





Abstract
Although phylogenetic inference of protein-coding sequences continues to dominate the literature, few analyses incorporate evolutionary models that consider the genetic code. This problem is exacerbated by the exclusion of codon-based models from commonly employed model selection techniques, presumably due to the computational cost associated with codon models. We investigated an efficient alternative to standard nucleotide substitution models, in which codon position (CP) is incorporated into the model. We determined the most appropriate model for alignments of 177 RNA virus genes and 106 yeast genes, using 11 substitution models including one codon model and four CP models. The majority of analyzed gene alignments are best described by CP substitution models, rather than by standard nucleotide models, and without the computational cost of full codon models. These results have significant implications for phylogenetic inference of coding sequences as they make it clear that substitution models incorporating CPs not only are a computationally realistic alternative to standard models but may also frequently be statistically superior.
Introduction
The growing abundance of available molecular sequence data has inspired research across ecology and evolutionary biology and continues to provide novel insights into a range of biological questions. While the questions are diverse, those using this information are linked by a common challenge: to provide the best estimate of the evolutionary history of their data. Probabilistic modeling of sequence evolution has become the norm in phylogenetic inference (Felsenstein 2001), dominated by maximum likelihood (ML) (Felsenstein 1981) and Bayesian (Yang and Rannala 1997) estimation. While this statistical revolution has clearly had a positive impact, the proliferation of sophisticated evolutionary models has placed a burden on researchers to select the model most appropriate for their data.
As has been repeatedly shown, an inappropriate choice of evolutionary model can affect the outcome of any phylogenetic analysis, for example, by incorrectly estimating tree topology (Penny et al. 1994; Bruno and Halpern 1999), influencing branch length estimation (Posada 2001), and biasing statistical support values (Buckley and Cunningham 2002). Recently, free software packages have provided a simple framework for choosing an evolutionary model (e.g., Modeltest [Posada and Crandall 1998] and variations). Using hierarchical likelihood ratio tests (Goldman 1993) or Akaike information criterion (AIC) tests (Posada and Buckley 2004), these programs iterate through a hierarchical set of evolutionary models to identify the most appropriate model for a data set.
Such objective techniques for model choice have been widely adopted in phylogenetics, to the extent that the use of Modeltest, in particular, is sometimes regarded as a prerequisite for publication of a phylogenetic analysis. While this is an improvement in phylogenetic practice, it has meant that the phylogenetics community has largely overlooked models not included in the Modeltest hierarchy. In particular, models such as GY94, proposed by Goldman and Yang (1994) and Muse and Gaut (1994) and which operate on codons rather than on individual nucleotides, are not considered by Modeltest. Full codon models such as GY94 are computationally expensive compared to standard nucleotide substitution models, which may have contributed to their underrepresentation in the phylogenetic literature. In fact, the use of codon-based models is essentially restricted to parameter estimation on fixed trees for purposes of detecting selection (Yang et al. 2000). Nevertheless, as computational power has increased, codon-based models are becoming a realistic alternative to nucleotide models for phylogenetic inference.
Codon-based models explicitly incorporate information about the genetic code and as such are arguably among the most biologically realistic models of the evolution of coding sequences. Codon models can be divided into two major categories: those like GY94 that specifically incorporate amino acid replacement rates (having codons rather than nucleotides as their states) and those that partition a nucleotide-based model into categories based on codon position (hereafter CP models). CP models are a specific form of the class of models that allow different models of substitution for different partitions of the data (e.g., Yang 1996). While full codon models like the GY94 model biological reality more closely, the CP models are much more computationally efficient.
To investigate whether CP models are more appropriate than standard nucleotide substitution models such as general time reversible with gamma distributed rate heterogeneity and a proportion of invariant sites (GTR + Γ + I), we conducted a comparative survey of nucleotide, CP, and codon substitution models on 283 multiple sequence alignments. Of these, 177 were RNA virus genes with alignments of 10–75 sequences (median 26) ranging from 468 to 2741 bp (median 940) in length (E. C. Holmes, personal communication). The remaining 106 were individual gene alignments constructed using the published genomes of seven Saccharomyces species and the outgroup Candida albicans (Rokas et al. 2003).
We compared six commonly used nucleotide substitution models to four CP models (table 1) and GY94 (see Methods). For each alignment analyzed, we used AIC as the model selection criterion to choose the best fitting of the nucleotide substitution models. We found that models that explicitly consider the genetic code were almost always superior to standard nucleotide models in both RNA virus and yeast protein-coding sequences. All four CP models performed better on average for the 283 alignments analyzed than did GTR + Γ + I. The HKY112 + CP112 + Γ112 model was the best nucleotide model for all but one of the 106 yeast genes, despite having two less parameters than GTR + Γ + I. The remaining yeast gene was best described by GTR + CP123. CP models were chosen above other nucleotide models in 174/177 virus alignments (99%; table 1). Taken together, this strongly suggests that biological information about CP should be used when choosing a substitution model for phylogenetic analysis of protein-coding sequences.
The Nucleotide Models Analyzed
Model | Parameters | CP Model? | In Modeltest? | Best Model (yeast) | Best Model (viruses) |
|---|---|---|---|---|---|
| HKY + I | 5 | No | Yes | 0 | 1 |
| HKY + Γ | 5 | No | Yes | 0 | 0 |
| HKY + Γ + I | 6 | No | Yes | 0 | 0 |
| HKY + CP123 | 6 | Yes | No | 0 | 5 |
| HKY112 + CP112 + Γ112 | 8 | Yes | No | 105 | 117 |
| GTR + I | 9 | No | Yes | 0 | 0 |
| GTR + Γ | 9 | No | Yes | 0 | 1 |
| GTR + Γ + I | 10 | No | Yes | 0 | 2 |
| GTR + CP123 | 10 | Yes | No | 1 | 23 |
| GTR + CP112 + Γ | 10 | Yes | No | 0 | 28 |
Model | Parameters | CP Model? | In Modeltest? | Best Model (yeast) | Best Model (viruses) |
|---|---|---|---|---|---|
| HKY + I | 5 | No | Yes | 0 | 1 |
| HKY + Γ | 5 | No | Yes | 0 | 0 |
| HKY + Γ + I | 6 | No | Yes | 0 | 0 |
| HKY + CP123 | 6 | Yes | No | 0 | 5 |
| HKY112 + CP112 + Γ112 | 8 | Yes | No | 105 | 117 |
| GTR + I | 9 | No | Yes | 0 | 0 |
| GTR + Γ | 9 | No | Yes | 0 | 1 |
| GTR + Γ + I | 10 | No | Yes | 0 | 2 |
| GTR + CP123 | 10 | Yes | No | 1 | 23 |
| GTR + CP112 + Γ | 10 | Yes | No | 0 | 28 |
The Nucleotide Models Analyzed
Model | Parameters | CP Model? | In Modeltest? | Best Model (yeast) | Best Model (viruses) |
|---|---|---|---|---|---|
| HKY + I | 5 | No | Yes | 0 | 1 |
| HKY + Γ | 5 | No | Yes | 0 | 0 |
| HKY + Γ + I | 6 | No | Yes | 0 | 0 |
| HKY + CP123 | 6 | Yes | No | 0 | 5 |
| HKY112 + CP112 + Γ112 | 8 | Yes | No | 105 | 117 |
| GTR + I | 9 | No | Yes | 0 | 0 |
| GTR + Γ | 9 | No | Yes | 0 | 1 |
| GTR + Γ + I | 10 | No | Yes | 0 | 2 |
| GTR + CP123 | 10 | Yes | No | 1 | 23 |
| GTR + CP112 + Γ | 10 | Yes | No | 0 | 28 |
Model | Parameters | CP Model? | In Modeltest? | Best Model (yeast) | Best Model (viruses) |
|---|---|---|---|---|---|
| HKY + I | 5 | No | Yes | 0 | 1 |
| HKY + Γ | 5 | No | Yes | 0 | 0 |
| HKY + Γ + I | 6 | No | Yes | 0 | 0 |
| HKY + CP123 | 6 | Yes | No | 0 | 5 |
| HKY112 + CP112 + Γ112 | 8 | Yes | No | 105 | 117 |
| GTR + I | 9 | No | Yes | 0 | 0 |
| GTR + Γ | 9 | No | Yes | 0 | 1 |
| GTR + Γ + I | 10 | No | Yes | 0 | 2 |
| GTR + CP123 | 10 | Yes | No | 1 | 23 |
| GTR + CP112 + Γ | 10 | Yes | No | 0 | 28 |
A strong log-log relationship was observed between the rate of evolution in the third position relative to the rest of the codon and the ratio of nonsynonymous to synonymous substitutions (ω) estimated under the GY94 model (fig. 1). The strength of this relationship demonstrates that the CP model of nucleotide evolution captures the essential characteristics of the full codon model. Further, it suggests that it may be possible to use this model as an approximate estimator of ω instead of the much more computationally demanding full codon models.
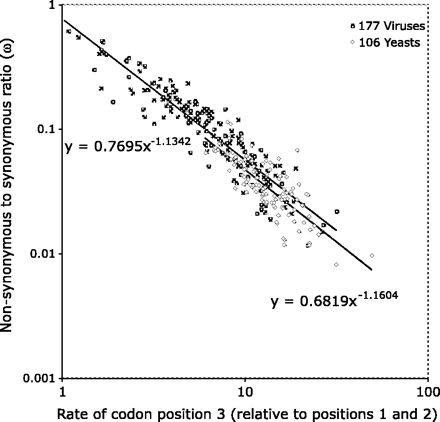
The relationship between ML estimates of the ω parameter of the GY94 model and the relative rate of the third CP in CP models. Closed squares represent virus gene alignments, and open diamonds represent yeast gene alignments. Both axes are plotted in log space, and the black lines are the best fitting power law for the two sets of data. This plot shows that the relative rate of the third CP is a good predictor of the ω parameter.
We have not attempted a systematic investigation of the full range of potential CP models, and it remains likely that superior models to those described here exist. Instead, we have attempted to demonstrate that biologically motivated CP models that are superior to the standard range of nucleotide substitution models can be constructed, with no increase in parametric complexity. We are aware that with only limited effort, the models introduced here can be used in Bayesian phylogenetic inference packages such as MrBayes (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003) and BEAST (available at http://evolve.zoo.ox.ac.uk/beast).
Although the performance of the CP models was impressive, one of the striking results of this analysis was the performance of the GY94 model, which was chosen ahead of all other models in all 106 yeast genes and in 119/177 (67%) virus genes analyzed (table 2). Unfortunately, the options for performing a phylogenetic analysis under the GY94 model are still quite limited. Currently, only MrBayes (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003) caters for this, and its speed is typically two orders of magnitude slower than for nucleotide models. Realistically, use of this model is currently feasible only for small data sets of fewer than 50 taxa. Nevertheless, in light of its excellent fit to real data, the GY94 model should be an obvious choice in situations where computational resources and data size permit.
The Classes of Substitution Models Analyzed and the Number of Data Sets for Which a Model in the Class Was the Best Fitting Model
Model Category | Best Model (yeast) | Best Model (viruses) | Total (%) |
|---|---|---|---|
| Non-CP | 0 | 3 | 1.1 |
| CP | 0 | 55 | 19.4 |
| Codon model | 106 | 119 | 79.5 |
Model Category | Best Model (yeast) | Best Model (viruses) | Total (%) |
|---|---|---|---|
| Non-CP | 0 | 3 | 1.1 |
| CP | 0 | 55 | 19.4 |
| Codon model | 106 | 119 | 79.5 |
The Classes of Substitution Models Analyzed and the Number of Data Sets for Which a Model in the Class Was the Best Fitting Model
Model Category | Best Model (yeast) | Best Model (viruses) | Total (%) |
|---|---|---|---|
| Non-CP | 0 | 3 | 1.1 |
| CP | 0 | 55 | 19.4 |
| Codon model | 106 | 119 | 79.5 |
Model Category | Best Model (yeast) | Best Model (viruses) | Total (%) |
|---|---|---|---|
| Non-CP | 0 | 3 | 1.1 |
| CP | 0 | 55 | 19.4 |
| Codon model | 106 | 119 | 79.5 |
Methods
Throughout the study, a single tree topology was assumed for each alignment. For virus data, ML phylogenies were estimated under the GTR + Γ + I model using PAUP* (Swofford 2003). For yeast genes, the tree estimated from the combined data and reported in Rokas et al. (2003) was used. We tested an array of nucleotide substitution models and GY94 (using codeml from the PAML package; Yang 1997). For each model and alignment, the branch lengths of the respective phylogeny were reestimated along with parameters of the substitution model. PAUP* was used to estimate a nested hierarchy of models commonly used for phylogenetic inference and which are found among those in Modeltest (Posada and Crandall 1998). These include combinations of the HKY85 (Hasegawa, Kishino, and Yano 1985) and GTR (Lanave et al. 1984) models of nucleotide substitution, the discrete gamma model of heterogeneity among sites (Yang 1994), and the invariant site model. We label these models by combining acronyms of the components, for example, GTR + Γ + I in the case of GTR with gamma rate heterogeneity and invariant sites. In addition, we considered models in which the CP are allowed different rates relative to each other, resulting in models we labeled HKY + CP123 and GTR + CP123, with the subscript indicating the rate category for each CP. Although these models are occasionally used in phylogenetic studies, often referred to as site-specific rate models, they are much less common than standard nucleotide substitution models.
Using baseml from the PAML package (Yang 1997), we constructed two additional models that we believed might better represent the biological reality of codon evolution. For these, we grouped first and second CPs into a single partition with respect to third CPs, allowing the latter to have a different rate. For the first model, we assumed that all three CPs shared the same GTR model of substitution and the same shape parameter (α) of the gamma model of rate heterogeneity. This model has exactly the same number of parameters as GTR + Γ + I and is labeled GTR + CP112 + Γ. The final nucleotide model was a combination of HKY + Γ with a different transition-transversion and α parameter along with a relative rate between partitions, resulting in fewer parameters than GTR + Γ + I. This model is labeled HKY112 + CP112 + Γ112. Table 1 summarizes the models investigated.
All authors contributed equally to this work.
Present address: Department of Computer Science, University of Auckland, Auckland, New Zealand.
Peter Lockhart, Associate Editor
We would like to thank E. Holmes for the collection and curation of the 177 virus data sets used in this analysis. This work was funded by the Wellcome Trust and the Royal Society.
References
Bruno, W. J., and A. L. Halpern.
Buckley, T. R., and C. W. Cunningham.
Felsenstein, J.
Goldman, N., and Z. H. Yang.
Hasegawa, M., H. Kishino, and T. Yano.
Huelsenbeck, J. P., and F. Ronquist.
Lanave, C., G. Preparata, C. Saccone, and G. Serio.
Muse, S. V., and B. S. Gaut.
Penny, D., P. J. Lockhart, M. A. Steel, and M. D. Hendy.
Posada, D.
Posada, D., and T. R. Buckley.
Posada, D., and K. A. Crandall.
Rokas, A., B. L. Williams, N. King, and S. B. Carroll.
Ronquist, F., and J. P. Huelsenbeck.
Swofford, D. L.
Yang, Z.
———.
Yang, Z., and B. Rannala.
Yang, Z. H.

{kind=link}