the host's initial physical and chemical barriers to infection (i.e., mucosal layer), influenza virus invades and replicates in lung epithelial cells. Mucus production offers some resistance to IAV infection, but resolution of infection requires innate immune responses (14). Indeed, viral replication in lung epithelial cells leads to cytokine production and activation of AMs that contribute to the initial control of infection (Figure 1B). Thus, mice lacking AMs had increased pulmonary viral burdens and mortality (15, 16). However, AMs are significantly reduced by 4 days after IAV infection (17, 18). Such reductions may partly be driven by cytolytic NK cells, which can recognize and target IAV-infected cells (19, 20). Indeed, activating natural cytotoxicity (NCRs) and other receptors on NK cells have been shown to bind IAV hemagglutinin proteins. When these proteins are expressed at the surface of virus-infected cells, this recognition can induce NK cells to lyse the infected target cell (19–21). Such lysis, and possibility other NK cell effector functions, contribute to early protection, since NK cell depletion increases lung damage and mortality (22). Chemokines produced by IAV-infected cells, such as CCL-1 (MCP), further protect the host by recruiting inflammatory phagocytes that help control infection (23, 24). Recruitment of neutrophils to the lung is further induced by IL-1 and plays an important role in reducing viral replication (25). Thus, effective induction of innate immune responses is critical for host resistance to IAV.
Excessive, inflammatory cell recruitment and the induction of a “cytokine storm” are hallmarks of more severe IAV infection and lung disease. These responses can be exacerbated by polymorphisms in TLRs or other PRRs. The cytokine proteins contributing to this storm include the type I IFNS, IL-1α, IL-1β, IL-6, IL-8, IL-10, IL-15, and the only type II IFN, IFNγ (25–28). Several of these cytokines appear to be beneficial to the host by contributing to host resistance. For example, mice deficient for the interferon alpha/beta receptor, IFNAR, that mediates cellular responses to type I IFNs show increased morbidity and mortality (26, 29). However, excessive production of specific cytokines drives excessive and detrimental inflammation. In particular, IFNγ can exacerbate disease severity during IAV infection. Indeed, mice lacking either expression of IFNγ or the ligand-binding subunit of its receptor, IFNGR1, showed increased survival following IAV challenge (18, 30) The increased survival in mice lacking IFNγ was shown to be associated with reduced immunopathology due to increased activity of type II innate lymphoid cells (ILC2s), which produce IL-5 and amphiregulin to promote tissue homeostasis (30). In this context, IL-5 elicited eosinophils and was required for enhanced survival of the mice lacking IFNγ. In mice lacking IFNGR1, inflammatory infiltrates and cytokine production were also reduced (18). However, in mice lacking IFNGR1 expression viral titers were reduced at 6-8 days after infection, whereas no differences in viral burden were observed up to 9 days after infection in these mice (18, 30) The reasons for this discrepancy are not clear, but could reflect the use of different IAV strains and infection timelines in these studies. Regardless, this collective data support the conclusion that IFNγ drives increased inflammation and lung damage during IAV infection (Figure 1B). Yet, other reports showed therapeutic effects of administering recombinant IFNγ early during IAV infection and demonstrated important protective roles for endogenous IFNγ against IAV during a recall infection or in mice lacking Nos2 expression (31–33). In one study, this protection was attributed to improved NK cell responses (33). Thus, IFNγ signaling to specific cell types and/or in specific settings can have both beneficial or detrimental roles in the response to IAV infection.
Given the potential detrimental effects of pro-inflammatory cytokines such as IFNγ, it is not surprising that anti-inflammatory cytokines are also key regulators of lung damage during IAV infection. IL-10 is a key anti-inflammatory cytokine implicated during IAV infection. A variety of immune cell types can produce IL-10 and respond to this cytokine through expression of the cognate receptor. IL-10 signaling activates STAT3 and other signaling pathways to suppress production of pro-inflammatory factors such as IL-12 and IFNγ (34, 35). In the context of murine IAV infection, IL-10 is important for dampening the pro-inflammatory cytokine response and subsequent pulmonary damage to increase survival of IAV-infected mice (26) (Figure 1B). Thus, the balance of IFNγ and IL-10 responses could be a key determinant of the outcome during IAV infection, with too little IL-10 tipping the balance to excessive IFNγ, inflammation and increased disease severity.
Streptococcus Pneumoniae Bacterial Infection in the Lung
Pulmonary infections are caused by both pathobiont (i.e., asymptomatically residing bacteria with pathogenic potential) and pathogenic (invasive) bacterial species, such as S. pneumoniae. S. pneumoniae (aka pneumococcus) transiently colonizes the nasopharynx asymptomatically in healthy humans with colonization rates highest in children3,4 However, this Gram-positive pathobiont causes ~50% of otitis media cases and is the most common cause of bacterial pneumonia in humans (36). S. pneumoniae can also establish invasive septicemia and meningitis with high mortality rates. In developed countries, pneumoccocal disease rates have dropped considerably in recent years due to vaccination. Nevertheless, nearly 900,000 people develop pneumococcal pneumonia each year in the United States and this remains an important cause of morbidity and mortality globally seen in immune compromised, elderly adults and particularly causing nearly 810,000 deaths in children under 53,4.
Pneumonia occurs when a colonizing S. pneumoniae strain gains access to the lower respiratory tract. Such access is promoted by inflammatory events, which likely contribute to increased density of colonizing S. pneumoniae. Consistent with this, polymorphisms in PRRs has been associated with increased colonization and/or invasive infection by S. pneumoniae (37, 38). Inflammation is thought to reflect an increased nutrient availability following inflammation-driven epithelial damage and increased access of bacteria to adhesion receptors such as those for platelet-activating factor (PAFr) or polymeric immunoglobulin (pIgR), which are upregulated in response to inflammatory cytokines (39–41). Inflammation thus increases the density of S. pneumoniae in the nasopharynx and thus provides an opportunity for increased aerosolization of bacteria into the lungs (for host-derived pneumonia) and environment (for transmission). These or other effects of inflammation may partly explain the increased incidence of pneumococcal pneumonia in individuals with a primary respiratory viral infection, elderly individuals, or other populations (see further information below) (42).
Despite the evidence that aspects of inflammation promote S. pneumoniae colonization, murine infection models have demonstrated protective roles for certain inflammation-associated responses. Two studies reported that the type I IFN response protects mice from colonization and invasive infection following intranasal infection by a serotype 2 strain of S. pneumoniae (43, 44). However, a third report using the different bacterial serotype 3 strain correlated type I IFNs with increased lung bacterial burdens (29). Whether these differing results indicate distinct roles for type I IFNs in protection of distinct tissues or reflect use of distinct S. pneumoniae isolates remains unclear. IFNγ also appears to protect mice against pulmonary S. pneumoniae. Early work found that mice lacking IFNγ were more susceptible to bacteremia and mortality following intranasal infection (45). Treatment with IL-12 was subsequently shown to induce NK cell production of IFNγ and protect mice against pulmonary S. pneumoniae (46). However, the overall impact of NK cells in this setting may not be beneficial as NK cell depletion lowered lung bacterial burdens in infected Scid mice with no effect on burdens in controls (47). NK cell depletion likewise reduced survival of mice infected systemically with another streptococcus strain, S. suis (48). Yet, a more recent study using a genetic diptheria toxin (DT)-based approach to deplete NKp46+ NK cells found that this manipulation reduced mouse survival following pulmonary S. pneumoniae infection (49). Effects of the DT-induced NK cell depletion on bacterial burdens was not reported in the latter study, but the authors showed a transfer of wildtype NK cells improved survival in mice lacking the four-and-a-half LIM-only protein 2 (FHL2) significantly better than transfer of IFNγ-deficient NK cells. Though NK cell specific IFNγ improved survival in this setting, the impact of NK cell IFNγ in wildtype mice is not yet clear. Overall, IFNγ appears to play important roles during S. pneumoniae infection (Figure 1C).
In humans and in murine models, vaccination against pneumococcal capsular polysaccharides or killed bacteria reduces colonization and transmission of S. pneumoniae. In mice, vaccine-induced protection was shown to be mediated by antibody or T cell immune responses (50, 51). Together with complement, opsonizing antibodies increase the ability of neutrophils and other phagocytes to engulf and kill encapsulated pneumococci. Consistent with the importance of neutrophils, IL-17 production and neutrophil recruitment to the lungs reduce bacterial burdens (52). Moreover, protection in IL-12 treated mice correlates with increased neutrophil recruitment or survival in S. pneumoniae-infected lungs (46). Nevertheless, excessive neutrophil recruitment can damage lung function and increase mortality. IL-10 has been shown to dampen the influx of these inflammatory cells, as well as production of pro-inflammatory cytokines such as TNF-α, to reduce tissue damage and mortality during infection (53, 54) (Figure 1C). Supporting the interpretation that this is a key role for IL-10, mortality was increased in mice deficient for IL-10 despite reduced bacterial burdens in the lung and reduced bacterial dissemination (53). Thus, therapeutic strategies that mimic or induce IL-10 may reduce damage to lungs or other vital host tissues, though at risk of increasing bacterial burdens.
Viral-Bacterial Co-Infection in the Lung
Influenza virus infection predisposes the host to severe disease outcomes during co-infection with S. pneumoniae. In this context, viral infection appears to both increase the incidence and the severity of secondary bacterial infections clinically. They are associated with high morbidity and mortality in the context of seasonal flu and were a major correlate of death during 1918 Spanish Flu and 2009 H1N1 pandemics (55–57).
A number of studies have modeled IAV/S. pneumoniae co-infection in mice. Importantly, IAV enhances susceptibility to multiple S. pneumoniae serotypes with more virulent strains exhibiting the highest susceptibility (58). Results of these studies suggest diverse mechanisms contribute to the enhanced susceptibility to secondary bacterial challenge following IAV infection. These mechanisms likely include damage to the lung epithelial barrier, which can permit increased bacterial crossing of the epithelium and may both increase nutrient availability and expose host adhesions such as PAFr or pIgR to increase bacterial numbers (59–61) (Figure 1D). A recent study additionally proposed that viral adherence to the S. pneumoniae surface promotes adhesion to respiratory epithelia (62). Other possible detrimental effects of IAV infection on airway physiology include altered mucus production, reduced ciliary beating and alteration of the host microbiome (63–66). Albeit, there have been conflicting results regarding the impact of influenza infection on the respiratory tract microbiome (67, 68). Yet, both murine and recent human studies agree that an initial influenza (or live-attenuated vaccine) exposure increases susceptibility to secondary (or colonizing) S. pneumoniae (69, 70).
IAV-driven alteration of lung immune defenses have also been implicated in increased susceptibility to S. pneumoniae. Susceptibility and severity of secondary bacterial infections might be impacted by TNF and IL-1β production, which increase expression of S. pneumoniae adhesion receptors such as PAFr and pIgR. However, in a mouse model system, pneumococci administered 7 days after IAV (when reductions in viral burdens were first observed) induced less TNF and IL1β compared to non-IAV-infected mice (71). This study attributed increased susceptibility to an impaired early bacterial clearance from the lung by AMs (71). Following IAV infection, these and other phagocytes showed reduced effectiveness at engulfing bacteria that correlated with the onset of T cell-dependent IFNγ production. Further, burdens of S. pneumoniae at 4 h after infection (9 days after IAV) were ~50% lower in lungs of IFNγ or IFNGR1-deficient mice than in co-infected control mice. Genetic deficiency for IFNγ or IFNGR1 or neutralization of IFNγ also improved survival from secondary pneumococcal infection. These effects correlated with IFNγ-dependent reductions in staining for MARCO on lavaged CD11c+ cells. The MARCO scavenger receptor was previously implicated in the engulfment of non-opsonized S. pneumoniae bacteria by AMs (72). However, while IFNγ stimulation of myeloid cells has been associated with increased phagocytic and bactericidal activity of other bacteria, it has not been demonstrated that detrimental effects of IFNγ in the IAV/S. pneumoniae co-infection model were due to IFNγ targeting of myeloid cells (73–75). Still, there are other lines of evidence supporting the notion that suppression of myeloid cell activity is an important mechanism driving increased susceptibility in IAV-infected animals. Specifically, the increased susceptibility correlates with increases in expression of CD200R, a negative regulator of myeloid cell function (63). Additionally, co-infection induces production of anti-inflammatory IL-10 that suppresses excessive neutrophil accumulation and host resistance (76) (Figure 1D). Type I IFNs also significantly increased the bacterial burdens following secondary exposure to S. pneumoniae, with little to no effect on viral burdens (29). Here, IFNAR expression correlated with reduced production of chemo-attractant CXCL2 and impaired recruitment of neutrophils to the lungs. Thus, inflammatory responses elicited by IAV infection and the induction of endogenous mechanisms for dampening these responses may collectively impair myeloid cell antibacterial activity to exacerbate pneumococcal infections.
Inflammation and Predisposition to Lung Infections
Altered or constitutive inflammatory responses are observed in elderly individuals and in individuals with genetic predispositions such as Down syndrome (DS). These responses may contribute to the high frequency and severity of IAV and S. pneumoniae infections in these populations (11, 77–80). Indeed, DS appears to accelerate aging-associated cellular processes and the health phenotype of individuals with DS overlaps with that of older non-DS individuals (81). Thus, improved understanding of inflammatory responses to lung infections in the context of DS may also provide insights into the causes and possible treatments for these infections in the elderly.
In the context of DS, production of inflammatory cytokines such as TNFα, IL-1β, IL-6, IL-8, IFNα, and IFNγ were elevated in blood samples from DS vs. sibling donors following an ex vivo treatment with IAV (82). In this study, expression of anti-inflammatory IL-10 was not altered in the DS cohort (82). However, IL-10 production was greater in DS blood cells following an ex vivo stimulation with S. pneumoniae (83). Thus, DS may predispose toward elevated IL-10 production during S. pneumoniae infection, which could dampen myeloid cell antibacterial functions and contribute to elevated bacterial burdens in DS patients, similarly observed in mice (54). However, IFNγ was also found to be increased in individuals with DS at specific time points following IAV stimulation (82, 84). This correlates with a trend toward basally increased IFNγ in DS individuals. Nevertheless, elevated production of IFNγ or other cytokines could have detrimental effects on resistance as described in the murine IAV-S. pneumoniae co-infection studies discussed above (Figure 1). Impairment of neutrophil function has also been shown in otherwise healthy individuals with DS (85) Thus, altered inflammatory responses could contribute to susceptibility to lung infections in DS (and elderly) individuals. However, it should be noted that other non-immune mechanisms may contribute to the susceptibility in the DS (and elderly) populations. For example, in the context of DS congenital abnormalities of the respiratory tract and altered ciliary function have also been reported (86).
Conclusions
Pulmonary infections caused by bacterial or viral pathogens are a serious clinical problem to the global human population. This clinical problem is even more concerning for specific susceptible groups including children, the elderly, and individuals with underlying genetic conditions that include Down syndrome. Co-infections of viral and bacterial pathogens can also increase susceptibility and disease severity in the broader immune-competent human population. Exacerbated or altered innate immune and inflammatory responses are characteristic of the above-mentioned susceptible groups and likely play important roles in defining disease outcome. Conflicting results and the difficulty of extrapolating from animal models of infection to human therapy remain and should be considered in the context of efforts to identify and implement specific and effective treatments. Thus, better defining the regulation of lung innate immune responses in susceptible populations and in the context of complex environmental elements (such as the microbiota) are needed to provide avenues for development of new treatments. It is also important to keep in mind the need to appropriately tune the immune and inflammatory mechanisms to minimize damage to lung tissue while ensuring adequate resistance to infections by various pathogen types.
Author Contributions
EA and LL wrote and edited the manuscript.
Funding
Our ongoing research on regulation of innate immune and inflammatory responses in the lung and other tissues was supported by NIH grants R21 AI140499 and R01AI131662 (to LL). LL received Grand Challenge grant funding from the Linda Crnic Institute for Down Syndrome Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We also thank our colleagues for stimulating conversations related to the topics raised in this manuscript.
Footnotes
1. ^https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
2. ^https://www.cdc.gov/pneumococcal/about/facts.html.
3. ^https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal).
4. ^https://www.who.int/en/news-room/fact-sheets/detail/pneumonia.
References
1. Hasenberg M, Stegemann-Koniszewski S, Gunzer M. Cellular immune reactions in the lung. Immunol. Rev. (2013) 251:189–214. doi: 10.1111/imr.12020
2. Tong Y, Lighthart B. The annual bacterial particle concentration and size distribution in the ambient atmosphere in a rural area of the Willamette Valley, Oregon. Aerosol Sci Technol. (2000) 32:393–403. doi: 10.1080/027868200303533
3. Huffnagle GB, Dickson RP. The bacterial microbiota in inflammatory lung diseases. Clin Immunol. (2015) 159:177–82. doi: 10.1016/j.clim.2015.05.022
4. Bustamante-Marin XM, Ostrowski LE. Cilia and mucociliary clearance. Cold Spring Harbor Persp Biol. (2017) 9:a028241. doi: 10.1101/cshperspect.a028241
5. Morris DE, Cleary DW, Clarke SC. Secondary bacterial infections associated with influenza pandemics. Front Microbiol. (2017) 8:1041. doi: 10.3389/fmicb.2017.01041
6. Thompson WW, Shay DK, Weintraub E, Cox N, Anderson LJ, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. J Am Med Assoc. (2003) 289:179–86. doi: 10.1001/jama.289.2.179
7. McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev Microbiol. (2014) 12:252–62. doi: 10.1038/nrmicro3231
8. Alshabani K, Haq A, Miyakawa R, Palla M, Soubani AO. Invasive pulmonary aspergillosis in patients with influenza infection: Report of two cases and systematic review of the literature. Exp Rev Resp Med. (2015) 9:89–96. doi: 10.1586/17476348.2015.996132
9. Kesh S, Mensah NY, Peterlongo P, Jaffe D, Hsu K, Van Den Brink M, et al. TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann N Y Acad Sci. (2005) 1062:95–103. doi: 10.1196/annals.1358.012
10. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
11. Hilton JM, Fitzgerald DA, Cooper DM. Respiratory morbidity of hospitalized children with Trisomy 21. J Paediatr Child Health. (1999) 35:383–6. doi: 10.1046/j.1440-1754.1999.00386.x
12. Taubenberger JK, Morens DM. The pathology of influenza virus infections. Ann Rev Pathol. (2008) 3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316
13. Thompson WW, Weintraub E, Dhankhar P, Cheng PY, Brammer L, Meltzer MIMI, et al. (2009). Estimates of US influenza-associated deaths made using four different methods. Influ Other Respir Viruses. (2008) 3:37–49. doi: 10.1111/j.1750-2659.2009.00073.x
14. Ehre C, Worthington EN, Liesman RM, Grubb BR, Barbier D, O'Neal WK, et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci USA. (2012) 109:16528–33. doi: 10.1073/pnas.1206552109
15. Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. (2014) 10:1004053. doi: 10.1371/journal.ppat.1004053
16. Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol. (2005) 79:14933–44. doi: 10.1128/JVI.79.23.14933-14944.2005
17. Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. (2013) 191:1250–9. doi: 10.4049/jimmunol.1300014
18. Nicol MQ, Campbell GM, Shaw DJ, Dransfield I, Ligertwood Y, Beard PM, et al. (2019). Lack of IFNγ signaling attenuates spread of influenza A virus in vivo and leads to reduced pathogenesis. Virology, 526, 155–164. doi: 10.1016/j.virol.2018.10.017
19. Achdout H, Meningher T, Hirsh S, Glasner A, Bar-On Y, Gur C, et al. Killing of avian and swine influenza virus by natural killer cells. J Virol. (2010) 84:3993–4001. doi: 10.1128/JVI.02289-09
20. Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. (2001) 409:1055–60. doi: 10.1038/35059110
21. Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Euro J Immunol. (2001) 31:2680–9. doi: 10.1002/1521-4141(200109)31:9<2680::AID-IMMU2680>3.0.CO;2-A
22. Stein-Streilein J, Guffee J. In vivo treatment of mice and hamsters with antibodies to asialo GM1 increases morbidity and mortality to pulmonary influenza infection. J Immunol. (1986) 136:1435–41.
23. Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol. (2000) 156:1951–9. doi: 10.1016/S0002-9440(10)65068-7
24. Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, et al. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol. (2006) 77:1817–24. doi: 10.4049/jimmunol.177.3.1817
25. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol. (2005) 79:6441–8. doi: 10.1128/JVI.79.10.6441-6448.2005
26. Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, et al. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res. (2013) 99:230–7. doi: 10.1016/j.antiviral.2013.05.007
27. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. (2008) 133:235–49. doi: 10.1016/j.cell.2008.02.043
28. To KKW, Hung IFN, Li IWS, Lee K, Koo C, Yan W, et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin Infect Dis. (2010) 50:850–9. doi: 10.1086/650581
29. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. (2009) 119:1910–20. doi: 10.1172/JCI35412
30. Califano D, Furuya Y, Metzger DW. Effects of influenza on alveolar macrophage viability are dependent on mouse genetic strain. J Immunol. (2018) 201:134–44. doi: 10.4049/jimmunol.1701406
31. Bot A, Bot S, Bona CA. Protective role of gamma interferon during the recall response to influenza virus. J Virol. (1998) 72:6637–45. doi: 10.1128/JVI.72.8.6637-6645.1998
32. Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD. Rapid interferon γ-dependent clearance of influenza a virus and protection from consolidating pneumonitis in nitric oxide synthase 2- deficient mice. J Exp Med. (1998) 188:1541–6. doi: 10.1084/jem.188.8.1541
33. Weiss ID, Wald O, Wald H, Beider K, Abraham M, Galun E, et al. IFN-γ treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J Interferon Cytokine Res. (2010) 30:439–49. doi: 10.1089/jir.2009.0084
34. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. (2008) 180:5771–7. doi: 10.4049/jimmunol.180.9.5771
35. Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. (2014) 122:177–210. doi: 10.1016/B978-0-12-800267-4.00005-5
36. Donkor ES. Understanding the pneumococcus: transmission and evolution. Front Cell Infect Microbiol. (2013) 4:7. doi: 10.3389/fcimb.2013.00007
37. Vuononvirta J, Toivonen L, Gröndahl-Yli-Hannuksela K, Barkoff AM, Lindholm L, Mertsola J, et al. Nasopharyngeal bacterial colonization and gene polymorphisms of mannose-binding lectin and toll-like receptors 2 and 4 in infants. PLoS ONE. (2011) 6:e26198. doi: 10.1371/journal.pone.0026198
38. Yuan FF, Marks K, Wong M, Watson S, De Leon E, McIntyre PB, et al. Clinical relevance of TLR2, TLR4, CD14 and FcγRIIA gene polymorphisms in Streptococcus pneumoniae infection. Immunol Cell Biol. (2008) 86:268–70. doi: 10.1038/sj.icb.7100155
39. Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. (1995) 377:435–8. doi: 10.1038/377435a0
40. Rijneveld AW, Weijer S, Florquin S, Speelman P, Shimizu T, Ishii S, et al. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor–deficient mice. J Infect Dis. (2004) 189:711–6. doi: 10.1086/381392
41. Zhang JR, Mostov KE, Lamm ME, Nanno M, Shimida S, Ohwaki M, et al. The polymeric immunoglobulin receptor translocates pneumococci across nasopharyngeal human epithelial cells. Cell. (2000) 102:827–37. doi: 10.1016/S0092-8674(00)00071-4
42. Puchta A, Naidoo A, Verschoor CP, Loukov D, Thevaranjan N, Mandur TS, et al. TNF drives monocyte dysfunction with age and results in impaired anti-pneumococcal immunity. PLoS Pathogens. (2016) 12:1005368. doi: 10.1371/journal.ppat.1005368
43. LeMessurier KS, Häcker H, Chi L, Tuomanen E, Redecke V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog. (2013) 9:e1003727. doi: 10.1371/journal.ppat.1003727
44. Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, et al. (2011). Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio, 2. doi: 10.1128/mBio.00016-11
45. Rubins JB, Pomeroy C. Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect Immunity. (1997) 65:2975–7. doi: 10.1128/IAI.65.7.2975-2977.1997
46. Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect Immunity. (2007) 75:1196–202. doi: 10.1128/IAI.01403-06
47. Kerr AR, Kirkham LAS, Kadioglu A, Andrew PW, Garside P, Thompson H, et al. Identification of a detrimental role for NK cells in pneumococcal pneumonia and sepsis in immunocompromised hosts. Microbes Infect. (2005) 7:845–52. doi: 10.1016/j.micinf.2005.02.011
48. Lemire P, Galbas T, Thibodeau J, Segura M. Natural killer cell functions during the innate immune response to pathogenic streptococci. Front Microbiol. (2017) 8:1196. doi: 10.3389/fmicb.2017.01196
49. Baranek T, Morello E, Valayer A, Aimar RF, Bréa D, Henry C, et al. FHL2 regulates natural killer cell development and activation during Streptococcus pneumoniae infection. Front Immunol. (2017) 8:123. doi: 10.3389/fimmu.2017.00123
50. Malley R, Trzcinski K, Srivastava A, Thompson CM, Anderson PW, Lipsitch M. CD4+ T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc Natl Acad Sci USA. (2005) 102:4848–53. doi: 10.1073/pnas.0501254102
51. Roche AM, Richard AL, Rahkola JT, Janoff EN, Weiser JN. Antibody blocks acquisition of bacterial colonization through agglutination. Mucosal Immunol. (2015) 8:176–85. doi: 10.1038/mi.2014.55
52. Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J Clin Invest. (2009) 119:1899–909. doi: 10.1172/JCI36731
53. Peñaloza HF, Nieto PA, Muñoz-Durango N, Salazar-Echegarai FJ, Torres J, Parga MJ, et al. Interleukin-10 plays a key role in the modulation of neutrophils recruitment and lung inflammation during infection by Streptococcus pneumoniae. Immunology. (2015) 146:100–12. doi: 10.1111/imm.12486
54. Van Der Poll T, Marchant A, Keogh CV, Goldman M, Lowry SF. Interleukin-10 impairs host defense in murine. J Infect Dis. (1996) 174:994–1000. doi: 10.1093/infdis/174.5.994
55. Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918 19 influenza pandemic. Emerg Infect Dis. (2008) 14:1193–9. doi: 10.3201/eid1408.071313
56. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:962–70. doi: 10.1086/591708
57. Shieh WJ, Blau DM, Denison AM, DeLeon-Carnes M, Adem P, Bhatnagar J, et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol. (2010) 177:166–75. doi: 10.2353/ajpath.2010.100115
58. Sharma-Chawla N, Sender V, Kershaw O, Gruber AD, Volckmar J, Henriques-Normark B, et al. Influenza A virus infection predisposes hosts to secondary infection with different Streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect Immun. (2016) 84:3445–57. doi: 10.1128/IAI.00422-16
59. Eisenhut M. Influenza Virus Amplifies Interaction Of Polymeric Immunoglobulin Receptor With Pneumococcal Surface Protein A, which mediates invasion by pneumococcus. J Infect Dis. (2010) 201:1272–3. doi: 10.1086/651432
60. McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. (2002) 186:341–50. doi: 10.1086/341462
61. Van Der Sluijs KF, Van Elden LJR, Nijhuis M, Schuurman R, Florquin S, Shimizu T, et al. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol. (2006) 172:7603–09. doi: 10.1152/ajplung.00050.2005
62. Rowe HM, Meliopoulos VA, Iverson A, Bomme P, Schultz-Cherry S, Rosch JW. Direct interactions with influenza promote bacterial adherence during respiratory infections. Nat Microbiol. (2019) 4:1328–36. doi: 10.1038/s41564-019-0447-0
63. Goulding J, Godlee A, Vekaria S, Hilty M, Snelgrove R, Hussell T. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J Infect Dis. (2011) 204:1086–94. doi: 10.1093/infdis/jir467
64. Hanada S, Pirzadeh M, Carver KY, Deng JC. (2018). Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front Immunol. (2011) 9:2640. doi: 10.3389/fimmu.2018.02640
65. Lee KH, Gordon A, Shedden K, Kuan G, Ng S, Balmaseda A, et al. The respiratory microbiome and susceptibility to influenza virus infection. PLoS ONE. (2019) 14:e207898. doi: 10.1371/journal.pone.0207898
66. Yildiz S, Mazel-Sanchez B, Kandasamy M, Manicassamy B, Schmolke M. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome. (2018) 6:9. doi: 10.1186/s40168-017-0386-z
67. Ding T, Song T, Zhou B, Geber A, Ma Y, Zhang L, et al. Microbial composition of the human nasopharynx varies according to influenza virus type and vaccination status. mBio. (2019) 10:4. doi: 10.1128/mBio.01296-19
68. Ramos-Sevillano E, Wade WG, Mann A, Gilbert A, Lambkin-Williams R, Killingley B, et al. The effect of influenza virus on the human oropharyngeal microbiome. Clin Infect Dis. (2019) 68:1993–2002. doi: 10.1093/cid/ciy821
69. de Steenhuijsen Piters WAA, Jochems SP, Mitsi E, Rylance J, Pojar S, Nikolaou E, et al. Interaction between the nasal microbiota and S. pneumoniae in the context of live-attenuated influenza vaccine. Nat Commun. (2019) 10:2981. doi: 10.1038/s41467-019-10814-9
70. Rylance Steenhuijsen Piters J, de WAA, Mina MJ, Bogaert D, French N, Ferreira DM. Two randomized trials of the effect of live attenuated influenza vaccine on pneumococcal colonization. Am J Resp Crit Care Med. (2019) 199:1160–3. doi: 10.1164/rccm.201811-2081LE
71. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat Med. (2008) 14:558–64. doi: 10.1038/nm1765
72. Arredouani M, Yang Z, Ning YY, Qin G, Soininen R, Tryggvason K, et al. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med. (2004) 200:267–72. doi: 10.1084/jem.20040731
73. Crisler WJ, Lenz LL. Crosstalk between type I and II interferons in regulation of myeloid cell responses during bacterial infection. Curr Opin Immunol. (2018) 54:35–41. doi: 10.1016/j.coi.2018.05.014
74. Eshleman EM, Delgado C, Kearney SJ, Friedman RS, Lenz LL. Down regulation of macrophage IFNGR1 exacerbates systemic L. monocytogenes infection. PLoS Pathog. (2017) 13:e1006388. doi: 10.1371/journal.ppat.1006388
75. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Ann Rev Immunol. (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
76. van der Sluijs KF, van Elden LJR, Nijhuis M, Schuurman R, Pater JM, Florquin S, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. (2004) 290:7603. doi: 10.4049/jimmunol.172.12.7603
77. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘Garb-aging.' Trends Endocrinol Metab. (2017) 28:199–212. doi: 10.1016/j.tem.2016.09.005
78. Franceschi C, Campisi J. Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. J Gerontol. (2014) 69:S4–9. doi: 10.1093/gerona/glu057
79. Pérez-Padilla R, Fernández R, García-Sancho C, Franco-Marina F, Aburto O, López-Gatell H, et al. Pandemic (H1N1) 2009 virus and down syndrome patients. Emerg Infect Dis. (2010) 16:1312–4. doi: 10.3201/eid1608.091931
80. Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population-based study. Lancet. (2002) 359:1019–25. doi: 10.1016/S0140-6736(02)08092-3
81. Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, et al. Accelerated epigenetic aging in Down syndrome. Aging Cell. (2015) 14:491–5. doi: 10.1111/acel.12325
82. Broers CJM, Gemke RJBJ, Weijerman ME, Van Der Sluijs KF, Van Furth AM. Increased pro-inflammatory cytokine production in down syndrome children upon stimulation with live influenza a virus. J Clin Immunol. (2012) 32:323–9. doi: 10.1007/s10875-011-9625-4
83. Broers CJM, Gemke RJBJ, Morré SA, Weijerman ME, Van Furth AM. Increased production of interleukin-10 in children with Down syndrome upon ex vivo stimulation with Streptococcus pneumoniae. Pediatr Res. (2014) 75:109–13. doi: 10.1038/pr.2013.173
84. Torre D, Zeroli C, Ferrario G, Casalone R, Broggini M, Agrifoglio L, et al. Serum levels of gamma interferon in patients with Down's syndrome. Infection. (1995) 23:66–7. doi: 10.1007/BF01710065
85. Barroeta O, Nungaray L, Lopez-Osuna M, Armendares S, Salamanca F, Kretschmer RR. Defective monocyte chemotaxis in children with down's syndrome. Pediatr Res. (1983) 17:292–5. doi: 10.1203/00006450-198304000-00013
Keywords: pulmonary inflammation, viral infection, bacterial infection, innate immunity, down syndrome
Citation: Aguilera ER and Lenz LL (2020) Inflammation as a Modulator of Host Susceptibility to Pulmonary Influenza, Pneumococcal, and Co-Infections. Front. Immunol. 11:105. doi: 10.3389/fimmu.2020.00105
Received: 01 November 2019; Accepted: 15 January 2020;
Published: 11 February 2020.
Edited by:
Nadeem Khan, University of North Dakota, United StatesReviewed by:
Muhammad Ammar Zafar, Wake Forest School of Medicine, United StatesSumit Ghosh, The Research Institute at Nationwide Children's Hospital, United States
Copyright © 2020 Aguilera and Lenz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurel L. Lenz, laurel.lenz@cuanschutz.edu