



Abstract
A nonsense mutation c.4250T>A (p.Leu1417X) in the dystrophin gene of a patient with an intermediate phenotype of muscular dystrophy induces partial in-frame skipping of exon 31. On the basis of UV cross-linking assays and pull-down analysis, we present evidence that the skipping of this exon is because of the creation of an exonic splicing silencer, which acts as a highly specific binding site (UAGACA) for a known repressor protein, hnRNP A1. Recombinant hnRNP A1 represses exon inclusion both in vitro and in vivo upon transient transfection of C2C12 cells with Duchenne muscular dystrophy (DMD) minigenes carrying the c.4250T>A mutation. Furthermore, we identified a downstream splicing enhancer in the central region of exon 31. This region functions as a Tra2β-dependent exonic splicing enhancer (ESE) in vitro when inserted into a heterologous splicing reporter, and deletion of the ESE showed that incorporation of exon 31 depends on the Tra2β-dependent enhancer both in the wild-type and mutant context. We conclude that dystrophin exon 31 contains juxtaposed sequence motifs that collaborate to regulate exon usage. This is the first elucidation of the molecular mechanism leading to exon skipping in the dystrophin gene and allowing the occurrence of a milder phenotype than the expected DMD phenotype. The knowledge of which cis-acting sequence within an exon is important for its definition will be essential for the alternative gene therapy approaches based on modulation of splicing to bypass DMD-causing mutations in the endogenous dystrophin gene.
INTRODUCTION
Splicing is carried out by the spliceosome, a large and dynamic complex of proteins and small RNAs (1,2). A crucial question in splicing, which has important consequences for its regulation, is how the splicing machinery recognizes authentic splice sites. Sequences around the splice junctions—the 5′ and 3′ splice site (5′ss and 3′ss)—are clearly important for splice site recognition, but they contain only part of the information required for exon and intron recognition in human transcripts (3,4). The sequence and/or structure context in the vicinity of the 5′ss and 3′ss, especially exonic and intronic enhancers and silencers, play a critical role in splice site recognition, particularly in regulating alternative splicing (5–7). Exonic splicing enhancer (ESE) elements are required for definition and/or efficient splicing of the exons in which they reside, whereas exonic splicing silencer (ESS) elements lead to exon skipping. ESEs are very prevalent and might be present in most, if not all, alternative exons, as well as in multiple constitutive exons. These elements have been identified through the analysis of disease alleles, by site-directed mutagenesis of minigene constructs, or by protocols based on functional SELEX to identify sequences with enhancer activity from a pool of random sequences (8,9). Several different families of ESEs have been recognized, including purine-rich, AC-rich and pyrimidine-rich elements (10,11). The best studied are purine-rich sequences that act by recruiting members of serine/arginine-rich (SR) proteins, a family of essential splicing factors which promote splicing by recruiting spliceosomal components to the correct splice sites (12,13). Under splicing conditions, each SR protein recognizes specific, albeit degenerate and partially redundant, sequence motifs (for review, see 13). In contrast, ESSs inhibit the use of adjacent splice sites, often acting through interactions with members of heterogeneous nuclear ribonucleoprotein (hnRNP) family. A large number of ESS elements has now been identified, but in contrast to ESE motifs, they share little obvious similarity (reviewed in 11,14).
Until recently, splicing mutations were thought to account for ~15% of the point mutations that result in human genetic diseases (15). It is now well established that nonsense, missense and even silent variations may also cause splicing defects, resulting in a disease phenotype (8,9,16). Therefore, a large fraction of disease-causing exonic mutations would in fact affect splicing and most of those, which do not directly alter splice sites are believed to abrogate ESEs (8). Recent data indicate that single-base changes can also function to create negative elements (17).
Duchenne muscular dystrophy (DMD, MIM# 310200) is a lethal X-linked recessive disorder that affects 1 in 3500 males (18) and is caused by mutations of the dystrophin gene (also known as DMD), which encodes a 14 kb mRNA that consists of 79 exons (19). Dystrophin is a cytoskeletal protein that is implicated in membrane stability and in communication between the extracellular matrix and the inner cytoskeleton (20). Approximately, 65% of dystrophin mutations are large duplications or deletions, whereas the remaining cases are because of point mutations (nonsense mutations, small deletions or insertions or splice site mutations) (21–24). The vast majority of mutations responsible for the severe DMD phenotype disrupts the dystrophin mRNA reading frame and/or introduces a stop codon that prematurely ends protein translation leading to a complete loss of functional dystrophin in skeletal muscle (25,26). Mutations in the dystrophin gene that do not disrupt the translational reading frame cause a less severe form of the disease, Becker muscular dystrophy (BMD, MIM#300376), which is found in 1 in every 20 000 newborn boys. In-frame mutations generate some functional gene products, albeit reduced quantity and/or quality, which contribute to the milder phenotype of BMD patients (27). A few cases of exonic mutations associated with exon skipping have been reported in the dystrophin gene. All are nonsense mutations (in exons 27, 29 and 72) that would normally cause a DMD-like phenotype. They have been identified through the absence of correspondence between the presence of a truncating mutation and the mildness of the phenotype in BMD patients, which showed partial skipping of the exon encoding the mutation in the dystrophin transcripts, thus producing an in-frame RNA (28–33). Exon skipping was attributed to the disruption of ESEs, however the detailed mechanisms of the events have not been elucidated.
We have previously reported the c.4250T>A nonsense mutation in the dystrophin gene of a patient with an intermediate phenotype (34). Transcript analysis has shown that this mutation causes partial skipping of exon 31. In this study, we are interested in understanding the molecular mechanisms underlying exon 31 splicing, and elucidating the pathway leading to exon skipping in the presence of the mutation.
RESULTS
The c.4250T>A mutation produces in-frame skipping of exon 31
We previously reported a nonsense exon skipping-associated mutation in the dystrophin gene of a patient with an intermediate phenotype (34). The patient carries a T>A transversion at nt 4250 in dystrophin exon 31 (c.4250T>A) producing an in-frame stop codon (p.Leu1417X) (Fig. 1A). By analysis of muscular transcripts, this mutation was found to induce exon skipping in a proportion of the dystrophin transcripts, thus eliminating the nonsense mutation (Fig. 1B). The skipped exon 31 is in-frame, consistent with the milder phenotype of this 13y11m old patient, who has difficulty in walking but is still ambulant. Accordingly, residual expression of dystrophin was detected both by immunofluorescence and western blot analysis using the Dys2 C-terminal antibody (unpublished data). Consistent with our observation, the c.4250T>A mutation has been found in one additional patient reported to have a BMD phenotype, however no other data is available (Leiden DMD database, http://www.dmd.nl). We quantitated the proportion of skipped transcripts in the patient using a semi-quantitative PCR approach. We found that the transcripts lacking exon 31 detected in our patient represent 17% that of the full-length transcripts according to the measured peak areas of the wild-type and mutated RT–PCR products (Fig. 1C). Mutations that generate premature termination codons can often reduce the steady-state level of mRNA via nonsense-mediated decay (NMD) (35), which could lead to an underestimate of exon inclusion and an exaggeration of the skipping phenotype. However, the full-length transcript containing the premature stop codon was detected in large excess in our patient suggesting that the nonsense mutation in exon 31 may not have a major impact on the level of dystrophin transcripts (see also Discussion). The mutation occurred close to the 5′ end of the exon, 17 base pairs downstream of an acceptor splice site that fits well to the consensus with a calculated consensus value (CV) score of 89.8%, whereas the CV value for the donor site is 74% (36) (Fig. 1A).
Based on the assumption that mutation-associated exon skipping has been mostly associated with ESE disruption, we attempted to predict if the c.4250T>A mutation lies in and eventually abrogates a high score ESE motif in the region encompassing the mutated residue. We analyzed the wild-type and mutant dystrophin exon 31 sequences with two available ESE-prediction softwares, ESEfinder (37) and RESCUE-ESE (3). However, the results were conflicting for the mutation, which was found to decrease the score (from 4.93 to 3.88) of an SRp40 motif without falling below the threshold value (2.13) according to ESEfinder, and to create a novel ESE hexamer according to RESCUE-ESE (Fig. 1D).
A protein of ∼35 kDa in size specifically interacts with the exon 31 mutant sequence
To identify cellular factors binding to the sequence surrounding the mutation, we performed UV cross-linking assays with RNAs containing either wild-type (31wt) or mutant (31mut) exon 31 sequences (Fig. 2A). No significant cross-linking was observed in the S100 cytoplasmic fraction (Fig. 2B). Among the proteins that could be cross-linked to the labelled RNAs in the nuclear extract, a product which migrates at ∼35 kDa interacted significantly more strongly with the 31mut than with the 31wt RNA probe (Fig. 2B, left). Increasing concentration of unlabelled 31mut competing transcripts reduced the intensity of several cross-linked bands, including mostly the 35 kDa protein (unpublished data and Fig. 2D), suggesting that this protein interacts specifically with the 31mut RNA probe. To test if the 35 kDa complex contained SR proteins, we complemented an S100 extract with a preparation of total SR proteins or individual recombinant SR proteins (ASF/SF2, SRp40, 9G8, SC35). As shown in Fig. 2C, the 31mut RNA probe did not show significant cross-linking to ASF/SF2, SRp40, SC35 protein or total SR proteins, whereas it cross-linked very weakly with recombinant 9G8. In striking parallel to what we observed in nuclear extract, cross-linking of the mutant RNA to the ∼35 kDa protein was greatly increased when compared with the wild-type probe in S100 complemented with a 20–40% ammonium sulfate fraction from nuclear extract (NF20–40), which does not contain SR protein (Fig. 2B). Taken together, these results show that the mutation in exon 31 strengthens a natural binding site for a nuclear protein of 35 kDa distinct from SR proteins. This supports the hypothesis that the mutation activates a silencer rather than disrupts an enhancer and that exon skipping is mediated by the recruitment of this nuclear trans-acting factor to the exon.
hnRNP A1 specifically associates with the exon 31 mutated sequence
To further investigate the identity of the 35 kDa protein complex, we tested the ability of various unlabelled RNAs to compete for complex formation (Fig. 2A and D). If we consider the apparent molecular weight of this protein, a likely candidate could be the hnRNP A1 protein, a well-known factor involved in the inhibition of splicing. Moreover, the mutated sequence in exon 31, UAGACA, (Fig. 2A) perfectly matches the hnRNP A1-dependent ESS recently identified in SMN2 exon 7 (17). We designed a 20 nt-RNA probe containing a tandem repeat of the consensus high-affinity hnRNP A1 SELEX motif, UAGGGA/U (38). In addition, we used cold RNA competitors containing SELEX-defined target RNA sequences for 9G8 which displayed weak cross-linking to the 31mut probe in complementation assay, and for two additional SR proteins of ∼35–40 kDa in size (SRp38 and SC35) whose target sequences show similarities with the exon 31 region in the dystrophin gene (Fig. 2A). As shown in Figure 2D, cross-linking of the 35 kDa protein to the 31mut probe was resistant to competition by SRp38, 9G8-102 and SC35-7 cold RNAs, but was strongly and specifically inhibited by the hnRNP A1 competitor. These results strongly suggest that the 35 kDa complex is formed by hnRNP A1, and not the other proteins tested. It is noteworthy that the SRp38 RNA probe which differs by only a single nucleotide from the mutated exon 31 sequence (see Fig. 2A), could not displace hnRNP A1 binding, supporting the essential role of the UAG triplet in the interaction, although it is not sufficient per se (see Discussion).
To unambiguously establish the identity of the 35 kDa protein, we performed RNA affinity experiments using the 31wt and 31mut probes and a non-specific (GST-80) probe (39). RNA-associated proteins were analyzed by SDS-PAGE and silver staining (Fig. 3A). Comparison of the proteins bound to 31mut and 31wt RNAs versus non-specific (GST80) RNA showed the presence of three abundant specific proteins which migrate between 34 and 36 kDa, two of which interacted significantly more strongly with the 31mut RNA than with the 31wt RNA (Fig. 3A). Mass spectrometry and database search with the three excised bands revealed that they were hnRNP A1, its close homologue hnRNP A2 and hnRNP B1. Western blot analysis of the same samples confirmed that hnRNP A1 binds to the 31mut sequence with a higher efficiency than to the 31wt sequence (Fig. 3B). Moreover, in accordance with the results of complementation assay (Fig. 2C), 9G8 bound to the 31wt RNA probe but the mutation (31mut) had no effect on the binding level (Fig. 3B). Contradicting the ESEfinder prediction, only background binding of the SR protein ASF/SF2 was detected with either the 31mut or 31wt sequences. Finally, the ability of hnRNP A1 to bind with the 31wt and 31mut RNA sequences was also confirmed using immunoprecipitations after UV cross-linking and RNase digestion. As expected, the 35 kDa polypeptide was the major cross-linked product immunoprecipitated by a monoclonal antibody specific for hnRNP A1 (Fig. 3C), whereas immunoprecipitation using an antibody specific for 9G8 did not give any detectable signal (data not shown). In conclusion, these data show that exon 31 is bound by hnRNP A1 but the hnRNP A1-binding site is dramatically strengthened by the c.4250T>A mutation.
hnRNP A1 represses exon 31 inclusion in vitro and in vivo
Dystrophin minigenes including exons 30, 31 (wt or mut) and 32 as well as surrounding intronic sequences were prepared as described in Figure 4A. Upon transient transfection of mouse myoblastic C2C12 cells with the pDMD31wt minigene, all the transcripts were correctly spliced (Fig. 4B, lane 1), whereas transfection of the pDMD31mut induced exon skipping (Fig. 4B, lane 2), recapitulating the exon-skipping effect of the c.4250T>A mutation observed in the patient's muscle. Consistent with the hypothesis of a hnRNP A1-mediated repression of exon 31 splicing, we observed that co-expression of hnRNP A1 enhanced exon skipping of dystrophin exon 31 carrying the c.4250T>A mutation, increasing the proportion of transcripts lacking exon 31 from an initial 33% (lane 2) to some 51% (Fig. 4B, lanes 3–5). We next examined whether the skipping of the mutated exon 31 was affected by other trans-acting factors. We concentrated on the two SR proteins SRp40 and 9G8. As shown in Figure 4B (lanes 7–9), SRp40 also acted as a strong exon inhibitor (1.8-fold increase in the level of mutated exon 31 skipped transcripts with the highest concentration of SRp40), whereas 9G8 had no effect on the amount of skipped transcripts (Fig. 4C). Note that no significant skipping event was detected in co-transfection experiments of hnRNP A1 or SRp40 with the pDMD31wt (data not shown). Therefore, these results indicated that both hnRNP A1 and SRp40 are able to inhibit splicing of the mutated exon 31. However, unlike hnRNP A1, binding of SRp40 to the 31mut RNA probe could not be detected in UV cross-linking experiments, suggesting that its effect results from its binding to a different region or that it is indirect.
Next, we tested the cis-acting properties of the two 31wt and 31mut RNA probes (as defined in Fig. 2A) by inserting them in the Sp1 exon 2-inverted reporter system (39), (Fig. 5A). In vitro splicing was carried out in S100 extract complemented with SR proteins, in the presence of decreasing amounts of nuclear extract (from 12 to 0%). First we observed that the 31wt sequence acts as an ESE when tested in the presence of nuclear extract (Fig. 5B, lane 1), compared with the natural inverted exon 2 sequence or to a neutral sequence, which are both unable to activate splicing in similar conditions (39 and unpublished data). However, the factor(s) that activate splicing via this sequence have not been identified. As expected, splicing of the transcript containing the 31mut sequence was reduced when compared with the 31wt, mainly in the presence of nuclear extract (compare lanes 1 and 3, 5 and 7). Interestingly, this comparison shows that the rate of inhibition correlates with the increase in nuclear extract concentration (compare the white bars of the histogram), a result that is in agreement with the presence of a negative factor (like hnRNP A1) in the nuclear extract. In line with this hypothesis, we observed that the addition of purified hnRNP A1 repressed the splicing of both 31wt and 31mut-containing transcripts in all conditions tested (Fig. 5B, lanes 2, 4, 6, 8, 10, 12), and also that the 31mut transcript is slightly more sensitive to hnRNP A1 than the 31wt transcripts (compare the ratios of inhibition in the presence of 12 or 6% nuclear extract).
Altogether, these in vivo and in vitro results demonstrated that the 35nt region flanking the c.4250T>A mutation contains an undefined splicing enhancer element but also an hnRNP A1-dependent silencer element whose activity is increased by the mutation. This correlates well with the increased association of hnRNP A1 with the mutated exon 31.
Scanning of dystrophin exon 31 by deletions uncovers a Tra2β-dependent exonic enhancer
The fact that binding of hnRNP A1 to the highly specific binding site (UAGACA) created by the c.4250T>A mutation only induced a moderate level of exon skipping in the pDMD31mut minigene (31–33%), as well as in the patient, supports the hypothesis that exon 31 might contain enhancer sequences capable of counteracting the inhibitory effect of hnRNP A1. In vitro splicing of the Sp1 substrates is also indicative of the presence of an ESE within the proximal region of exon 31, but no specific sequence could be clearly identified. To localize further the sequences necessary for exon 31 splicing regulation, we divided the sequence corresponding to the 31wt RNA probe (as in Fig. 2A) in two separate fragments, regions A and B, excluding the ESS motif (Fig. 6A). Furthermore, examination of the exon 31 sequence downstream of region B disclosed the presence of a repetition of a GAAGAAA motif separated by one thymidine (region C as defined in Fig. 6A) which resembles known purine-rich ESEs.
We tested the three sequences as defined by regions A, B and C in another well-characterized ESE-dependent substrate, IgM M1-M2 pre-mRNA. Because of the presence of a repressor sequence at the 3′ end of exon 2, this substrate requires the presence of a strong ESE to be activated (40). Regions A and B failed to activate splicing of the IgM substrates (Fig. 6B). Together with the results of Figure 5, which showed that the first 35nt of exon 31 have an ESE activity, this suggests that the putative enhancer sequence could lie over the A+B region and overlap with the ESS motif. In clear contrast with regions A and B, the purine-rich region C acted as a strong activator in the IgM pre-mRNA as shown in Figure 6B by the time-course analysis and in comparison with the splicing of a positive control (IgM E1), in which the E1 sequence is an SRp40-dependent ESE (41).
To verify the existence of this ESE in vivo, we constructed a series of deletions as described in Figure 6A, consisting in either the deletion of the first 38 nt of exon 31 (31 ΔA-B) or the deletion of region C (nt +39 to +56) in the wild-type (31wt ΔC) and the mutant context (31mut ΔC). The deletion mutants were transiently expressed in C2C12 cells and RNA were analyzed by RT–PCR (Fig. 6C). The splicing of exon 31 was not affected by deletions of the first 38 nt (31 ΔA-B), as only exon 31-containing transcripts were detected. In contrast, the ΔC mutation caused a dramatic exclusion of exon 31 in both the wild-type (24%) and mutant (70%) contexts, suggesting that the function of this ESE is dominant over that of other potential elements.
Possible candidate that bind to the purine-rich region C are ASF/SF2 or Tra2, which are known to bind with similar sequences (42–45). We co-transfected these two factors (and other previously tested trans-acting factors) with the pDMD31mut minigene, to be able to monitor an activation of exon inclusion (Fig. 7A). As previously shown, hnRNP A1 and SRp40 both promoted exon skipping of the mutated exon 31 (Fig. 7A, lanes 5 and 6). Only the recombinant GFP-Tra2β had an antagonistic effect and was able to stimulate exon 31 inclusion reducing the exon skipping level from 35 to 26% (Fig. 7A, lane 3), whereas 9G8 like ASF/SF2 did not have any effect (Fig. 7A, lanes 2 and 4). The transfection of the total pDMD31mut minigene along with increasing amounts of GFP-Tra2β confirmed that Tra2β increased exon 31 inclusion (from 35 to 23% skipping, data not shown). To determine if Tra2β binds to the purine-rich C region of exon 31, we performed UV cross-linking with whole-cell extracts (WCE) prepared from 293-EBNA cells over-expressing the GFP-Tra2β plasmid or GFP alone (42) (Fig. 7B). This experiment clearly showed a specific interaction between Tra2β and the wild-type C sequence (DMD31-Cwt), which is completely abolished when the purine-rich tract is interrupted by pyrimidines (DMD31-Cmut, see sequences in Materials and Methods). The interaction was equivalent to that observed with a previously identified Tra2β-specific ESE in the HipK3 testis-specific exon (42).
Finally, we tested whether Tra2β was able to increase the splicing of the region C-containing IgM pre-mRNA in vitro. Figure 7C shows the results of a splicing assay performed in a limiting amount of nuclear extract complemented with SR proteins. In these conditions (right panel) splicing of the IgM-C substrate is inefficient but is greatly and specifically enhanced upon the addition of the GFP-Tra2β-enriched WCE, and not upon the addition of a control GFP-expressing WCE. In contrast, the control SRp40-responsive IgM-E1 substrate (left panel) was spliced efficiently and was not further activated by GFP-Tra2β (in fact splicing was slightly reduced in a non-specific manner upon the addition of both WCE extracts, compare lanes 2 and 3 to lane 1 in Fig. 7C). Therefore, these data strongly suggest that Tra2β is required for ESE-dependent activation of DMD exon 31 in vitro as well as in vivo.
DISCUSSION
The main objective of this work was to analyze the exact mechanism by which the nonsense mutation c.4250T>A occurring in exon 31 of the dystrophin gene alleviates the severity of the disease by inducing partial skipping of this exon. The first expected consequence of such mutation was the triggering of the NMD, a pathway which takes place in the cytoplasm and specifically degrades the mRNA containing a premature stop codon (35,46). However, the full-length dystrophin mRNAs carrying the nonsense mutation was still present in large excess when compared with the skipped in-frame species in our BMD patient (Fig. 1C). In addition, the quantification of exon skipping in experiments using minigenes which are not submitted to NMD (Fig. 4) gave results which were at least equivalent to what was observed in the patient. Therefore, these results suggest that NMD does not affect significantly the mutant mRNAs. A detailed study of NMD has been carried out on BRCA1 mRNA bearing various premature stop codons (47). Interestingly, this study shows that NMD efficiency is comprised between 30 and 70% for most of the mutations located within the central region of the mRNA, suggesting that NMD could be only moderately efficient in genes containing a large number of exons (22 exons for BRCA1).
Studies of constitutively spliced exons suggest that most exons contain at least one functional ESE (8,10). Nucleotide substitution in ESEs can result in decreased binding of SR proteins or other splicing factors to the ESE, leading to a failure to recognize the sequence as exonic by the spliceosome and to exon skipping (8,48–50). The phenomenon of exon skipping induced by nonsense mutations has been reported in a few cases in the dystrophin gene (28–33). These mutations that would be expected to cause a severe DMD phenotype were found in BMD patients for whom the elimination of the mutation by exon skipping restores a correct translational reading frame. Although hypothesized, disruption of an ESE in the dystrophin gene was demonstrated in only one case, in an in vitro splicing study (28), but the trans-acting factor that binds to this ESE was not identified.
In contrast to the prediction that the c.4250T>A substitution reported in our patient down-regulates binding of SRp40 (Fig. 1D), we have experimentally demonstrated that the single-base change creates a splicing silencer that represses DMD exon 31 splicing. Whereas the 5′ region of the wt exon 31 interacts only poorly with hnRNP A1, the mutation c.4250T>A creates a strong binding site for hnRNP A1, as evidenced by RNA affinity and UV cross-linking experiments (Fig. 3). hnRNP A1 has been implicated in a variety of cellular and viral splicing silencing mechanisms through its cooperative recognition of UAGGG[U/A] and related motifs (reviewed in 51). The hnRNP A1-responsive motif (UAGACA) that we identified is similar to high-affinity sequences identified by SELEX (UAGGGA) (38), and is identical or very similar to other human hnRNP A1-specific silencer elements identified in exon 7 of SMN2 gene (UAGACA) (17), CD44 exon v5 (UAGACA) (52), HIV tat exon 2 or 3 (UAGACU, UAGUGA) (53,54) or in FGFR2 K-SAM exon (UAGGGC) (55). Although the first UAG triplet, present in each of these motifs, is essential for splicing silencing (17,56,57), it is not sufficient per se to constitute a high-affinity site for hnRNP A1. Indeed, RNA sequences which bind to 9G8 or SC35 but contain a UAGAA or a UAGGGU sequence, respectively, did not compete efficiently the binding of hnRNP A1 to the mutated exon 31 motif (Fig. 2D), and a sequence with a UAGGCU motif had an affinity for hnRNP A1 decreased by a factor of 20 relatively to those containing a UAGGGA motif (38).
Surprisingly, although the hnRNP A1-responsive element created by the c.4250T>A mutation efficiently recruited hnRNP A1, it had only a limited impact on the splicing of exon 31, as exon skipping was observed with an efficiency of only 17–33% in the patient or in transfected cells (Figs 1, 4 and 7). However, evidence that the hnRNP A1-binding site is a strong ESS was given by the deletion of the Tra2β-responsive ESE (Fig. 6C), as exon 31 skipping became predominant in this context.
In light of what has been shown in other models, it is obvious that the consequences of the presence of hnRNP A1-responsive elements in exons are highly variable. This may depend on the type of splicing involved, on the number and position of the binding sites in the exon and on the presence of other splicing regulatory elements. For instance, while the unique hnRNP A1-responsive element of the mutated exon 31 in the dystrophin gene has a limited impact on exon 31 splicing, the outcome is very different for the exon 7 of SMN2 gene. In this example, the presence of a Tra2β-specific ESE (like in dystrophin exon 31) does not efficiently counteract the effect of the hnRNP A1-responsive ESS, as the UAGACA motif results in nearly complete skipping of exon 7 (17). Whereas both splicing systems seem comparable, several reasons could explain the differences in splicing. In SMN2 exon 7, the hnRNP A1-binding site is very close to both the 3′ss and the Tra2β-specific ESE (+6 and –8, respectively) so that the binding of a single hnRNP A1 molecule may interfere strongly with the binding of U2AF to the 3′ss and of Tra2β, and/or may impair the interactions between these two factors, as suggested by Kashima and Manley (17). In contrast, in dystrophin exon 31, the hnRNP A1-binding site is located further apart from both the 3′ss and the Tra2β ESE (+16 and −18, respectively) so that hnRNP A1-binding may be less detrimental for the interactions occurring in the flanking regions. Furthermore, the situation is more complicated in SMN2 exon 7, as the C>T mutation could create an extended inhibitory environment near the 3′ss of the exon (58) and appears to affect also the binding of ASF/SF2, in support for an enhancer-loss model (59,60).
In both examples described earlier (dystrophin exon 31 and SMN2 exon 7), the repression of splicing might involve the binding of a single hnRNP A1 molecule to the ESS. In other cases, however, several motifs have been identified in a restricted region, sometimes close to ESS responding to other repressors (61). In the HIV tat exon 3, a cooperativity between at least two hnRNP A1-specific motifs has been demonstrated, which is required to efficiently repress splicing of the upstream intron (56,62–64). In this example, the initial binding of one protein onto a high-affinity site could nucleate the formation of a chain of hnRNP A1 molecules along the RNA, possibly bordered by the next high-affinity binding site (62). Another attractive model is based on the bridging activity of hnRNP A1 showed by Blanchette and Chabot (65), whereas the binding of hnRNP A1 to a unique site appears to be highly reversible. Its binding to distant specific sites followed by the homodimerization of the proteins through their RGG domains could stabilize their association with RNA.
In our study, we also hypothesized that the inhibitory effect of hnRNP A1 may be overcome by a nearby or overlapping ESE. Deletion analysis of dystrophin pre-mRNA sequences showed that the region from +39 to +56 of exon 31, downstream of the ESS, is a critical element for constitutive inclusion of DMD exon 31 in vivo (Fig. 6). We found that only the SR-like protein Tra2β, but none of the SR proteins tested (ASF/SF2, 9G8, SRp40), favors the inclusion of the mutated exon 31 in vivo and that the purine-rich ESE can induce splicing of an heterologous IgM reporter in vitro in a Tra2β-dependent manner (Fig. 7). We also showed that Tra2β directly binds with ESE. Therefore, it is likely that Tra2β is the primary activator of exon 31 splicing. There are multiple examples of human genes whose splicing is controlled through the binding of Tra2 to a specific sequence (42). The mechanism by which Tra2β activates the inclusion of dystrophin gene exon 31 is not clear. The results from our experiments let us formulate speculative models of splicing regulation of DMD exon 31 in both the wild-type and mutant context (Fig. 8). It is likely that Tra2β does not work alone, as several groups pointed out a requirement for cofactors in Tra2-mediated splicing activation (42,66–71). Two major families of factors have been identified as modulating the activity of human Tra2 proteins. First, human Tra2 proteins interact directly with several SR proteins, including SRp30c, SRp55 and 9G8 (68,71,72). A cooperation between human Tra2 and SR proteins was observed in several examples of splicing regulation (42,67,68,71,73). Secondly, hnRNP G and its two testis-specific paralogues hnRNP G-T and RBMY also interact directly with Tra2β and affect its activity, either positively (66,73) or negatively (74). This last example is interesting, because hnRNP G and Tra2β exert opposite effects on tropomyosin exon SK splicing through their antagonistic binding to non-overlapping binding sites within the exon (74). This functional antagonism between one hnRNP protein on one side and one RS domain-containing protein on the other side can be compared with what has been described about the opposite functions of hnRNP A1 and ASF/SF2 in the regulation of alternative splicing (62,75).
Although purine-rich ESEs were initially identified by their ability to activate upstream 3′ss (reviewed in 10), one report recently demonstrated that the binding of human Tra2β to the tau exon 10 ESE increases U1 snRNP binding to the 5′ss (76), and the Drosophila Tra2 protein was shown to regulate the use of alternative 5′ss in the fruitless and tra2 pre-mRNAs (77,78). Therefore, it appears that, like ASF/SF2, Tra2 proteins can influence the early recognition of the 5′ss by the splicing machinery. Despite the central position of the Tra2β-responsive ESE (+39 to +56) within the 111 bp-long DMD exon 31, it is possible that an efficient binding of U1 snRNP to the suboptimal 5′ss (consensus value, 74%, Fig. 1A) requires Tra2β and possibly other cofactors (Fig. 8). However, we cannot rule out an alternative model of RS-domain-mediated interactions between the 3′ss region and the Tra2β-dependent ESE (Fig. 8).
Efficient splicing is the result of a plethora of complex and often antagonistic interactions between different splicing factors, each binding to its proper target sequence. It is therefore difficult to evaluate the effect of a single nucleotide substitution whenever composite exonic regulatory elements of splicing (CERES) are present as previously described in CFTR exon 9 and 12 (79,80). We speculate that binding of hnRNP A1 to the 5′ part of the mutated DMD exon 31 may prevent the function of other splicing factors. Besides the results discussed earlier, our in vitro splicing data using Sp1 (Fig. 5B) and IgM substrates (Fig. 6B) argue for the presence of an as yet unidentified ESE motif that would overlap with the UAGACA silencer or be present at proximity. Consistent with this hypothesis, another nonsense mutation (c.4240C>T) located 10 bp upstream of the c.4250T>A mutation analyzed in this study has been reported in exon 31 of a BMD patient in the Leiden DMD database (http://www.dmd.nl). The phenotype of this patient suggests an effect on splicing, but this mutation does not seem to create an hnRNP A1-specific binding site, although no study has been performed so far to demonstrate it formally. Thus, it will be interesting to analyze whether this mutation also induces exon 31 skipping and if it disrupts an ESE sequence. Finally, SRp40 was found to strongly inhibit splicing of the mutant DMD exon 31 in co-transfection experiments (Fig. 4), although no direct binding of SRp40 could be observed in our UV cross-linking assays (Fig. 2). We hypothesized either that SRp40 exerts its inhibitory effect through an interaction with other splicing factors or that the SRp40 binding site does not lie within the sequences that we used in our experiments (the first 35 bp of exon 31). Hence, inhibition of splicing of the DMD exon 31 harboring the c.4250T>A mutation reflects a competition between positive and negative factors, with Tra2β and possibly other SR or SR-like proteins functioning through ESEs and hnRNP A1 through the ESS.
Previous data and the present study show that a milder muscular dystrophy phenotype than expected resulted from partial elimination of a nonsense mutation from dystrophin mRNA by exon skipping. This observation supports the development of alternative gene therapy approaches based on the modulation of splicing to bypass DMD-causing mutations in the endogenous dystrophin gene. Antisense oligonucleotides (AONs), designed to anneal to motifs involved in exon definition and/or splicing, are used to induce the specific skipping of exons in order to correct the reading frame of a mutated transcript so that it can be translated into a partially functional protein (81,82). This approach has great potential for treatment of DMD caused by mutations within non-essential regions of the dystrophin gene. To date target sites within the dystrophin pre-mRNA have been mostly defined using an empirical approach including donor and acceptor splice sites, as well as in silico predicted ESEs. However, the complexity of the various mechanisms leading to the recognition of constitutive exons suggests that there is not only one specific splicing motif that can be universally targeted. The choice of the target site within an exon will probably be a major determinant of the effectiveness of an exon skipping strategy. Our data on exon 31 splicing provide essential insights into the complexity of constitutive exon splicing in the dystrophin gene and support the idea that antisense-based therapy would require to experimentally assess the best motifs on an exon-to-exon basis.
MATERIALS AND METHODS
Dystrophin minigenes constructs
Minigenes containing exons 30, 31 and 32 of the dystrophin gene and surrounding intronic boundaries were constructed as follows. The genomic DNAs of the patient carrying the c.4250T>A mutation and of a normal control were used as templates for three separate PCR amplifications to synthesize intron-truncated DNA fragments: the first PCR amplified the exon 30 and its flanking intronic sequences using primers M30M1 (forward, 5′ GAATGAGTGCCAGGAAGCTG 3′) and M30M2 (reverse, 5′ CCAAGAGTGGTGGTTCACACCGTGAGGTCATACAAACAAAAA 3′); the second PCR amplified the exon 31 and flanking intronic sequences using primers M31M3 (forward, 5′ GTGTGAACCACCACTCTTGG 3′) and M31M4 (reverse, 5′ TTCCATTCATCCAACACAGGGGTCCTAAATCCAATCTTGCCAA 3′); the third PCR amplified the exon 32 and flanking intronic sequences using primers M32M5 (forward, 5′ CCCTG TGTTGGATGAATGGAA 3′) and M32M6 (reverse, 5′ GTTCTCAATCTTAAAATTACATAGT 3′). After agarose gel purification, the PCR products were combined and amplification was performed using primers M30M1 and M32M6. Final PCR products were cloned into the pGEM-T vector (Promega) and sequence-verified. This overlap-extension PCR generated two DMD minigenes, pDMD31wt and pDMD31mut with shortened introns, but with otherwise natural intronic splicing signals. Finally, the minigenes were EcoRI-excised from pGEM-T and transferred into the EcoRI-digested and phosphorylated mammalian expression vector pSI (Promega) driven by the SV40 promoter, which contains the late SV40 polyadenylation signal. The DMD minigenes pDMD31delA-B and pDMD31delC were also generated by overlap-extension PCR using primers 31delA-B (reverse) (5′ ATGTTTCTTCATTTCTTCTATTTTCTGTTGGGAGGATA 3′) and 31delA-B (forward) (5′ AGAAGAAATGAAGAAA CAT 3′), or primers 31delC (reverse) (5′ CAGCCTCC TTCCCCTGATTAACTGATCTCATGACTTGT 3′) and 31delC (forward) (5′ TAATCAGGGGAAGGAGGCTG 3′) in combination with the primers described earlier.
In vivo splicing assay
We transiently transfected or co-transfected wild-type and mutant DMD minigene constructs (1.5 µg) in six-well plates and, where indicated, protein expression plasmids into C2C12 with PolyFect (Qiagen) as recommended by the manufacturer. After 24–48 h, we isolated total RNA and reverse-transcribed in cDNAs using 500 ng RNA, 300 ng of random primers (Invitrogen) and M-MLV reverse transcriptase (Invitrogen) at 42°C for 40 min. We amplified cDNA from the transfected minigenes with a plasmid-specific forward primer (PSIF, 5′ AGTTCAATTACAGCTCTTAAGGC 3′) and a DMD-specific reverse primer located in exon 32 (32R, 5′ CAATGATTTAGCTGTGACTGTACTA 3′). As an internal control, the hypoxanthine phosphoribosyltransferase (HPRT) gene was simultaneously amplified using the primers HPRTF (forward, 5′ TGTAATGACCAGTCAACAGGG 3′) and HPRTR (reverse, 5′ TGACCAAGGAAAGCAAAGTCTG 3′). We carried out PCR (35 cycles; 94°C, 30 s; 52°C, 30 s; 72°C, 1 min) using the Multiplex PCR Master mix (Qiagen). The PCR products were resolved on 1.5% agarose gel and visualized by staining with ethidium bromide. For semi-quantitative analysis, a Fam-labelled PSIF primer was used and PCR reactions were terminated during the linear phase (25 cycles). A 1 µl sample of the PCR mixture was electrophoresed in a 6% denaturing polyacrylamide gel on a ABI377 sequencer. Quantification of the fluorescently labelled products was performed with Applied Biosystem 672 Genescan software and the areas of the peaks and the size of the products were obtained. The ratios of exclusion of exon 31 were quantified and expressed as percentages of exclusion relative to total intensities. The percentages of exon 31-included and exon 31-excluded were calculated from an average of three independent experiments.
In vitro splicing assays
Splicing assays using both 32P-labelled IgM and adenovirus Sp1 pre-mRNA substrates were performed using various mixtures of HeLa nuclear extract and S100 cytoplasmic fractions, as specified in the figures, in the following buffer: 12 mm Hepes pH 7.9, 60 mm KCl, 3.2 mm MgCl2, 1.5 mm ATP, 25 mm creatine phosphate, 0.3 mm DTT, 0.12 mm EDTA, 3% poly-vinyl alcohol, 40 U RNase inhibitor and 10% glycerol. When reactions were performed in limiting conditions (low concentration of nuclear extract), they were supplemented with around 500 ng of purified total SR proteins. For the time-course experiment, a splicing assay equivalent to three individual samples was assembled and three aliquots were taken after a 15, 30 or 90 min incubation and analyzed as in regular reactions. Regular splicing assays were carried out for 90 min at 31°C, followed by a 30 min-proteinase K treatment at 31°C, phenol extraction and ethanol precipitation. RNA products of the reactions were analyzed by Urea-PAGE (6% acrylamide/bisacrylamide 30/1) and autoradiography. To test the activity of the Tra2β protein on IgM-derived RNA substrates, we used WCE from 293-EBNA cells transfected with plasmids encoding either a GFP-Tra2β fusion protein or GFP alone, as described in Venables et al. (42).
Quantifications of splicing assays were made using a Typhoon 8600 imager and the ImageQuant software (Amersham Pharmacia Biotech). After correction of the band counts for the background, splicing efficiency for each reaction was calculated as the ratio mRNA/(pre-mRNA+mRNA). The inhibition rate of splicing by hnRNP A1 was calculated for each transcript as the ratio between the splicing efficiency in the absence of hnRNP A1 to the splicing efficiency in the presence of hnRNP A1.
UV cross-linking and immunoprecipitation assays
UV cross-linking experiments showed in Figures 2 and 3, were carried out as previously described (83). Briefly, sense and antisense primers corresponding to each probe were annealed and then digested with EcoRI/PstI and subcloned in the same restriction sites of the PGEM-3Z vector (Promega). Plasmids were linearized by digestion with HindIII prior to in vitro transcription using the Riboprobes In Vitro Transcription Systems (Promega). [α-32P]UTP-labelled RNA probes were incubated for 15 min at 30°C with 15 µg of nuclear extract in binding buffer. In the competition experiments, cold competitor RNAs were added at the same time as the labelled RNAs. Reaction mixtures were pre-chilled on ice and irradiated with UV light (254 nm) for 8 min. The samples were digested with RNase A and RNase T1 at 37°C for 30 min, and run on 10% SDS-PAGE. The gels were fixed, dried and subjected to autoradiography. The sequences of the competitor RNAs hnRNP A1, SRp38, 9G8-102 and SC35-7 are from references (38,39,84). For immunoprecipitation, following the RNase treatment, samples were incubated in 150 µl of immunoprecipitated (IP) buffer (20 mm Tris–HCl, pH 8.0, 300 mm NaCl, 1 mm EDTA and 0.25% Nonidet P-40) and the hnRNP A1 antibody 4B10 (gift by G. Dreyfuss) was added to each cross-linked sample and incubated for 2 h at 4°C on a rotator wheel. Afterwards, 30 µl of protein A/G Plus-agarose beads (Santa Cruz Biotechnology) was added and incubated overnight at 4°C. The RNA-labelled proteins retained on the beads after several washes were eluted and resolved by SDS-PAGE.
The experiment showed in Fig. 7B was performed as follows. The wild-type (5′ AGAAGAAATGAAGAAACA 3′, DMD31Cwt) and mutated (5′ AGTAGTAATGTAGTAACA 3′, DMD31Cmut) purine-rich C region of DMD exon 31 were cloned into KpnI and BamHI sites of pBSSK (Stratagene). The purine-rich region from the HipK3-T exon (5′ GGGAGGAAGAAATAGAAGATGCAGAAGAG 3′) (42) was used as a positive control. Plasmids were linearized by XbaI and highly [α32P]ATP-labelled RNAs were in vitro-transcribed with T7 RNA polymerase. UV cross-linking assays were performed essentially as previously described (42), using 3 µl of WCE from 293-EBNA cells transfected with GFP or GFP-Tra2β and 400 000 cpm of RNA, in the presence of 0.2 µg E. coli tRNA.
RNA affinity and mass spectrometry
The control non-specific sequence (GST-80) used in the RNA affinity assay was described previously (39). The different RNAs were transcribed using T7 RNA polymerase. Covalent immobilization of RNA on beads and RNA affinity were performed as previously described (42), except that 1.8 mg of HeLa nuclear extract (225 µl) was used for 300 pmol of immobilized RNA probe. Pulled-down proteins were analyzed by western-blotting with an anti-hnRNP A1 antibody (4B10), an anti-9G8 monoclonal antibody and an affinity-purified polyclonal antibody directed against the N-terminal part of ASF/SF2. Mass spectrometry analysis was performed as described in (42).
ACKNOWLEDGEMENTS
We gratefully acknowledge Javier Caceres for coding plasmids for SRp40, ASF/SF2 and 9G8, Emmanuele Buratti for the coding plasmid for hnRNP A1, Julian Venables and David Elliott for the coding plasmid for GFP-Tra2β, and Christiane Branlant for the purified hnRNP A1 protein. We thank Gideon Dreyfuss for the generous gift of anti-hnRNP A1 antibodies and Manuela Argentini (IGBMC, Illkirch) for her help with mass spectrometry. We thank Jamal Tazi and Johann Soret for their help in preparing HeLa nuclear extracts, and Adrian Krainer for providing the IgM constructs. Finally, we are grateful to Liliane Kister for her excellent technical assistance and her contribution to part of the in vitro splicing experiments presented here and Céline Saquet for the semi-quantitative analysis of muscle transcripts in the patient. We thank Julian Venables for the critical reading of the manuscript. A.D. was supported by an AFM predoctoral training grant. This work was also supported by an AFM grant to S.T.G., and an ARC grant for C.F.B. and J.S.
Conflict of Interest statement. None declared.
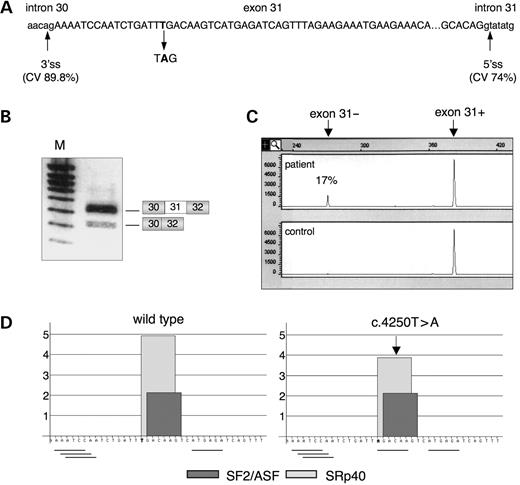
Figure 1. Dystrophin transcripts carrying the c.4250T>A mutation are prone to exon skipping. (A) Schematic diagram of exon 31 (capital letters) and flanking intronic sequences (lower-case letters). The position of the c.4250T>A is indicated. 3′ss, 3′ splice site; 5′ss, 5′ splice site. CV score calculated according to Shapiro and Senapathy (36). (B) Muscle dystrophin transcripts from the patient harboring the mutant allele causing exon skipping were amplified by RT–PCR using primers located in exons 30 (forward) and 32 (reverse). Two products are seen after agarose gel electrophoresis: the upper band (384 bp) includes exon 31, whereas the lower band (273 bp) lacks this exon, as indicated on the right. M, molecular weight marker. (C) Proportion of exon 31-lacking transcripts in the patient (upper panel) quantified by semi-quantitative analysis of Fam-labelled RT–PCR products (Fam-32R primer) on a capillary electrophoresis ABI-PRISM automatic sequencer. Control individual (lower panel). (D) Effect of the c.4250T>A mutation on SR proteins matrix score identified by ESEfinder (http://rulai.cshl.edu/tools/ESE/) (37). The arrow indicates the decrease of the SRp40 motif from 4.93 to 3.88. The ASF/SF2 motif (2.13) is unchanged. The presence of hexamers predicted by RESCUE-ESE (http://genes.mit.edu/burgelab/rescue-ese/) (3) to function as ESE elements is indicated by horizontal lines below the wild-type and mutant exon 31 sequences.
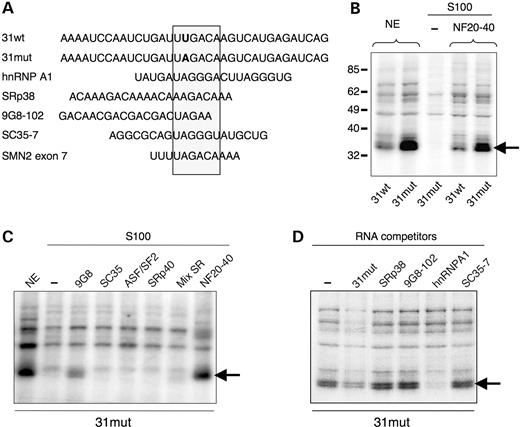
Figure 2. A protein from the nuclear fraction 20–40 specifically interacts with the mutated exon 31 sequence. (A) Comparison of the first 35 nucleotides of dystrophin exon 31 with sequences of the cold competitor RNAs used in the UV cross-linking experiments and with the ESS of SMN2 exon 7. The position of the mutation in exon 31 is in bold. Sequences of hnRNP A1, SRp38, 9G8-102 and SC35-7 competitor RNAs, obtained by SELEX, contain high-affinity binding motifs for these proteins (38,39,84). hnRNP A1 and SRp38 RNA probes contain tandem repeats of the SELEX motif. (B) Radioactively labelled wild-type (31wt) or mutant (31mut) RNA were incubated with nuclear extract or in a cytoplasmic S100 fraction complemented or not with a 20–40% ammonium sulfate precipitation fraction from nuclear extract (NF20–40). After cross-linking and RNase digestion, the reactions were analyzed by SDS-PAGE and autoradiography. The arrow indicates the position of the 35 kDa complex. The positions of molecular mass markers (in kiloDaltons) are shown on the left. (C) UV cross-linking with purified SR proteins. The RNA probe (31mut) was incubated in the presence of nuclear extract or in S100 complemented or not with 250 ng of different recombinant SR proteins (9G8, SC35, ASF/SF2, SRp40), a mixture of total SR proteins (Mix SR) or a NF20–40 fraction lacking SR proteins. The arrow indicates the position of the 35 kDa complex. (D) A hnRNP A1 cold competitor RNA specifically inhibits the 35 kDa complex formation. Competition analysis using labelled 31mut RNA incubated with HeLa nuclear extract following the addition of 30 pmol of various cold RNAs. The 35 kDa complex (arrow) shows specific competition with the hnRNP A1 SELEX motif.
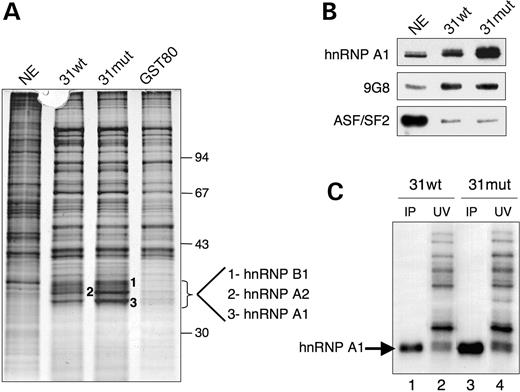
Figure 3. Recruitment of hnRNP A1 by the c.4250T>A mutation in exon 31. (A) RNA affinity experiment using immobilized 31wt, 31mut as well as non-specific (GST-80) RNA sequences. The first lane shows the input HeLa nuclear extract used in the experiment. Three bands that strongly bound to the 31mut RNA were excised from the silver-stained gel and identified by MALDI-TOFF mass spectrometry as hnRNPs A1, A2 and B1. Shown on the right are the mobilities of protein size markers (in kiloDaltons). (B) Western blot analysis of the same RNA affinity experiment as in (A) with specified antibodies. (C) UV cross-linking/immunoprecipitation experiment. The 31wt or 31mut labelled RNA probe were incubated with HeLa nuclear extract followed by cross-linking. A 5% input was loaded directly (lanes 2 and 4), whereas lanes 1 and 3 show subsequent immunoprecipitation of cross-linked proteins by anti-hnRNP A1 antibodies. Position of hnRNP A1 is indicated on the right.
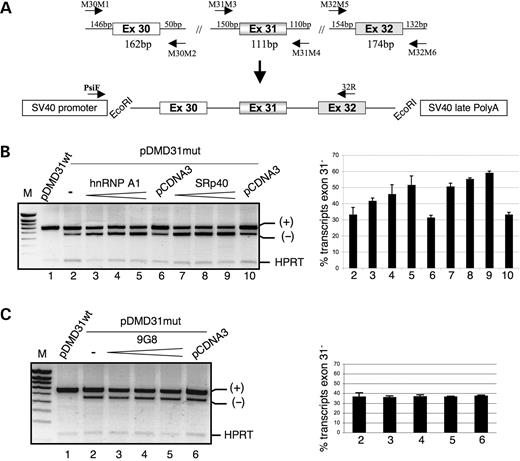
Figure 4. hnRNP A1 and SRp40 modulate the in vivo splicing pattern of dystrophin minigenes harboring the c.4250T>A mutation. (A) Schematic diagram of dystrophin minigenes and strategy for cloning exons, as detailed in Materials and Methods. Both a wild-type (pDMD31wt) and a mutant (pDMD31mut) minigene carrying the c.4250T>A mutation in exon 31 were generated. The approximate position of primers PsiF and 32R used for RT–PCR analysis is indicated (lower panel). (B) A dose-response curve of exon 31 skipping by hnRNP A1 and SRp40. The indicated plasmid, wild-type (pDMD31wt) or mutant (pDMD31mut) was introduced into C2C12 cells by transfection either in presence of 100 ng of the empty vector pCDNA3 (control) or with increasing amounts (50, 100 and 200 ng) of hnRNP A1 and SRp40 plasmids. The RT–PCR products analysis is shown on the left. The products corresponding to exon 31 skipping (−) and inclusion (+) are indicated. HPRT is an internal control showing that each assay contained roughly equivalent amounts of RNA. M, molecular weight marker. The proportion of exon 31-lacking transcripts was determined by semi-quantitative analysis of fluorescently labelled transcripts. Co-transfection of the pDMD31mut minigene with hnRNP A1 (lanes 3–5) and SRp40 (lanes 7–9) coding plasmids further increases the level of exon 31 skipping. Standard bars based on three independent transfection experiments are shown. (C) In vivo splicing of the pDMD31mut minigene in the presence of 100 ng of the empty vector pcDNA (control) or increasing amounts (50, 100 and 200 ng) of plasmids overexpressing 9G8 in C2C12 cells. Quantitation of exon skipping was done as in B.
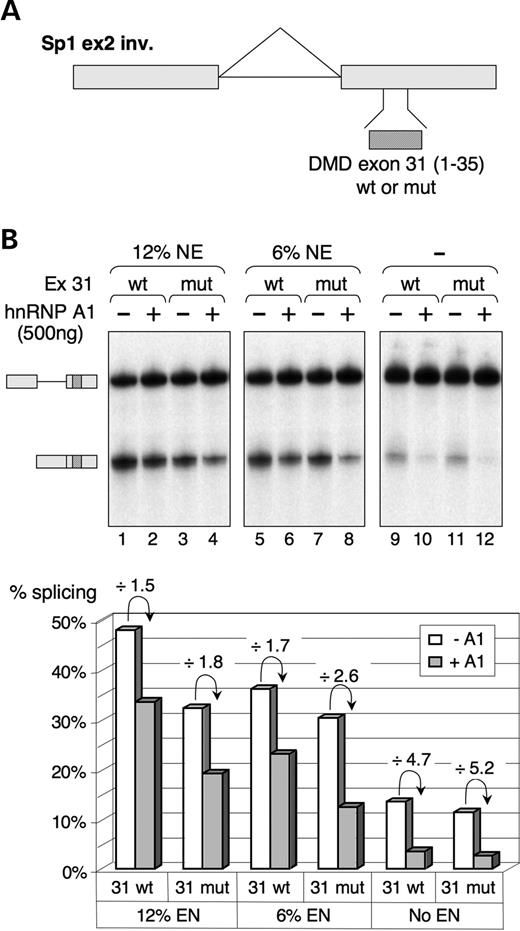
Figure 5. hnRNP A1 represses splicing of 31wt- or 31mut-containing pre-mRNA substrates in vitro. (A) Schematic representation of Sp1 ex2 inv-derived plasmids containing two exons (boxes) separated by an intron (line). Sequences to be tested (31wt and 31mut as in Fig. 2A) were inserted within the inverted exon 2 of this splicing-deficient substrate in order to test their capacity to act as an ESE. (B) (Upper panel) In vitro splicing of Sp1 pre-mRNAs containing the indicated sequence (31wt or 31mut) in HeLa S100 extract complemented with 12% nuclear extract (lanes 1–4), 6% nuclear extract (lanes 5–8) or with a mixture of total SR proteins (lanes 9–12). In vitro splicing assays were performed in the presence (+) or in the absence (−) of 500 ng recombinant hnRNP A1 protein for 1.5 h, and spliced products were analyzed by urea-PAGE on a 6% polyacrylamide gel. The pre-mRNA and the spliced mRNA are symbolized on the side of the gel. (Lower panel) Quantification of splicing efficiencies in the different experimental conditions without added (open bars) or with added (grey bars) hnRNP A1. The inhibition of splicing induced upon addition of hnRNP A1 is indicated for each pair of samples. Note that because of the low splicing efficiency in lanes 9–12, the quantification could not be made very accurately and the observed differences cannot be considered as significant.
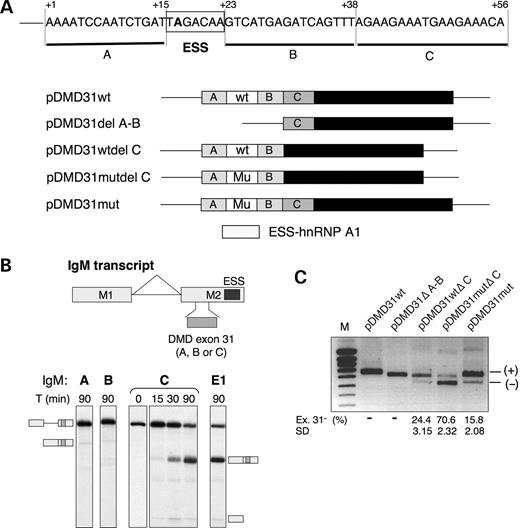
Figure 6. Identification of a positive splicing regulatory sequence in the central region of dystrophin exon 31. (A) Sequence and structure of exon 31 in the wild-type and mutated minigenes used for transfection assays. The ESS is boxed and the surrounding regions were delimited in regions A (nt +1 to +15), B (nt +23 to +38), or C (nt +39 to +56). The various deletion constructs are given below the sequence of the mutated exon 31. (B) In vitro splicing of IgM M1-M2 pre-mRNA substrates containing various exon 31 sequences. The characterized natural ESE in mouse IgM exon 2 was replaced by regions A, B or C of the dystrophin exon 31 or by an SRp40-specific control ESE (E1: CGAGGAAUAUAAAGGUGGGA) (41) by overlap-extension PCR. Splicing assays were carried out in standard conditions of nuclear extract and a time-course analysis of in vitro splicing is shown for region C (nt +39 to +56) of dystrophin exon 31. The pre-mRNA, spliced mRNA and intermediate product exon 1 are symbolized on the side of the gel. (C) The influence of exon 31 deletions on exon 31 splicing in vivo. Reporter constructs shown in (A) were transiently transfected into C2C12 cells and RNA splicing variants were detected by RT–PCR and analyzed on 1.5% agarose gel. Quantitation of exon skipping was done as in Figure 4. Exon 31 inclusion (+) and exclusion (−) are indicated. M, molecular weight marker. Numbers below the lanes indicate the percentage (%) of exon 31-lacking transcripts. The mean of three independent transfection experiments and standard deviation (SD) are given.
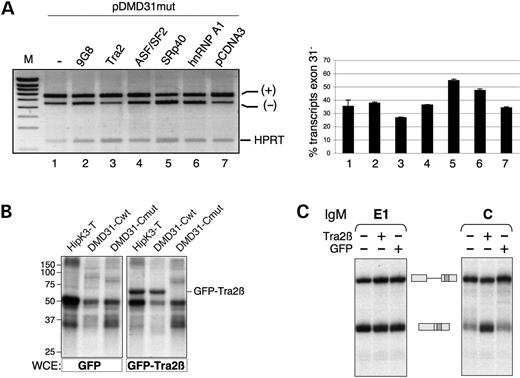
Figure 7. Tra2β stimulates ESE-dependent inclusion of dystrophin exon 31. (A) Effect of several splicing factors on exon 31 inclusion in vivo. C2C12 cells were transfected with the DMD31mut minigene and with 100 ng of the empty pCDNA3 or of coding plasmid for different splicing factors (9G8, GFP-Tra2β, ASF/SF2, SRp40, hnRNP A1). Analysis and quantitation of exon skipping was as in Figure 4B. (B) The purine-rich region C of dystrophin exon 31 is recognized specifically by Tra2β. The wild-type (DMD31-Cwt) or mutated (DMD31-Cmut) sequences were incubated with WCE from cells expressing GFP or GFP-Tra2β and UV-irradiated. The Tra2β-responsive HipK3-T ESE (42) was used as a positive control. Note that the 50 and 35 kDa bands are likely to correspond to hnRNP H and hnRNP A/B proteins, respectively, on the basis of previous results (42). (C) Ability of Tra2β to activate splicing in vitro through the region C of exon 31. In vitro splicing assays of radiolabelled IgM pre-mRNAs containing either the region C from dystrophin exon 31 or the SRp40-specific control ESE (E1) in splicing limiting conditions. WCE from cells transfected with GFP alone (GFP) or GFP-Tra2β (Tra2β) was added. The pre-mRNA and the spliced mRNA are symbolized on the side of the gel.
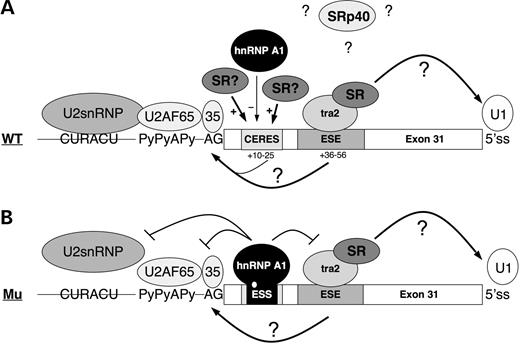
Figure 8. Speculative model of splicing regulation of dystrophin exon 31 in the wild-type and mutant context. (A) In the wild-type context, binding of Tra2β to the ESE promotes exon definition by recruiting either U1 snRNP to the downstream 5′ss or the splicing factor U2AF to the upstream 3′ss (or both) so that exon 31 is constitutively included. Tra2β is likely to function with other SR or SR-related proteins. Other ESEs in the exon could mediate weak exon inclusion. Indeed the exonic region +10 to 25 is believed to act as a composite exonic regulatory element of splicing (CERES) (79,80) containing a weak binding site for hnRNP A1 and a putative enhancer sequence that may recruit SR proteins or other activatory factors. The SRp40 protein is believed to repress exon 31 inclusion, however the mechanism of inhibition of this protein is unknown. (B) The c.4250T>A mutation creates a high-affinity binding site for hnRNP A1. The inhibitor displaces the binding of activatory factors to the CERES and sterically interferes with the binding of U2AF on the 3′ss. The binding of Tra2β on the ESE is preserved, and still stimulates or stabilizes the interactions of general splicing factors at the 5′ss consistent with the weak level of exon skipping observed in the patient.
References
Stamm, S., Ben-Ari, S., Rafalska, I., Tang, Y., Zhang, Z., Toiber, D., Thanaraj, T.A. and Soreq, H. (
Jurica, M.S. and Moore, M.J. (
Fairbrother, W.G., Yeh, R.F., Sharp, P.A. and Burge, C.B. (
Lim, L.P. and Burge, C.B. (
Black, D.L. (
Buratti, E. and Baralle, F.E. (
Matlin, A.J., Clark, F. and Smith, C.W. (
Cartegni, L., Chew, S.L. and Krainer, A.R. (
Caceres, J.F. and Kornblihtt, A.R. (
Blencowe, B.J. (
Zheng, Z.M. (
Bourgeois, C.F., Lejeune, F. and Stevenin, J. (
Pozzoli, U. and Sironi, M. (
Krawczak, M., Reiss, J. and Cooper, D.N. (
Faustino, N.A. and Cooper, T.A. (
Kashima, T. and Manley, J.L. (
Emery, A.E. (
Koenig, M., Hoffman, E.P., Bertelson, C.J., Monaco, A.P., Feener, C. and Kunkel, L.M. (
O'Brien, K.F. and Kunkel, L.M. (
Flanigan, K.M., von Niederhausern, A., Dunn, D.M., Alder, J., Mendell, J.R. and Weiss, R.B. (
Buzin, C.H., Feng, J., Yan, J., Scaringe, W., Liu, Q., den Dunnen, J., Mendell, J.R. and Sommer, S.S. (
Tuffery-Giraud, S., Saquet, C., Chambert, S., Echenne, B., Cuisset, J.M., Rivier, F., Cossee, M., Philippe, C., Monnier, N., Bieth, E. et al. (
Mendell, J.R., Buzin, C.H., Feng, J., Yan, J., Serrano, C., Sangani, D.S., Wall, C., Prior, T.W. and Sommer, S.S. (
Hoffman, E.P., Brown, R.H. and Kunkel, L.M. (
Roberts, R.G., Gardner, R.J. and Bobrow, M. (
Bushby, K.M., Gardner-Medwin, D., Nicholson, L.V., Johnson, M.A., Haggerty, I.D., Cleghorn, N.J., Harris, J.B. and Bhattacharya, S.S. (
Shiga, N., Takeshima, Y., Sakamoto, H., Inoue, K., Yokota, Y., Yokoyama, M. and Matsuo, M. (
Fajkusova, L., Lukas, Z., Tvrdikova, M., Kuhrova, V., Hajek, J. and Fajkus, J. (
Ginjaar, I.B., Kneppers, A.L., Meulen, J.D., Anderson, L.V.B., Bremmer-Bout, M., van Deuketom, J.C.T., Weegenaar, J., den Dunnen, J.T. and Bakker, E. (
Barbieri, A.M., Soriani, N., Ferlini, A., Michelato, A., Ferrari, M. and Carrera, P. (
Melis, M.A., Muntoni, F., Cau, M., Loi, D., Puddu, A., Boccone, L., Mateddu, A., Cianchetti, C. and Cao, A. (
Politano, L., Nigro, G., Nigro, V., Piluso, G., Papparella, S., Paciello, O. and Comi, L.I. (
Tuffery-Giraud, S., Saquet, C., Thorel, D., Disset, A., Rivier, F., Malcolm, S. and Claustres, M. (
Maquat, L.E. (
Shapiro, M.B. and Senapathy, P. (
Cartegni, L., Wang, J., Zhu, Z., Zhang, M.Q. and Krainer, A.R. (
Burd, C.G. and Dreyfuss, G. (
Cavaloc, Y., Bourgeois, C.F., Kister, L. and Stevenin, J. (
Kan, J.L. and Green, M.R. (
Liu, H.X., Zhang, M. and Krainer, A.R. (
Venables, J.P., Bourgeois, C.F., Dalgliesh, C., Kister, L., Stevenin, J. and Elliott, D.J. (
Hofmann, Y., Lorson, C.L., Stamm, S., Androphy, E.J. and Wirth, B. (
Stoilov, P., Daoud, R., Nayler, O. and Stamm, S. (
Tacke, R., Tohyama, M., Ogawa, S. and Manley, J.L. (
Wilusz, C.J., Wormington, M. and Peltz, S.W. (
Perrin-Vidoz, L., Sinilnikova, O.M., Stoppa-Lyonnet, D., Lenoir, G.M. and Mazoyer, S. (
Ars, E., Serra, E., Garcia, J., Kruyer, H., Gaona, A., Lazaro, C. and Estivill, X. (
Fackenthal, J.D., Cartegni, L., Krainer, A.R. and Olopade, O.I. (
Moseley, C.T., Mullis, P.E., Prince, M.A. and Phillips, J.A. (
Chabot, B., LeBel, C., Hutchison, S., Nasim, F.H. and Simard, M.J. (
Matter, N., Marx, M., Weg-Remers, S., Ponta, H., Herrlich, P. and Konig, H. (
Tange, T.O., Damgaard, C.K., Guth, S., Valcarcel, J. and Kjems, J. (
Zahler, A.M., Damgaard, C.K., Kjems, J. and Caputi, M. (
Del Gatto-Konczak, F., Olive, M., Gesnel, M.C. and Breathnach, R. (
Caputi, M., Mayeda, A., Krainer, A.R. and Zahler, A.M. (
Hou, V.C., Lersch, R., Ponthier, J.L., Lo, A.J., Wu, M., Turck, C.W., Koury, M., Krainer, A.R., Mayeda, A. and Conboy, J.G. (
Singh, N.N., Androphy, E.J. and Singh, R.N. (
Cartegni, L. and Krainer, A.R. (
Cartegni, L., Hastings, M.L., Calarco, J.A., de Stanchina, E. and Krainer, A.R. (
Han, K., Yeo, G., Burge, C.B. and Grabowski, P.J. (
Zhu, J., Mayeda, A. and Krainer, A.R. (
Marchand, V., Mereau, A., Jacquenet, S., Thomas, D., Mougin, A., Gattoni, R., Stevenin, J. and Branlant, C. (
Damgaard, C.K., Tange, T.O. and Kjems, J. (
Blanchette, M. and Chabot, B. (
Hofmann, Y. and Wirth, B. (
Young, P.J., DiDonato, C.J., Hu, D., Kothary, R., Androphy, E.J. and Lorson, C.L. (
Wang, Y., Wang, J., Gao, L., Lafyatis, R., Stamm, S. and Andreadis, A. (
Seong, J.Y., Han, J., Park, S., Wuttke, W., Jarry, H. and Kim, K. (
Tacke, R., Tohyama, M., Ogawa, S. and Manley, J.L. (
Park, E., Han, J., Son, G.H., Lee, M.S., Chung, S., Park, S.H., Park, K., Lee, K.H., Choi, S., Seong, J.Y. and Kim, K. (
Tran, Q., Coleman, T.P. and Roesser, J.R. (
Venables, J.P., Elliott, D.J., Makarova, O.V., Makarov, E.M., Cooke, H.J. and Eperon, I.C. (
Nasim, M.T., Chernova, T.K., Chowdhury, H.M., Yue, B.G. and Eperon, I.C. (
Eperon, I.C., Makarova, O.V., Mayeda, A., Munroe, S.H., Caceres, J.F., Hayward, D.G. and Krainer, A.R. (
Jiang, Z., Tang, H., Havlioglu, N., Zhang, X., Stamm, S., Yan, R. and Wu, J.Y. (
Lam, B.J., Bakshi, A., Ekinci, F.Y., Webb, J., Graveley, B.R. and Hertel, K.J. (
Chandler, D.S., Qi, J. and Mattox, W. (
Pagani, F., Buratti, E., Stuani, C. and Baralle, F.E. (
Pagani, F., Stuani, C., Tzetis, M., Kanavakis, E., Efthymiadou, A., Doudounakis, S., Casals, T. and Baralle, F.E. (
Aartsma-Rus, A., Janson, A.A., Kaman, W.E., Bremmer-Bout, M., van Ommen, G.J., den Dunnen, J.T. and van Deutekom, J.C. (
Wilton, S.D. and Fletcher, S. (
Disset, A., Michot, C., Harris, A., Buratti, E., Claustres, M. and Tuffery-Giraud, S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}