






Abstract
The febrile response is thought to be mediated by endogenous mediators, generically called “endogenous pyrogens.” In the classical model of pathogenesis, induction of fever is mediated by the release of pyrogenic cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, and interferons into the bloodstream in response to exogenous pyrogens. These mediators act at the level of the organum vasculosum of the lamina terminalis in the central nervous system (CNS), inducing synthesis of prostaglandins, which are the central mediators of the coordinated responses leading to fever. However, analysis of recent data suggests that multiple pathways may be involved in the induction of fever by cytokines, such as local cytokine production leading to signaling through vagal fibers, release of cytokine-induced circulating mediators at the tissue level, the use of membrane-bound cytokines as mediators, or the local release of cytokines in the hypothalamus by circulating activated monocytes. In addition, certain bacterial products can stimulate cytokine production directly at the level of hypothalamus, probably by activation of Toll-like receptors. A multipathway mechanism for the induction of fever is therefore suggested.
Fever is one of the most frequent clinical signs encountered in human pathology, especially during infections. When a microorganism invades a host and enters into its bloodstream, stimulation of leukocytes and of other cell types determine the synthesis and release of a group of molecules that can induce fever. The nature of these mediators, called endogenous pyrogens (EP), has been unknown until recently. Studies performed at the end of the 1970s demonstrated that leukocytes, when stimulated with bacterial products, synthesize protein mediators called cytokines, some of which have potent EP-like properties [1]. It thus became apparent that the EP activity in the plasma is represented by several of these cytokines with proinflammatory properties: IL-1 [2], TNF and lymphotoxin [3], IL-6 [4], and IFNs [5]. These proinflammatory cytokines reach the CNS where, through induction of central mediators such as prostaglandins (PGs), they are able to increase the temperature set point and cause fever [6, 7]. The increase in body temperature has several advantages during infections: it results in inhibition of bacterial growth, increased bactericidal activities of neutrophils and macrophages, stimulation of acute-phase protein synthesis, iron sequestration, anorexia, and somnolence [6]. Through these physiological changes, fever has an important adaptive role in the host's survival during infectious episodes [8].
Classical Model for the Pathogenesis of Fever
The initial step in the cascade of events leading to fever is considered to be stimulation of leukocytes by various exogenous stimuli, called exogenous pyrogens. These stimuli are mainly represented by bacterial products, toxins, or microbes themselves. Live and heat-killed gram-negative [9] and gram-positive [10] bacteria elicit fever when administered to animals. Various bacterial products released during infection are also potent inducers of cytokine production and fever, including the lipopolysaccharide component of the gram-negative bacteria [11], peptidoglycans, and their muramyl peptide derivatives [12, 13], bacterial toxins such as enterotoxins of Staphylococcus aureus and group A streptococcal erythrogenic toxins [14], the mannan and glucan components of pathogenic fungi such as Candida albicans [9, 15], and viral components such as double-stranded RNA [16, 17].
Stimulation of the cells of the immune system, especially monocytes/macrophages and neutrophils, by the various exogenous pyrogens leads to synthesis of proinflammatory mediators that act as EP for the stimulation of fever [1]. EP released into circulation are transported to the brain vasculature, where either through active transport across the blood-brain barrier [18] or by passive transport through the fenestrated capillaries in the circumventricular organs [19], they reach the CNS. A key structure for EP recognition in the CNS is thought to be organum vasculosum of the lamina terminalis (OVLT) [19, 20]. At this level, the EP induces the release of central mediators such as PG, especially of the PG class E2 (PGE2), which mediates the elevation in the temperature set point [6, 7]. A long-standing debate regarding the need for PG in the induction of fever, or of other mediators such as corticotrophin-releasing factor or platelet-activating factor [6], has been recently settled by showing that mice lacking the PGE receptor subtype EP3 are not able to develop a febrile response [21]. Passive diffusion of PG, PG stimulation of neurotransmitters such as cyclic adenosine monophosphate (cAMP) in the hypothalamus, or both result in the elevation of the thermostatic set point, which in turn activates the coordinated endocrine, autonomic, and behavioral responses leading to fever [7].
Circulating Cytokines as EP
The role of proinflammatory cytokines in the pathogenesis of fever has been discovered at the end of the 1970s and the beginning of the 1980s, with the purification of IL-1 and demonstration of its pyrogenic properties [1]. Later it became apparent that other cytokines such as TNF, IFNs, and IL-6 are also able to induce fever and can be considered independent EP [1, 6, 14] (table 1).

Proinflammatory cytokines that act as endogenous pyrogens.
IL-1α and IL-1β are considered to be the most important EPs. Injection of IL-1 into experimental animals has potent pyrogenic effects, and administration of an excess dose of IL-1 receptor antagonist (IL-1Ra) can prevent fever [22]. The pyrogenic effects of IL-1 are mediated through the IL-1 receptor (IL-1R) type I and IL-1R accessory protein (IL-1RAcP) [23, 24]. In humans, IL-1 appears to be the most potent pyrogenic cytokine. After injection of recombinant IL-1α and IL-1β into patients with solid tumors or patients who had undergone bone marrow transplantation, chills and fever were observed in almost all patients [25]. The febrile response increased in magnitude with increasing doses [26, 27], and fever was reduced by administration of indomethacin [28]. More controversial are the data regarding the role of IL-1 in mediating fever induced by lipopolysaccharides (LPS). Although some studies reported decreased LPS-induced fever after treatment with IL-1Ra [29] or anti–IL-1 antiserum [30] and inhibition of LPS-induced fever in IL-1β−/− mice [31], others were not able to reproduce these data in IL-1RAcP knockout mice [24] and even reported hyperresponsive febrile reactions after LPS challenge in IL-1β knockout mice [32]. The reasons for these discrepancies are unclear. In contrast, IL-1β seems to be the major mediator of fever during infection with a poxvirus [33] or after injection of turpentine [34].
TNF is a proinflammatory cytokine that shares many biologic properties with IL-1 [35]. TNF injection induces a typical fever in rabbits that is indistinguishable from IL-1 [3]. Moreover, TNF induces a second fever peak 3–4 h after challenge, which is suggested to be mediated through induction of endogenous IL-1 production [1, 3]. Recombinant human TNF is highly pyrogenic in humans, and the fever induced is rapid and associated with generalized malaise and joint pain [1]. It is of note that when mice challenged with LPS were treated with TNF-soluble receptors or anti-TNF antiserum, they responded with higher temperature peaks [36]. Similarly, TNF double receptor knockout mice responded with exacerbated febrile responses to LPS, showing that TNF has cryogenic properties in this model [37].
IL-6 is a cytokine initially described as a potent acute-phase protein inducer [38]. It has been shown that IL-6 elicited fever when injected into rabbits and that IL-6 concentrations were correlated with fever in human patients with burns [39]. Although one of the most potent proinflammatory properties of IL-6 is the stimulation of PG synthesis, IL-6 induction of fever in rabbits requires 50- to 100-fold higher amounts of IL-6 than of IL-1 [1]. The pivotal role played by IL-6 in the induction of fever was recently demonstrated in mice deficient in IL-6, which were unresponsive to the pyrogenic effects of either LPS, IL-1β, or TNF [40, 41]. Given that IL-6 expression is under the control of TNF and IL-1 [42], it has been proposed that IL-6 is a downstream mediator of fever from IL-1 and TNF [41]. Figure 1 depicts this cytokine cascade of fever induction, in which initial stimulation of IL-1 and TNF by bacterial products induces secondary synthesis of IL-6, with subsequent induction of PG synthesis in the CNS and fever.
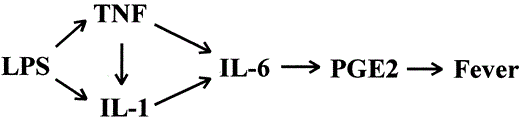
The cascade of cytokines acting as endogenous pyrogens in the pathogenesis of fever induced by bacterial products such as lipopolysaccharide (LPS). PGE2, prostaglandin E2.
IFNs were described as antiviral substances, with potent immunostimulatory activities such as enhanced expression of class I and II major histocompatibility complex antigens and stimulation of natural killer activity. When injected into rabbits, IFN-α induces a monophasic fever that peaks 80–90 min after the injection, which is mediated through induction of PG synthesis in the brain [5]. IFNs were the first cytokines to be administered to humans, and fever was a constant side effect encountered with treatments with IFN-α [43], IFN-β [44], or IFN-γ [45, 46]. Species specificity made it difficult to study the pyrogenic properties of IFNs in animals, and it is still unclear whether some of the EP properties of IFNs are due to indirect stimulation of IL-1, TNF, or both.
In addition to the proinflammatory cytokines acting as direct EP, other endogenous inducers can have indirect pyrogenic properties through their capacity to induce synthesis of EP. Infusion of recombinant IL-2 into humans induces fever that starts 3–4 h after injection [47]. However, the fact that by the time of fever, the IL-2 concentrations in the patients are low [48], and considering the capacity of IL-2 to induce production of IL-1 and TNF [49, 50], together suggest that the pyrogenic properties of IL-2 are mediated through induction of IL-1 and TNF. Other potentially important endogenous inducers are granulocyte-macrophage-colony stimulating factor, immune complexes, and uric acid crystals, all molecules with pyrogenic potential through their capacity to induce production of IL-1 and TNF [51]. An important role as endogenous inducer of fever may be reserved to the complement system. On the one hand, this is due to cytokine stimulation by the complement factors C3a and C5a [52, 53]. On the other hand, the fever response after LPS challenge is blocked in guinea pigs with low complement levels obtained by pretreatment with cobra venom factor, suggesting a direct role of the complement system in the induction of fever [54].
Circulating versus Local EP
A key event in the induction of fever in the classical model of pathogenesis described above is the release of pyrogenic cytokines into the bloodstream to mediate the signal leading to a febrile response from the site of inflammation to the thermoregulatory center in the hypothalamus (figure 2, panel I). However, there are aspects in the induction of fever that contradict this dogma. Experimental studies have shown that after LPS injection, fever precedes the appearance of cytokines in the circulation [55]. Clinical studies have failed in finding detectable levels of EP in specific patient groups with febrile conditions such as typhoid fever [56], fever of unknown origin [57], and Pneumocystis carinii pneumonia [58]. In severe infections with septic shock, proinflammatory cytokines are detected in the circulation only for a short time compared with the period the duration of fever [59]. In addition, release of high amounts of anti-inflammatory cytokines has a strong inhibitory effect on the action of pyrogenic cytokines. Moreover, continuous infusion of cytokines in animals leads to transient rather than ongoing fever [60]. Although it is conceivable that a combination of cytokines acting at very low concentrations may provide the signal for the induction of fever or that as-yet unidentified pyrogenic cytokines act as circulating EP, there are data in the literature that suggest that alternative pathways besides circulating pyrogenic cytokines may efficiently induce fever in response to peripheric stimulation.
![Panel I, the classical model for the induction of fever, in which circulating pyrogenic cytokines represent the key event. Panels II and III, an alternative model in which local production of proinflammatory cytokines at the level of infected tissues stimulates either the release of secondary mediators into circulation (panel II) or primary vagal terminals in the liver (panel III). The secondary mediators (e.g., soluble phospholipase A2 [PLA2]) induce prostaglandin release in the organum vasculosum of the lamina terminalis (OVLT) and subsequently fever. Alternatively, signals through vagal afferent fibers and A1/A2 noradrenergic cell groups in brain stem reach the OVLT, where the febrile response is induced. Panels IV and V, local cytokine production and release at the endothelial level in OVLT, the key event in the induction of fever. The cytokines are either released upon adherence to the endothelium by activated monocytes in which expression of pyrogenic cytokines mRNA is high (panel IV) or produced directly by endothelial cells stimulated by exogenous pyrogens circulating in the bloodstream (panel V). Panel VI, the possible involvement of cell-associated cytokines on the membrane of activated monocytes for the stimulation of endothelial cells in OVLT.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cid/31/Supplement_5/10.1086_317513/1/m_31-Supplement_5-S178-fig002.gif?Expires=1716319284&Signature=Wtap3Lrahq8mY-o6h0a-jMSIJd4naKsUHf2qx32UlTrHXPjlSUPOCZVxev9oKREoMYYdIl5Y4eASiRwT8Y5cDM-joYhVj4ft~9LJH2h0BpgBieOiQgsypzkXasG-uMba4LPJeeoHF6tu7~3azI4W6bNFt5XaTh5c7WXy1YkofsunAx1hb8G01YIvtTM-hhGVPTON2UGu6-57EJmSMPfAORdZvGSERojgsUccmS6cdGoPoTPR8F0DZWF96QKXkqsxkVcrOZr3a0v0tGe2MnK1jvk-e6XdIQGrCavPNxn3-6JYsq4XsZg0Pom40GCYrZdVNdrMIwYlWT2jFDrVf5R1lA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Panel I, the classical model for the induction of fever, in which circulating pyrogenic cytokines represent the key event. Panels II and III, an alternative model in which local production of proinflammatory cytokines at the level of infected tissues stimulates either the release of secondary mediators into circulation (panel II) or primary vagal terminals in the liver (panel III). The secondary mediators (e.g., soluble phospholipase A2 [PLA2]) induce prostaglandin release in the organum vasculosum of the lamina terminalis (OVLT) and subsequently fever. Alternatively, signals through vagal afferent fibers and A1/A2 noradrenergic cell groups in brain stem reach the OVLT, where the febrile response is induced. Panels IV and V, local cytokine production and release at the endothelial level in OVLT, the key event in the induction of fever. The cytokines are either released upon adherence to the endothelium by activated monocytes in which expression of pyrogenic cytokines mRNA is high (panel IV) or produced directly by endothelial cells stimulated by exogenous pyrogens circulating in the bloodstream (panel V). Panel VI, the possible involvement of cell-associated cytokines on the membrane of activated monocytes for the stimulation of endothelial cells in OVLT.
Production of cytokines at the tissue level has been suggested as an alternative pathway for the induction of the signal leading to fever, rather than in circulation. Local production of proinflammatory cytokines in the infected tissues may induce release of secondary mediators with pyrogenic properties (figure 2, panel II). One possible candidate for this role may be soluble type II phospholipase A2 [61, 62]. Phospholipase A2 can stimulate PG synthesis at the OVLT level, and it has been suggested to function as EP [62, 63]. However, the arguments for its involvement in the pathogenesis of fever are circumstantial, and infusion of recombinant phospholipase A2 into rabbits have failed to induce fever [64].
An additional mechanism through which local cytokine production may induce fever is through stimulation of vagal terminals in the liver [20] (figure 2, panel III). These signals are transported through vagal afferent fibers and noradrenergic neurons in the brain stem, reaching the OVLT, where the release of noradrenaline induces PG release and fever [20]. The importance of these mechanisms for the induction of fever is sustained by experiments showing that subdiaphragmatic vagal transection abrogates the preoptic PGE2 release induced in guinea pigs by LPS [65] and reduces fever induced by IL-1β [66].
Release of EP at the CNS Level
In addition to production and release of proinflammatory cytokines in the peripheral tissues and in the circulation, release of these mediators at the level of the OVLT endothelium may also represent an important mechanism in the induction of fever. In this model, activation of monocytes may result in little or no production of cytokines. However, the activated monocytes may subsequently enter the bloodstream and adhere to the endothelium in the circumventricular organs [67], where release of EP from either the monocytes themselves or from endothelial cells can induce the signal leading to fever (figure 2, panel IV). Indeed, activated monocytes are present in the bloodstream with low or absent circulating cytokine concentrations [68–70], and production of cytokines by perivascular cells in the circumventricular organs has been demonstrated [71].
An important pathway through which exogenous pyrogens such as LPS may induce fever is through direct induction of proinflammatory cytokine production by endothelial cells in the circumventricular organs (figure 2, panel V). It has been recently discovered that the signaling chain of the LPS receptor is a member of the Toll-like receptor (TLR) family. In humans there are at least 5 TLRs [72], and it appears that TLR4 is involved in mediating the intracellular signals to LPS [73]. Although LPS has a relatively low affinity for TLR4, its binding is greatly enhanced by CD14 in either membrane or soluble form. An additional important factor for the ligand-receptor interaction is a plasma factor, LPS-binding protein, which transfers LPS from the circulating micellae to the receptor complex [74]. Interestingly, an intracytoplasmic domain of the TLRs is homologous to a segment of the IL-1R type I [75], resulting in striking similarities in the intracellular signals induced by LPS and IL-1, respectively (figure 3). In terms of fever, injection of a small amount of LPS into rabbits induces a monophasic fever identical to that observed after IL-1 or TNF [11], probably through intermediary production of IL-6 [40]. These findings provide the theoretical basis for the hypothesis that certain bacterial products can circumvent the need of stimulating a circulating EP in order to be able to induce fever [7].
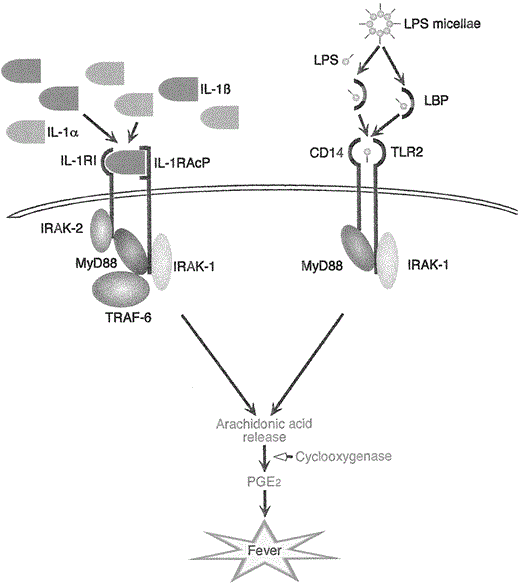
IL-1 (IL-1α and β) binds to its cellular receptor type I (IL-1RI) and the IL-1 receptor accessory protein (IL-1RacP). This leads to signal transduction via receptor associated proteins IRAK-1 and -2, MyD88 and TRAF-6 with release of arachidonic acid and prostaglandin E2 (PGE2) and finally induction of fever. Lipopolysaccharides (LPS) complex with LPS-binding protein, which enables binding of LPS to CD14 and Toll-like receptor 2 (TLR-2). Thereafter, the signal transduction pathway is very similar to that of the IL-1 receptor. Here too, MyD88 and IRAK-1 are activated, arachidonic acid is released, and fever induced through PGE2.
Membrane-Bound Cytokines as Cell-Associated EP
In addition to inducing secretion of soluble forms of proinflammatory cytokines, live bacteria and their products also stimulate the synthesis of cell-associated cytokines, which are known to be highly biologically active [76, 77]. Stimulation of endothelial cells by membrane-bound IL-1α and TNF on the surface of activated monocytes has been documented [78–80], and the importance of membrane-bound forms of TNF for endothelial cell activation in cerebral malaria and acute respiratory distress syndrome has been recently demonstrated [81]. It is therefore tempting to speculate that activated monocytes in the circulation, expressing high levels of membrane-bound cytokines, are able to activate endothelial cells in the OVLT and to induce fever in the absence of detectable concentrations of secreted cytokines (figure 2, panel VI). Indeed, several studies have reported increased concentrations of cell-associated cytokines in patients with severe infections and trauma [82, 83], and Muñoz et al. [83] have shown that membrane-bound TNF, IL-1α, and IL-1β are present in the bloodstream of septic patients for longer periods than are secreted forms.
Multipathway Mechanism for the Induction of Fever
Fever is an adaptive mechanism that plays an important role in the survival of the host during infection with pathogenic microorganisms. Studies performed in the last century, and especially in the last few decades, have greatly contributed to the understanding of this complex process and the mechanisms responsible for its induction. Circulating proinflammatory cytokines acting as EPs play a pivotal role in transmitting the signal responsible for inducing a febrile response from the peripheral tissues to the CNS. However, additional mechanisms may have an important role in the pathogenesis of fever, including local production of cytokines in the tissues, release of proinflammatory cytokines at the CNS level, direct induction of fever by exogenous pyrogens such as LPS, and membrane-bound cytokines acting as cell-associated EP [64]. It is possible, even probable, that different combinations of these mechanisms are involved in certain situations. The study of their relative importance in various clinical disorders will be a further step forward in understanding the pathogenesis of fever.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments