




Abstract
Telomere dynamics are a critical component of both aging and cancer. Telomeres progressively shorten in almost all dividing cells and most human cells do not express or maintain sufficient telomerase activity to fully maintain telomeres. There is accumulating evidence that when only a few telomeres are short, they form end-associations, leading to a DNA damage signal resulting in replicative senescence (a cellular growth arrest, also called the M1 stage). In the absence of cell-cycle checkpoint pathways (e.g. p53 and or p16/Rb), cells bypass M1 senescence and telomeres continue to shorten eventually resulting in crisis (also called the M2 stage). M2 is characterized by many ‘uncapped’ chromosome ends, end-fusions, chromosome breakage fusion–bridge cycles, mitotic catastrophe and a high fraction of apoptotic cells. In a rare M2 cell, telomerase (a cellular reverse transcriptase) can be reactivated or up-regulated, resulting in indefinite cell proliferation. This cellular immortalization is a potentially rate-limiting step in carcinogenesis that is important for the continuing evolution of most advanced cancers. In this perspective we will present our views on the evidence for telomere dysfunction in aging and in cancer progression. We will argue that telomere shortening in the absence of other alterations may be a potent tumor suppressor mechanism and we will discuss the evidence for and against the major molecular mechanisms proposed to initiate replicative senescence.
Received July 1, 2004; revised and accepted September 27, 2004
Evolutionary considerations in aging and cancer
Knowledge of the pathogenesis of cancer includes not only dominant changes that accelerate growth (oncogenes), but, just as importantly, recessive changes involving growth inhibition (tumor suppressors, or gatekeeper genes), elements that control the stability of DNA and chromosomes (caretakers or longevity assurance genes), and programmed cell death pathways (apoptosis genes) ( 1 ). One could speculate that cell division is potentially a risky process, and organisms with renewable tissues have evolved mechanisms to limit the maximal number of permissable divisions in order to prevent the occurrence of genomic instability and premature onset of cancer yet permit appropriate cellular DNA repair and maintenance ( 2 – 11 ).
Cancer cells must accumulate many mutations before acquiring malignant characteristics. Each mutation probably requires at least 20–30 cell divisions: the cell in which an initial mutation occurs must expand to perhaps 1 million cells before there is a reasonable probability of a second mutation occurring. Furthermore, as most mutations are recessive, an additional clonal expansion is required to eliminate the remaining wild-type allele (usually through loss of heterozygosity). Limiting the number of available cell divisions to less than 100 would thus prevent pre-malignant cells from dividing after accumulating only a few mutations, and thus block their progression ( 10 , 11 ). Obviously the most efficient tumor-prevention strategy would be to have few or no available divisions, but this is clearly incompatible with the growth, maintenance and repair needs of the body of long-lived species. How then does one ‘set’ the maximal number of permitted divisions? Having many more divisions than one needs for an average lifespan would increase the risk of cancer without any benefit. The number of permitted divisions has thus probably been reduced to the point of providing ‘optimal’ cell turnover for one's expected lifespan in the wild (e.g. Stone Age conditions for humans). As modern improvements in sanitation, vaccines, antibiotics and other modern medical interventions have extended the average lifespan beyond that, we may now expect that proliferation limits may adversely affect the function of some tissues, especially in situations of chronic diseases involving increased cell turnover.
Senescent cells, while not dividing, remain metabolically active and produce many secreted factors, some of which stimulate and others inhibit the growth of tumors ( 12 – 16 ). This cellular arrest of proliferation is accompanied by changes in cell function (such as changes in secretory pathways, expression of proteases, extracellular matrix components and inflammatory cytokines). In some contexts, a threshold of senescent stromal cells could potentially provide a permissive environment for adjacent pre-malignant epithelial cells to survive, migrate and divide ( 11 ). These alterations in gene expression in senescent cells may change tissue homeostasis and impact on both aging and tumorigenesis in the elderly ( 17 – 21 ). There are good theoretical reasons for believing a regulated and restricted proliferative capacity contributes to declining tissue homeostasis with increasing age. Although the presence of telomere shortening (see next section) provides strong circumstantial evidence that replicative senescence occurs in vivo ( 14 – 40 ), thus far there is only very limited direct evidence for actual physiologic effects of replicative senescence.
The telomere and telomerase connection to aging and cancer
The ends of linear eukaryotic chromosomes contain specialized structures called telomeres. Human telomeres consist of tandem repetitive arrays of the hexameric sequence TTAGGG, with overall telomere sizes ranging from ∼15 kb at birth to sometimes <5 kb in chronic disease states. The telomeric repeats help maintain chromosomal integrity and provide a buffer of potentially expendable DNA. The ends of telomeres are protected and regulated by telomere-binding proteins and form a special lariat-like structure called the t-loop ( 41 ). This packaging or protective cap at the end of linear chromosomes is thought to mask telomeres from being recognized as broken or damaged DNA, thus protecting chromosome termini from degradation, recombination and end-joining reactions ( 41 – 77 ).
The inability of DNA polymerase to replicate the end of the chromosome during lagging strand synthesis (‘end replication problem’) ( 74 ) coupled with possible processing events in both leading and lagging daughters, results in the losses of telomeric repeats each time a cell divides and ultimately leads to replicative senescence. The ability to bypass replicative senescence is thought to be one critical rate-limiting step in the evolution of most malignancies. While there is substantial correlative evidence that there is telomere attrition in pre-cancerous tissues ( 13 – 16 , 22 – 26 , 29 – 31 , 34 – 40 ), the direct evidence that most pre-cancerous cells senesce, and that senescence is a potent tumor suppressor pathway, remains elusive. Most malignant tumors must have a mechanism for bypassing senescence to have the unlimited proliferative capacity that appears to be required for advanced cancers. The loss of cell-cycle checkpoint pathways leads to an extended lifespan but continued telomere losses. This eventually leads to crisis or the M2 stage of replicative senescence. To escape M2, a rare human cell (about 1 in 10 million) ( 8 ) can reactivate or up-regulate telomerase activity ( 78 – 106 ), even though in certain cancer types up-regulation of telomerase can occur at an earlier stage ( 97 ). Even more rarely, a cell may engage an alternative to telomerase for maintaining telomeres ( 107 – 114 ) that appears to involve DNA recombination between telomere sister chromatids (but this pathway will not be covered in this perspective).
Telomerase is a cellular ribonucleoprotein enzyme responsible for adding telomeric repeats onto the 3′ ends of chromosomes ( 57 , 78 ). It has two major components (protein and RNA): an enzymatic human telomerase reverse transcriptase catalytic subunit, hTERT ( 80 , 89 ), and an RNA component (hTR or hTERC) ( 83 ). Telomerase uses its integral RNA component (which contains an 11-bp sequence complementary to the telomeric single stranded overhang) as a template in order to synthesize telomeric DNA (TTAGGG) n , directly onto the ends of chromosomes. After adding six bases, the enzyme pauses while it repositions (translocates) the template RNA for the synthesis of the next 6 bp repeat (i.e. telomerase is processive). This extension of the 3′ DNA template eventually permits additional replication of the C-rich strand, thus compensating for the end-replication problem. The enzyme is expressed in embryonic cells ( 103 ) but the hTERT gene undergoes silencing and the enzyme activity is repressed. Telomerase is present in adult male germline cells, but is undetectable in most normal somatic cells except for proliferative cells of renewal tissues where there is regulated telomerase activity (e.g. hematopoietic proliferating stem-like cells, activated lymphocytes, proliferative transit amplifying cells of the epidermis, proliferative endometrium and intestinal crypt cells) ( 115 , 116 ). In normal somatic cells, even including stem-like cells expressing telomerase, progressive telomere shortening is observed, eventually leading to greatly shortened telomeres and to a limited ability to continue to divide. This implies that the functional telomerase activity in these stem-like cells may be enough to slow but not prevent telomere shortening.
Direct evidence linking telomere shortening to replicative senescence can be demonstrated by producing telomerase activity in telomerase-negative cells following the introduction of only the hTERT catalytic component (normal cells constitutively express the RNA component of telomerase). Normal human cells stably expressing transfected telomerase can divide indefinitely, providing direct evidence that telomere shortening controls replicative senescence ( 79 ). Furthermore, elongating telomeres with telomerase and then excising the exogenous gene results in a greatly extended lifespan, demonstrating that it is the length of the telomere rather than telomerase itself that is responsible for the proliferative limits ( 101 ). The introduction of hTERT either before M1 or in between M1 and M2 results in direct immortalization, thus demonstrating the importance of telomeres in both stages of replicative senescence ( Figure 1 ). Cells with introduced telomerase maintain a normal chromosome complement for a considerable period and continue to grow in a normal manner ( 91 ). These observations provide direct evidence for the hypothesis that telomere length determines the proliferative capacity of human cells.
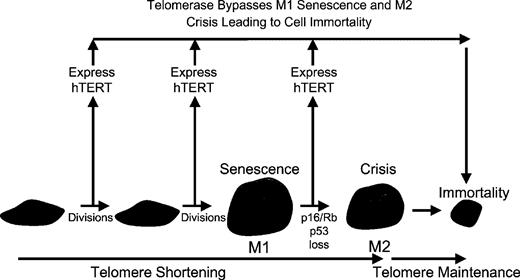
The ectopic expression of the catalytic subunit of hTERT results in immortalization of human cells if telomeres are rate-limiting for continued cell proliferation. Telomeres are thus important in both senescence (M1) and crisis (M2) as hTERT introduction either before M1 or after M1 results in cell immortalization. If the introduction of hTERT does not result in immortalization this reflects another type of growth arrest that is telomere-independent and is likely to reflect inadequate culture conditions leading to STASIS or what has been termed premature senescence or culture shock.
Senescence and STASIS
In addition to progressive telomere shortening (leading to replicative senescence), telomere dysfunction can be initiated by a change of state (‘uncapping’) that leads to a rapid induction of growth arrest that has also been termed senescence ( 10 , 117 – 133 ). When the telomeric DNA structure or sequence is altered, or telomere proteins are depleted or mutated, cells undergo chromosome end-associations and fusions leading to growth arrest or death. This growth arrest is similar to telomere-based replicative senescence in most, but not all, regards. For example, in both types of growth arrest cells cannot divide even if stimulated by mitogens, cells remain metabolically active, and cells show characteristic changes in morphology.
It has been demonstrated that growth inhibitory genes can be activated in cell culture and in vivo due to a variety of environmental stresses in a process variously termed, premature senescence, culture shock, stress-induced senescence and STASIS ( 10 , 119 – 121 , 123 ). As this type of growth arrest has received many names that are easily confused with telomere-based replicative senescence, the term ‘STASIS’ (STress or Aberrant Signaling Induced Senescence) is gaining acceptance ( 123 ). While cells undergoing replicative senescence can be immortalized by expression of hTERT to maintain telomere homeostasis, this does not occur in cells undergoing growth arrest due to STASIS ( 124 – 129 ). Thus, one working (if not circular) definition for replicative senescence would be as follows: growth arrest under adequate culture conditions is replicative senescence if telomeres are rate-limiting for continued cell proliferation and hTERT can directly immortalize the cells. It is important to make this distinction as the triggering agents are different (short telomeres versus a stress or damage-induced signaling pathway that may or may not involve telomeres).
STASIS may be an evolutionarily conserved mechanism that helps guard cells against oncogenic insults. It would be advantageous to prevent normal and pre-cancerous cells from proliferating if placed in an inappropriate environment (e.g. not receiving the proper mitogens or other signals from their neighbors), or following genotoxic stresses likely to induce multiple mutations ( 10 ). Treatment of most types of tumor cells with conventional anticancer therapies activates DNA damage-signaling pathways and can induce a rapid onset of STASIS. Other examples include the growth arrest elicited in normal cells in response to oncogenic Ras ( 120 ) or Raf ( 122 ), transfection of oligonucleotides, and inadequate culture conditions (plastic dishes, oxidative damage, etc.) ( 10 , 121 ). In these instances, the expression of hTERT does not result in the bypass of STASIS, thus demonstrating that this type of growth arrest does not involve counting cell replications (e.g. telomere-based replicative senescence) ( 117 – 133 ).
In both replicative senescence and STASIS, the initiating event can be triggered by similar mechanisms including recognition by cellular sensors of DNA double-strand breaks leading to the activation of cell-cycle checkpoint responses and recruitment of DNA repair foci (see next section). A fundamental area of recent investigations is to understand the diverse signaling pathways that cause cells, in some contexts, to undergo replicative senescence and in other contexts to initiate STASIS or apoptotic signaling programs. In summary, it is generally believed that somatic cells in organisms with renewable tissues have evolutionarily conserved defence mechanisms that guard against unrestrained proliferation. In some instances, the cellular proliferation control pathways may be potent anticancer protection mechanisms (tumor suppressor genes) so when there are sufficient acute stresses (or damage) cells immediately growth arrest or undergo cell death. Thus, normal cells in the context of genotoxic insults may have innate and probably highly conserved defence mechanisms that initiate signal transduction cascades leading to growth arrest or apoptosis ( 10 ). Having cellular mechanisms that cause cells to stop growing or to die in the face of acute damage would be highly advantageous.
What induces replicative (M1) senescence?
There is persuasive evidence for progressive telomere shortening being the counting mechanism of replicative senescence ( 8 , 79 ), but there remain some uncertainties about the actual initiating events that ‘triggers’ senescence. While there are many models of senescence that are difficult to test experimentally, the current ideas that are being tested include, telomere position effects ( 134 – 136 ), DNA damage signaling from short telomeres ( 137 – 141 ) and loss of the 3′ G-rich telomere single-strand overhangs ( 69 ).
Telomere position effects
One model combining telomere progressive shortening and changes in gene expression is called telomere position effects (TPE). TPE is dependent on telomere length and is characterized by an ‘all or nothing’ effect that is heritable and semi-stable. The idea is that when cells have long telomeres, genes near telomeres may be silenced due to chromatin effects near telomeres ( Figure 2 ). As cells age (and telomeres are shorter), there may be some de-repression of genes near telomeres eventually leading to reactivation of other previously silenced genes. This could occur on all or only a subset of chromosome ends. The dependence of TPE on telomere length provides a mechanism for the modification of gene expression throughout the replicative lifespan of human cells ( Figure 2 ) ( 134 ). A number of proteins have been reported to change in expression level as a function of the replicative age of the cell. The existence of TPE in mammalian cells ( 134 – 136 ) raises the possibility that some pre-senescent changes could be ‘programmed’ by the progressive shortening of telomeres with ongoing cell division, leading to altered patterns of gene expression that might affect both cell and organ function. While this is a feasible mechanism for downstream effects of telomere shortening, a biologically relevant role for TPE has not yet been demonstrated in higher organisms. Thus, it will be important to identify endogenous genes whose expression is influenced by telomere length in order to determine whether TPE actually influences the physiology of aging or cancer. As the hTERT gene is only a few hundred kilobases from the end of chromosome 5p, one could speculate that TPE (silencing) of hTERT limits the maximal length of human telomeres during embryogenesis ( 136 ). Finally, the recent evidence that the actual signal for growth arrest is a result of DNA damage signaling from ‘too-short’ telomeres ( Figure 2 ) is strong and argues against TPE as a proximate cause of senescence.
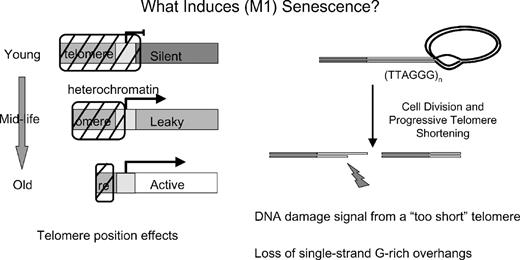
The leading models for the induction of replicative senescence are TPE (left side) and a DNA damage signal from perhaps only one or two short telomeres (right side). It is now known that there is not one sentinel short telomere that initiates replicative senescence. Instead there are about 10 short telomeres and in any given cells a single short telomere is sufficient to induce replicative senescence. An alternative hypothesis is that the loss of most of the G-rich single-strand overhangs induces senescence.
DNA damage signal from a ‘too short’ telomere
The responses of cells to progressive telomere shortening (replicative senescence) versus acute uncapping of telomeres may or may not be similar. Since loss of telomere function can lead to cell-cycle arrest or cell death, one possibility is that DNA damage/repair responses may be involved in both programs. Recently, however, the molecular mechanism(s) by which a single or a few short telomeres signal the growth arrest caused by replicative senescence is starting to emerge ( 137 – 140 ).
Several studies have demonstrated that DNA damage signals from telomeres at senescence have typical DNA damage foci containing factors such as gamma H2AX (a phosphorylated variant of histone 2A that associates with DNA double-strand breaks). Phosphorylation of gamma H2AX occurs on H2AX histones that are located around DNA breaks and co-localizes with breast cancer susceptibility gene-1, MDC1, MRE11, Nijmegen breakage syndrome 1, RAD50 and 53BP1 ( 137 – 143 ). It has also been shown that telomere-induced foci are rapidly observed by over-expressing a dominant-negative telomere repeat binding factor 2 protein (TRF2 that protects telomeres) ( 10 , 139 , 140 , 142 ). In this type of telomere-induced damage, the uncapping of telomeres is rapid, growth arrest occurs quickly, and there is also the loss of the single-stranded G-rich telomeric overhang ( 142 ).
Thus, experimental procedures that unfold the lariat-like telomere end-protection lead to an ‘immediate senescence’ or STASIS and this has led to the concept that the end-fusions and chromosome breakage–fusion cycles are also the cause of telomere-based replicative senescence ( 12 , 16 , 51 , 142 ). Whether this rapid uncapping of telomeres reflects replicative senescence (M1) or crisis (M2) is not clear ( 141 ). While many of the details are yet to be determined, it now appears that at M1 senescence there are weak telomere initiated end-associations ( Figure 3 ) leading to a few gamma H2AX-induced DNA repair foci, and this may be sufficient to trigger replicative senescence (without activating a cell death program) ( 138 ). In the absence of telomerase or another mechanism to maintain telomeres, the replicative senescence pathway is engaged and cells can remain in a non-proliferative state for years. The timing of senescence is dependent on the shortest telomeres ( 61 , 93 , 101 , 137 , 138 ). There is not one sentinel telomere but approximately 10 telomere probe-signal-free chromosome termini that can potentially form end-associations with each other at senescence ( 138 ). However, in any given cell it is only one or two of the shortest telomeres that produce gamma H2AX repair foci ( 137 , 138 ) and end-associations ( 138 ) and it is dependent on signaling pathways involving ataxia-telangiectasia mutated kinase, p53 and p21 CIP1 but not p16 INK4a ( 141 ).
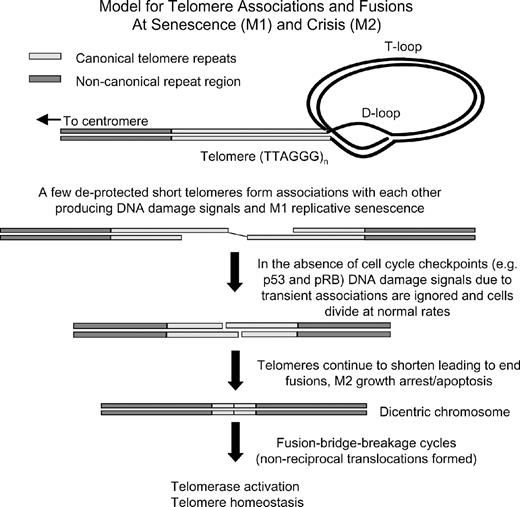
There is mounting evidence that the end of telomeres form a special lariat-like structure that protect the end of chromosomes from being recognized as broken DNA needing repair. Perhaps when one or two short telomere ends within a cell become deprotected, they can form associations with each other and this can lead to senescence. Insults that rapidly lead to telomere deprotection can also result in a growth arrest (STASIS) but this is likely to be a different molecular mechanism of growth arrest (as many telomeres form end fusions) when compared with telomere-based replicative senescence (where only one or two ends form associations). In the absence of important cell-cycle checkpoints, replicative senescence is bypassed or abrogated and this will eventually lead to end fusions, apoptosis, fusion bridge–breakage cycles and genomic instability.
In the absence of p53 function, cells divide beyond M1 senescence (extended lifespan period) while telomeres continue to shorten until mitotic catastrophe or apoptosis become the dominant response of cells attempting to divide ( Figure 3 ) ( 8 ). There may be a difference between the phenotype of end-associations (M1) when a few short telomeres become metastable and induced DNA damage checkpoint responses and M2 when so many telomeres become sufficiently short (unprotected) that the frequency of dicentrics/breakage-fusion events leads to cell death ( Figure 3 ).
Does loss of 3′ G-rich single-strand overhangs cause M1 senescence?
A final model for the initiating event in replicative senescence is that it is the loss of the 3′ G-rich single-strand overhangs that trigger senescence ( 69 ). This idea is based on the observation that 60–85% of the 3′ G-rich overhangs are eroded at senescence and that the remaining overhangs progressively shorten after cells bypass M1. This model raises an important question of whether the activation of cellular DNA damage response by dysfunctional telomeres initiates telomere 3′ overhang shortening, or is it that shortened overhangs activates DNA damage responses? If only one or two telomere ends initiate replicative senescence, then this model of global loss of overhangs is likely to be a secondary event. One should not see a 60–85% reduction in all overhangs using an assay that measures all 92 ends if only one or two ends are limiting for growth within a given cell. A more likely explanation for the results is that loss of overhangs is a secondary, epi-phenomenon, perhaps caused by end-processing events following activation of stress/DNA damage response and is not the direct cause of replicative senescence.
Summary/perspective
Telomere positional effects exist in human cells but there is no direct evidence for regulating the onset of replicative senescence. Loss of the 3′ G-rich overhang is probably not the proximal cause of replicative senescence but is likely a secondary phenomenon due to culture conditions. Finally, the timing of senescence is dependent on the shortest telomeres and there is not one sentinel short telomere but approximately 10 short chromosome termini that form the vast majority of end-associations with each other at senescence. In any given cell it is only one or two of the shortest telomeres that produce gamma H2AX repair foci and end-associations ( 138 ).
There are also many outstanding questions concerning the transition between the M1 (senescence) and M2 (crisis) and the unlimited proliferation during oncogenesis. The genomic instability from breakage–fusion cycles at M2 presumably contributes to the reactivation/up-regulation of telomerase that allows immortalization of the cells and telomere maintenance. However, once telomerase has been activated, do short telomeres continue to contribute to additional genomic instability in cancer cells? If the biological purpose of replicative senescence is to block the ability of pre-malignant cells to divide, why is there an increased incidence of cancer in the genetic disease dyskeratosis congenita? Some cases of this disease are caused by mutations in the RNA component of telomerase that result in decreased telomerase activity during development and in stem-like cells, which results in telomeres being much shorter than expected ( 22 – 26 ). If short telomeres in DKC contribute to genomic instability and cancer, how do short telomeres in normal individuals prevent the progression of pre-cancerous cells rather than promoting their genomic instability and accelerating their progression? Would elongating telomeres in chronic diseases such as ulcerative colitis (where telomeres have become very short) ( 144 ) or in dyskeratosis congenita ( 22 – 26 ) be beneficial by preventing the genomic instability that leads some of the cells to become malignant, or be harmful by removing the block to progression of the pre-cancerous cells already present? Resolving these apparent contradictions will be essential in designing the best therapeutic approaches for exploiting our increasing knowledge of telomere biology for the treatment of both aging and cancer.
We acknowledge support from P50 CA070907 and AG07992.
References
Vogelstein,B. and Kinzler,K.W. (
Faragher,R.G.A. and Kipling,D. (
Kapahi,P., Boulton,M.E. and Kirkwood,T.B. (
Hayflick,L. (
Hayflick,L. and Moorhead,P.S. (
Campisi,J. (
Wright,W.E. and Shay,J.W. (
Wright,W.E. and Shay,J.W. (
Shay,J.W. and Roninson,I.B. (
Maser,R.S. and DePinho,R.A. (
Wong,K.K. and DePinho,R.A. (
Meeker,A.K. and De Marzo,A.M. (
Meeker,A.K., Hicks,J.L., Iacobuzio-Donahue,C.A., Montgomery,E.A., Westra,W.H., Chan,T.Y., Ronnett,B.M. and De Marzo,A.M. (
Sharpless,N.E. and DePinho,R.A. (
Slagboom,P.E., Droog,S. and Boomsma,D.I. (
Benetos,A., Okuda,K., Lajemi,M., Kimura,M., Thomas,F., Skurnick,J., Lobat,C., Bean,K. and Aviv,A. (
Brouilette,S., Singh,R.K., Thompson,J.R., Goodall,A.H. and Samani,N.J. (
Okuda,K., Bardequez,A., Gardner,J.P., Rodriguez,P., Ganesh,V., Kimura,M., Skurnick,J., Awad,G. and Aviv,A. (
Graakjaer,J., Bischoff,C., Korsholm,L., Holstebroe,S., Vach,W., Bohr,V.A., Christensen,K. and Kolvraa,S. (
Vulliamy,T., Marrone,A., Goldman,F. et al . (
Mitchell,J.R., Wood,E. and Collins,K. (
Shay,J.W. and Wright,W.E. (
Shay,J.W. and Wright,W. E. (
Cawthon,R.M., Smith,K.R., O'Brien,E., Sivatchenko,A. and Kerber,R.A. (
Lindsey,J., McGill,N., Lindsey,L., Green,D. and Cooke,H. (
Wu,X. Amos,C.I., Zhu,Y., Zhao,H., Grossman,B.H., Shay,J.W., Swan,G.E., Benowitz,N.L., Luo,S. and Spitz,M.R. (
Kitada,T., Seki,S., Kawakita,N., Kuroki,T. and Monna,T. (
Wiemann,S.U., Satyanarayana,A., Tsahuridu,M. et al . (
Samani,N.J., Boultby,R., Butler,R., Thompson,J.R. and Goodall,A.H. (
Obana,N., Takagi,S., Kinouchi,Y. et al . (
Hastie,N.D., Dempster,M., Dunlop,M.G., Thompson,A.M., Green,D.K. and Allshire,R.C. (
Chadeneau,C., Hay,K., Hirte,H.W., Gallinger,S. and Bacchetti,S. (
Engelhardt,M., Drullinsky,P., Guillem,J. and Moore,M.A. (
Tang,R., Cheng,A.J., Wang,J.Y. and Wang,T.C. (
Yan,P., Saraga,E.P., Bouzourene,H., Bosman,F.T. and Benhattar,J. (
Rudolph,K.L., Millard,M., Bosenberg,M.W. and DePinho,R.A. (
Herrera,E., Samper,E., Martin-Caballero,J., Flores,J.M., Lee,H.W. and Blasco,M.A. (
de Lange,T. (
Allshire,R.C., Dempster,M. and Hastie,N.D. (
Baird,D.M., Coleman,J., Rosser,Z.H. and Royle,N.J. (
Baird,D.M., Rowson,J., Wynford-Thomas,D. and Kipling,D. (
Blasco,M. (
Blasco,M.A., Lee,H.W., Hande,M.P., Samper,E., Lansdorp,P.M., DePinho,R.A. and Greider,C.W. (
Blasco,M.A. (
Broccoli,D., Smogorzewska,A., Chong,L. and de Lange,T. (
Chan,S.R. and Blackburn,E.H. (
der-Sarkissian,H., Bacchetti,S., Cazes,L. and Londono-Vallejo,J.A. (
Ferreira,M.G., Miller,K.M. and Cooper,J.P. (
Goytisolo,F.A. and Blasco,M.A. (
Griffith,J.D., Comeau,L., Rosenfield,S., Stansel,R.M., Bianchi,A., Moss,H. and de Lange,T. (
Greider,C.W. and Blackburn,E.H. (
Harley,C.B. (
Harley,C.B., Fletcher,A.B. and Greider,C.W. (
Hemann,M.T., Strong,M.A., Hao,L.Y. and Greider,C.W. (
Huffman,K.E., Levene,S.D., Tesmer,V.M., Shay,J.W. and Wright,W.E. (
Keys,B., Serra,V., Saretzki,G. and von Zglinicki,T. (
Levy,M.Z., Allsopp,R.C., Futcher,A.B., Greider,C.W. and Harley,C.B. (
Londono-Vallejo,J.A., DerSarkissian,H., Cazes,L. and Thomas,G. (
Martens,U.M., Chavez,E.A., Poon,S.S., Schmoor,C. and Lansdorp,P.M. (
Moyzis,R.K., Buckingham,J.M., Cram,L.S. et al . (
Olovnikov,A.M. (
Stewart,S.A., Ben-Porath,I., Carey,V.J., O'Connor,B.F., Hahn,W.C. and Weinberg,R.A. (
Suda,T., Fujiyama,A., Takimoto,M., Igarashi,M., Kuroiwa,T., Waguri,N., Kawai,H., Mita,Y. and Aoyagi,Y. (
van Steensel,B., Smogorzewska,A. and de Lange,T. (
von Zglinicki,T. (
von Zglinicki,T., Saretzki,G., Docke,W. and Lotze,C. (
Wright,W.E. and Shay,J.W. (
Wright,W.E. and Shay,J.W. (
Smith,S., Giriat,I., Schmitt,A. and de Lange,T. (
Bodnar,A.G., Ouellette,M., Frolkis,M., Holt,S.E., Chui,C.-P., Morin,G.B., Harley,C.B., Shay,J.W., Lichsteiner,S. and Wright,W.E. (
Bryan,T.M. and Cech,T.R. (
Chan,S.W.-L. and Blackburn,E.H. (
Coursen,J.D., Bennett,W.P., Gollahon,L., Shay,J.W. and Harris,C.C. (
Feng,J., Funk,W.D. and Wang,S.-S. et al . (
Hahn,W.C., Counter,C.M., Lundberg,A.S., Beijersbergen,R.L., Brooks,M.W. and Weinberg,R.A. (
Holt,S.E. and Shay,J.W. (
Holt,S.E., Wright,W.E. and Shay,J.W. (
Jiang,X.R., Jimenez,G., Chang,E. et al . (
Kim,N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L., Coviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (
Lingner,J., Cooper,J.P. and Cech,T.R. (
Masutomi,K., Yu,E.Y., Khurts,S. et al . (
Morales,C.P., Holt,S.E., Ouellette,M., Kaur,K.J., Yan,Y., Wilson,K.S., White,M.A., Wright,W.E. and Shay,J.W. (
Ouellette,M.M., McDaniel,L.D., Wright,W.E., Shay,J.W. and Schultz,R.A. (
Ouellette,M.M., Liao,M., Herbert,B.S., Johnson,M., Holt,S.E., Liss,H.S., Shay,J.W. and Wright,W.E. (
Shay,J.W. (
Shay,J.W. (
Shay,J.W. and Wright,W.E. (
Shay,J.W. and Bacchetti,S. (
Shay,J.W. and Wright,W.E. (
Shay,J.W. (
Granger,M.P., Wright,W.E. and Shay,J.W. (
Steinert,S., Shay,J.W. and Wright,W.E. (
Vaziri,H. and Benchimol,S. (
Wright,W.E., Piatyszek,M.A., Rainey,W.E., Byrd,W. and Shay,J.W. (
Wright,W.E., Brasiskyte,D., Piatyszek,M.A. and Shay,J.W. (
Wyllie,F.S., Jones,C.J., Skinner,J.W., Haughton,M.F., Wallis,C., Wynford-Thomas,D., Faragher,R.G. and Kipling,D. (
Yang,J., Chang,E., Cherry,A.M., Bangs,C.D., Oei,Y., Bodnar,A., Bronstein,A., Chiu,C.P. and Herron,G.S. (
Bryan,T.M., Marusic,L., Bacchetti,S., Namba,M. and Reddel,R.R. (
Bryan,T.M., Englezou,A., Gupta,J., Bacchetti,S. and Reddel,R.R. (
Dunham,M.A., Neumann,A.A., Fasching,C.L. and Reddel,R.R. (
Reddel,R.R. (
Ford,L.P., Zou,Y., Pongracz,K, Gryaznov,S.M., Shay,J.W. and Wright,W.E. (
Bechter,O.E., Shay,J.W. and Wright,W.E. (
Bechter,O, Zou,Y., Walker,W., Wright,W.E. and Shay,J.W. (
Bechter,O.E., Zou,Y., Shay,J.W. and Wright,W.E. (
Aisner,D.L., Wright,W.E. and Shay,J.W. (
Forsyth,N.R, Wright,W.E. and Shay,J.W. (
Ramirez,R.D., Morales,C.P., Herbert,B.S., Rohde,J.M., Passons,C., Shay,J.W. and Wright,W.E. (
Robles,S.J. and Adami,G.R. (
Serrano,M. and Blasco,M. (
Serrano,M., Lin,A.W., McCurrach,M.E., Beach,D. and Lowe,S.W. (
Sherr,C.J. and DePinho,R.A. (
Zhu,J., Woods,D., McMahon,M. and Bishop,J.M. (
Drayton,S. and Peters,G. (
Jones,C.J., Kipling,D., Morris,M. et al . (
Dickson,M.A., Hahn,W.C., Ino,Y., Ronfard,V., Wu,J.Y., Weinberg,R.A., Louis,D.N., Li,F.P. and Rheinwald,J.G. (
Farwell,D.G., Shera,K.A., Koop,J.I., Bonnet,G.A., Matthews,C.P., Reuther,G.W., Coltrera,M.D., McDougall,J.K. and Klingelhutz,A.J. (
Forsyth,N.R., Evans,A.P., Shay,J.W. and Wright,W.E. (
Jarrard,D.F., Sarkar,S., Shi,Y. et al . (
Kiyono,T., Foster,S.A., Koop,J.I., McDougall,J.K., Galloway,D.A. and Klingelhutz,A.J. (
Ramirez,R.D., Herbert,B.-S., Vaughn,M.B, Zou,Y., Gandia,K., Morales,C.P., Wright,W.E. and Shay,J.W. (
Dimri,G.P., Lee,X., Basile,G. et al . (
Busutil,R.A., Rubio,M., Dolle,M.E.T., Campisi,J. and Vijg,J. (
Naka,K., Tachibana,A., Ikeda,K. and Motoyama,N. (
Baur,J.A., Zou,Y., Shay,J.W. and Wright,W.E. (
Koering,C.E., Pollice,A., Zibella,M.P. et al . (
Shay,J.W. and Wright,W.E. (
d'Adda di Fagagna,F., Reaper,P.M., Clay-Farrace,L., Fiegler,H., Carr,P., Von Zglinicki,T., Saretzki,G., Carter,N.P. and Jackson,S.P. (
Zou,Y., Sfeir,A., Shay,J.W. and Wright,W.E. (
Sedelnikova,O.A., Horikawa,I., Zimonjic,D.B., Popescu,N.C., Bonner,W.M. and Barrett,J.C. (
Gire,V., Roux,P., Wynford-Thomas,D., Brondello,J.M. and Dulic,V. (
Herbig,U., Jobling,W.A., Chen,P.B.C., Chen,D.J. and Sedivy,J.M. (
Takai,H., Smogorzewska,A. and de Lange,T. (
Shay,J.W. and Wright,W.E. (

{kind=link}
{kind=link}
{kind=link}