
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the cause of the ongoing coronavirus disease 2019 (COVID-19) pandemic1. To understand the pathogenicity and antigenic potential of SARS-CoV-2 and to develop therapeutic tools, it is essential to profile the full repertoire of its expressed proteins. The current map of SARS-CoV-2 coding capacity is based on computational predictions and relies on homology with other coronaviruses. As the protein complement varies among coronaviruses, especially in regard to the variety of accessory proteins, it is crucial to characterize the specific range of SARS-CoV-2 proteins in an unbiased and open-ended manner. Here, using a suite of ribosome-profiling techniques2,3,4, we present a high-resolution map of coding regions in the SARS-CoV-2 genome, which enables us to accurately quantify the expression of canonical viral open reading frames (ORFs) and to identify 23 unannotated viral ORFs. These ORFs include upstream ORFs that are likely to have a regulatory role, several in-frame internal ORFs within existing ORFs, resulting in N-terminally truncated products, as well as internal out-of-frame ORFs, which generate novel polypeptides. We further show that viral mRNAs are not translated more efficiently than host mRNAs; instead, virus translation dominates host translation because of the high levels of viral transcripts. Our work provides a resource that will form the basis of future functional studies.
Similar content being viewed by others

Main
SARS-CoV-2 is an enveloped virus consisting of a positive-sense, single-stranded RNA genome of around 30 kb. Two overlapping ORFs, ORF1a and ORF1b, are translated from the positive-strand genomic RNA and generate continuous polypeptides, which are cleaved into a total of 16 nonstructural proteins (NSPs). The translation of ORF1b is mediated by a −1 frameshift that allows translation to continue beyond the stop codon of ORF1a. Negative-strand RNA intermediates are produced from the viral genome and serve as templates for the synthesis of positive-strand genomic RNA and subgenomic RNAs5. The subgenomic RNAs contain a common 5′ leader fused to different segments from the 3′ end of the viral genome, a 5′-cap structure and a 3′ poly(A) tail6,7. These distinct fusions occur during negative-strand synthesis at 6–7 nucleotide (nt) core sequences called transcription-regulating sequences (TRSs), which are located at the 3′ end of the leader sequence as well as preceding each viral ORF. The different subgenomic RNAs encode four conserved structural proteins—spike (S), envelope (E), membrane (M) and nucleocapsid (N)—and several accessory proteins. On the basis of sequence similarity to other betacoronaviruses, and specifically to SARS-CoV, the current annotation of SARS-CoV-2 includes predictions of six accessory proteins (3a, 6, 7a, 7b, 8 and 10, NC_045512.2), but not all have been experimentally confirmed8,9.
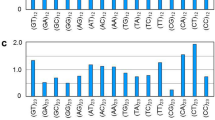
To capture the full coding capacity of SARS-CoV-2, we applied a range of ribosome-profiling approaches to Vero E6 cells infected with SARS-CoV-2 for 5 or 24 h (Fig. 1a). At 24 h post-infection (hpi) the vast majority of cells were infected and cells were still intact (Extended Data Fig. 1). For each time point, we prepared three ribosome-profiling libraries (Ribo-seq), each one in two biological replicates. To facilitate mapping of translation initiation sites, we prepared two Ribo-seq libraries by treating cells with lactimidomycin (LTM) or harringtonine (Harr), two drugs with distinct mechanisms that prevent elongation at 80S ribosomes at translation initiation sites. These treatments lead to excessive accumulation of ribosomes precisely at the sites of translation initiation and depletion of ribosomes over the body of the ORF (Fig. 1a). The third Ribo-seq library was prepared from cells treated with the translation elongation inhibitor cycloheximide (CHX), and provides a snapshot of actively translating ribosomes across the body of the translated ORF (Fig. 1a). In parallel, RNA-sequencing (RNA-seq) was applied to map viral transcripts. Analysis of cellular genes from the different Ribo-seq libraries revealed the expected distinct profiles in both replicates. Ribosome footprints displayed a strong peak at the translation initiation site, which, as expected, is more pronounced in the Harr and LTM libraries; the CHX library also exhibited a distribution of ribosomes across the entire coding region, and the mapped ribosome footprints were enriched in fragments that align to the translated frame (Fig. 1b, Extended Data Fig. 2a). As expected, the RNA-seq reads were uniformly distributed across coding and non-coding regions (Fig. 1b). The footprint profiles of viral coding sequences at 5 hpi fit the expected profile of translated sequences (Fig. 1c, Extended Data Fig. 2b) and the footprint densities were highly reproducible between biological replicates, at single-nucleotide resolution (Extended Data Fig. 2c). Notably, the footprint profile over the viral genome at 24 hpi, did not fit the expected profile of translating ribosomes and was generally not affected by Harr or LTM treatments (Extended Data Fig. 2b). To further examine the characteristics of the footprints, we applied a fragment length organization similarity score (FLOSS) that measures the magnitude of disagreement between the footprint distribution on a given transcript and the footprint distribution on canonical coding sequences (CDSs)10. At 5 hpi, protected fragments from SARS-CoV-2 ORFs did not differ from highly expressed cellular transcripts (Fig. 1d). However, reads at 24 hpi could be clearly distinguished from cellular CDSs (Fig. 1e). We conclude that the footprint data from 5 hpi constitutes robust and reproducible ribosome footprint information but that viral protected fragments at 24 hpi may reflect additional interactions with viral RNA that occur at late time points in infection.

a, Vero E6 and Calu3 cells infected with SARS-CoV-2 were collected at 5, 24 (Vero E6) and 7 (Calu3) hpi for RNA-seq, and for Ribo-seq using LTM, Harr or CHX treatments. b, Metagene analysis of read densities relative to the maximal signal of each gene around the start and stop codons of cellular CDSs at 5 hpi. The read densities are shown with different colours indicating the three frames (red, 0; black, +1; grey, +2). c, Metagene analysis around the start codon, as described in b, for viral ORFs at 5 hpi. d, e, FLOSS score for cellular and SARS-CoV-2 ORFs at 5 hpi (d) and 24 hpi (e).
A global view of RNA and CHX-footprint reads mapping to the viral genome at 5 hpi demonstrates that RNA levels are constant across ORFs 1a and 1b, and steadily increase towards the 3′ end, reflecting the cumulative abundance of these sequences due to the nested transcription of subgenomic RNAs (Fig. 2a). Increased coverage is also seen at the 5′ untranslated region (UTR), reflecting the presence of the 5′ leader sequence in all subgenomic RNAs as well as the genomic RNA. Reduction in footprint density between ORF1a and ORF1b reflects the proportion of ribosomes that terminate at the ORF1a stop codon instead of frameshifting into ORF1b (Extended Data Fig. 3). By dividing the footprint density in ORF1b by the density in ORF1a we estimate a frameshift efficiency of 57% ± 12%. This value is similar to the frameshift efficiency measured by Ribo-seq of mouse hepatitis virus (MHV) (48–75%)3. Similar to observations in MHV and avian infectious bronchitis virus (IBV)3,11, we did not observe noticeable ribosome pausing before or at the frameshift site, but we identified several potential pausing sites within ORF1a and ORF1b (Extended Data Fig. 3).

a, RNA-seq (green) and Ribo-seq CHX (red) read densities (number of reads) at 5 hpi along the SARS-CoV-2 genome. SARS-CoV-2 canonical ORFs are labelled. b, Transcript abundance relative to ribosome densities of each SARS-CoV-2 canonical ORF at 5 hpi. c, Scatter plot of the abundance of reads that span canonical leader-dependent junctions (red), non-canonical leader-dependent junctions (green), non-canonical leader-independent junctions (purple) or genomic deletions (cyan) at 5 and 24 hpi.
Besides ORF1a and ORF1b, all other canonical viral ORFs are translated from subgenomic RNAs. As raw RNA-seq densities represent the cumulative sum of genomic and subgenomic RNAs, we calculated transcript abundance using two approaches: deconvolution of RNA densities, in which RNA expression of each ORF is calculated by subtracting the RNA-read density of cumulative densities upstream of the ORF region; and relative abundances of RNA reads spanning leader–body junctions of each of the canonical subgenomic RNAs. For the majority of the ORFs, there was high correlation between these two approaches (Pearson’s R = 0.897; Extended Data Fig. 4a), and in both approaches the N transcript was the most abundant transcript, in agreement with other studies9,12. We next compared footprint densities to RNA abundance. For the majority of viral ORFs, transcript abundance correlated almost perfectly with footprint densities (Fig. 2b), indicating that these viral ORFs are translated with similar efficiencies (probably owing to their almost identical 5′ UTRs); however, three ORFs were outliers. The translation efficiencies of ORF1a and ORF1b were considerably lower. This may stem from distinct features in their 5′ UTR (discussed below) or from under estimation of their true translation efficiency, as some of the full-length RNA molecules may serve as template for replication or packaging and are thus not part of the translated mRNA pool. The third outlier is ORF7b, for which we identified very few body–leader junctions; nevertheless, it exhibited relatively high translation, probably owing to ribosome leaky scanning of the ORF7a transcript, as was suggested for SARS-CoV13.
Many transcripts derived from non-canonical junctions have been identified for SARS-CoV-29,12. These junctions contain either the leader combined with 3′ fragments at unexpected sites in the middle of ORFs (leader-dependent noncanonical junction) or fusions between sequences that do not show similarity to the leader (leader-independent junction). We estimated the frequency of junction-spanning reads in our RNA libraries and obtained excellent agreement between our replicates (Extended Data Fig. 4b, c, Supplementary Table 1) and significant correlation with previous data from Vero E6 cells12 (Pearson’s R = 0.81; Extended Data Fig. 4d), illustrating that many of these junctions are reproducible between experimental systems. We also identified five abundant leader-independent junctions that, to our knowledge, were unique to our data (Supplementary Table 2). We noted that three of these junctions represent short in-frame deletions in the spike protein that overlap deletions in the furin-like cleavage site that were recently described9 (Extended Data Fig. 4e). The recurrence of the same genomic deletion supports the conclusion that this deletion is being selected for during passage in Vero E6 cells. To examine whether additional non-canonical junctions are derived from genomic deletions, we sequenced the genomic RNA of the virus we used in our infections. In addition to the deletions at the furin-like cleavage site, we identified an 8 amino acid (aa) deletion in ORF-E in 2.3% of the genomic RNA (Fig. 2c, Supplementary Table 2). When we compared the frequency of junctions between 5 h and 24 h time points, the leader-dependent junctions and the genomic deletions showed good correlation, but the leader-independent junctions were specifically increased at 24 hpi (Fig. 2c). These data show that a small number of the leader-independent junctions represent genomic deletions and a larger subset increases at late stages of infection when genome replication is dominant and therefore probably do not substantially affect viral transcripts and translated ORFs.
Examination of SARS-CoV-2 translation as reflected by the diverse Ribo-seq libraries, revealed unannotated translated ORFs. We detected in-frame internal ORFs (iORFs) within existing ORFs, resulting in N-terminally truncated products. These include relatively long truncated versions of canonical ORFs, such as the one found in ORF6 (Fig. 3a, Extended Data Fig. 5a), or very short truncated ORFs that may serve as an upstream ORF (uORF), such as truncated ORF7a, which might regulate ORF7b translation (Fig. 3b, Extended Data Fig. 5b). We also detected internal out-of-frame translations, which would yield novel polypeptides, such as ORFs within ORF3a (41 aa and 33 aa in size, respectively; Fig. 3c, Extended Data Fig. 5c) and within ORF-S (39 aa; Fig. 3d, Extended Data Fig. 5d) or short ORFs that probably serve as uORFs (Fig. 3e, Extended Data Fig. 5e). Additionally, we observed a 13-aa extended ORF-M, in addition to the canonical ORF-M, which is predicted to start at the near-cognate codon AUA (Fig. 3f, Extended Data Fig. 5f).

a–i, Ribosome density profiles (number of reads) of CHX, Harr and LTM samples at 5 hpi. Densities are shown with different colours indicating the frame relative to the main ORF (red, 0; black, +1; grey, +2). Filled and open rectangles indicate the canonical and novel ORFs, respectively. ORFs starting in a near cognate codon are labelled with stripes. a, b, In-frame iORFs within ORF6 (a) and within ORF7a (b). c–e, Out-of-frame internal initiations within ORF-3a, within ORF-S (d) and within ORF-M (e). f, An extended version of ORF-M. g, An uORF that overlaps ORF10 initiation and an in-frame internal initiation generating truncated ORF10. h, Two uORFs embedded in the ORF1a 5′ UTR. i, Non-canonical CUG initiation upstream of the TRS leader. Reads that were cut to fit the scale are indicated with horizontal lines.
The presence of the annotated ORF10 was recently called into question, as almost no subgenomic reads were found for its corresponding transcript12,14. Although we also did not detect subgenomic RNA designated for ORF10 translation (Supplementary Table 1), the ribosome footprint densities indicate the presence of a translation initiation signal in ORF10 (Fig. 3g, Extended Data Fig. 5g). Of note, we additionally detected two putative ORFs in this region, an upstream out-of-frame ORF that overlaps ORF10 initiation and an in-frame internal initiation that leads to a truncated ORF10 product. Further research is needed to delineate how ORFs in this region are translated and whether they have any functional roles.
Finally, we detected four distinct initiation sites in the SARS-CoV-2 5′ UTR. Three of these encode for uORFs that are located just upstream of ORF1a; the first initiating at an AUG (uORF1) and the other two initiating at near-cognate codons (uORF2 and extended uORF2; Fig. 3h, Extended Data Fig. 5h). These uORFs are in line with findings in other coronaviruses3,15. The fourth site is the most prominent peak in the ribosome footprint densities on the SARS-CoV-2 genome and is located on a CUG codon at position 59, just 10 nucleotides upstream of the TRS leader (Fig. 3i, Extended Data Fig. 5i). The reads mapped to this site have the tight length-distribution characteristic of ribosome-protected fragments (Extended Data Fig. 6a). The occupancy at the CUG is higher than the downstream translation signal (Fig. 3i), implying that this peak might reflect ribosomal pausing. Notably, potenial ribosome pauses located just upstream of the TRS leader were also identified in MHV and IBV genomes3,11. Owing to its location upstream of the TRS leader, footprints mapping to this site could potentially derive from any of the subgenomic or genomic RNAs. Therefore, to view this initiation in its context, we aligned the footprints to the genomic RNA or to the most abundant subgenomic N transcript. On the genome and on ORF-N transcript, this initiation results in translation of uORFs, which, on the genome would generate an extension of uORF1 (Extended Data Fig. 6b, c). To assess the distribution of footprints at this initiation on the different viral transcripts, viral transcripts were divided into three groups on the basis of their sequence similarity downstream of the leader–junction site (to enable unique alignment) (Extended Data Fig. 6d). Of note, substantially more footprints were mapped to the group that includes the genomic RNA and the subgenomic E and M transcripts, than would be expected from their relative RNA abundance (Extended Data Fig. 6e). When only footprints that allow unique mapping to genomic RNA or subgenomic M and E transcripts are used (sizes 31–33 base pairs (bp) to discriminate M from genome or E transcript, and sizes 32–33 bp to discriminate E from the genome) a strong enrichment of footprints that originate from the genome is observed (Extended Data Fig. 6f, g). This footprint enrichment on genomic RNA suggests that ribosome pausing might be more prominent on the genome or that ribosomes engage with genomic RNA differently than with subgenomic transcripts. The proximity of this pause to the leader TRS, which seems to be conserved in MHV and IBV3,11, together with the relative enrichment on the viral genome, raises the possibility that a ribosome at this position might affect discontinuous transcription either by sterically blocking the TRS-L site or by affecting RNA secondary structure. In addition, ribosomes initiating at the CUG have the potential to generate uORFs or ORF extensions in the different subgenomic transcripts (Supplementary Table 3).
To systematically define the SARS-CoV-2 translated ORFs we used PRICE and ORF-RATER, two computational methods that rely on a combination of translation features to predict novel translated ORFs from ribosome-profiling measurements16,17. After application of a minimal expression cut-off and manual curation on the predictions, these classifiers identified 25 ORFs, these included 10 out of the 11 canonical translation initiations and 15 novel viral ORFs. In addition, ORF-RATER identified three putative ORFs that originate from the CUG initiation and extend to the subgenomic transcripts of S, M and ORF6 (Supplementary Table 3). The majority (85%) of the classifier-identified ORFs where independently identified in each of the biological replicates (Supplementary Table 4). Visual inspection of the ribosome-profiling data suggested eight additional putative novel ORFs, some of which are presented above (Fig. 3a, b, g, Supplementary Table 4). Overall, we identified 23 putative ORFs, in addition to the 12 canonical viral ORFs that are currently annotated in the NCBI database and 3 additional potential ORFs that stem from the CUG initiation upstream of the leader.
To confirm the robustness of these annotations, we extended these experiments to human cells. We first examined the infection efficiency of several human cell lines that were used to study SARS-CoV-2 infection: Calu3, A549 and Caco-2. Infection of Calu3 was most efficient and the presence of trypsin increased infection efficiency by at least twofold (Extended Data Fig. 7a). We infected Calu3 with a different SARS-CoV-2 isolate, which was sequenced to confirm its integrity. The same set of Ribo-seq techniques were applied to cells at 7 hpi, each in two biological replicates, in parallel with RNA-seq. The different Ribo-seq libraries showed the expected distinct profiles in both replicates, confirming the overall quality of these libraries (Extended Data Fig. 7b). We examined the translation of the new viral ORFs; all 23 novel ORFs we identified as being translated in Vero E6 cells also showed evidence of translation in infected Calu3 cells and 16 were annotated by PRICE and ORF-RATER (Extended Data Fig. 8, Supplementary Table 4). ORF-RATER also identified the same three ORFs that originate from the CUG initiation upstream of the leader (Supplementary Table 3). LTM-induced ribosome accumulation at the canonical and predicted initiation sites was highly reproducible between biological replicates as well as between Calu3 and Vero E6 cells (Extended Data Fig. 9a–c). Furthermore, ribosome-protected footprints displayed a 3-nt periodicity that was in phase with the predicted start site in both Vero E6 and Calu3 cells, providing further evidence for the active translation of the predicted ORFs (Extended Data Fig. 9d). We conclude that 23 unannotated ORFs are reproducibly translated from SARS-CoV-2 independently of the host cell and the viral origin, and additional ORFs may be translated from the CUG initiation located upstream of the TRS leader.
Ribosome density also enables accurate quantification of viral protein production. We first quantified the relative expression levels of canonical viral ORFs on the basis of the non-overlapping regions. ORF-N shows the highest expression in Vero E6 and Calu3 cells, followed by the other viral ORFs, with some differences in the relative expression between the two cell types (Fig. 4a). To quantify the expression of out-of-frame iORFs, we computed the contribution of the iORF to the frame periodicity signal relative to the expected contribution of the main ORF. For in-frame iORF quantification, we subtracted the coverage of the main ORF in the non-overlapping region. We also used ORF-RATER, which uses a regression strategy to calculate relative expression of overlapping ORFs, resulting in largely similar estimates of translation levels (Extended Data Fig. 10a, b). These measurements show that many of the novel ORFs we annotated are expressed at similar levels to the canonical ORFs (Fig. 4b, Supplementary Table 5). Furthermore, the relative expression of viral proteins seems to be mostly independent of the host cell type (Fig. 4c).

a, Translation levels of canonical viral ORFs in Vero E6 cells at 5 hpi and in Calu3 cells at 7 hpi. ORFs are ordered on the basis of their genomic location. b, Viral ORF translation levels, as calculated from ribosome densities for Vero E6 cells at 5 hpi. Solid fill represents canonical ORFs, and striped fill represents novel ORFs. c, Scatter plot of viral ORF expression in Vero E6 cells at 5 hpi and Calu3 cells at 7 hpi. Points representing canonical ORFs are outlined in black. d–f, Relative transcript abundance versus ribosome densities for each host and viral ORF at 5 hpi (d) and 24 hpi (e) in Vero E6 cells and at 7 hpi in Calu3 cells (f). Transcript abundance was estimated by counting the reads that span the corresponding junction and footprint densities were calculated from the CHX sample.
Of the novel ORFs we identified, 14 are very short (up to 20 codons) or located in the 5′ UTR of the genomic RNA and therefore likely have a regulatory role, and three are extensions or truncations of canonical ORFs (M, 6 and 7a). We examined the properties of the six out-of-frame iORFs that are longer than 20 aa: one of these ORFs is ORF9b and its truncated version (size 97 aa and 90 aa; Extended Data Fig. 10c, d). ORF9b appears in UniProt annotations and was detected previously8 in proteomic measurements. Together with our translation measurements this indicates that the product of ORF9b is a bona fide SARS-CoV-2 protein. In addition, we detected an iORF at the 5′ of ORF-S and its truncated version (39 aa and 31 aa; Fig. 3d), and two iORFs within ORF3a (41 aa and 33 aa; Fig. 3c). Mining proteomic measurements of SARS-CoV-2 infected cells8,9 did not detect peptides that originate from these out-of-frame ORFs, probably owing to challenges in detecting trypsin-digested products from short coding regions16. Indeed, two canonical SARS-CoV-2 proteins, ORF7b (43 aa) and ORF-E (75 aa) were also not detected by mass-spectrometry8,9, and our ribosome-profiling data are the first to show that these SARS-CoV-2 proteins are indeed expressed.
S.iORF1 and 3a.iORF1 are predicted to contain a transmembrane domain (Extended Data Fig. 10e, f) and 3a.iORF2 contains a predicted signal peptide (Extended Data Fig. 10g). Analysis of the conservation of these out-of-frame iORFs in SARS-CoV and in related viruses (sarbecoviruses) revealed that 3a.iORF1 is highly conserved (Supplementary Table 6). This ORF was also identified by three independent comparative genomic studies, which demonstrate that it has a purifying selection signature, implying that it is a functional polypeptide18,19,20. In combination, these findings indicate that 3a.iORF1 encodes a functional transmembrane protein, which is conserved throughout sarbecoviruses, and should thus be named ORF3c19,20. The second iORF overlapping ORF3a (3a.iORF2) and the iORF overlapping S (S.iORF1) are not conserved in most sarbecoviruses19 (Supplementary Table 6). The expression level of 3a.iORF2 is much lower compared to those of ORF3a and ORF3c (Fig. 4b, Extended Data Fig. 9d). A protein corresponding to an extended version of this ORF was pulled down21 and shown to elicit an antibody response22, but we found that translation of the truncated version predominated (Extended Data Fig. 10h, i). The internal S-ORF (S.iORF1) is situated just downstream of ORF-S AUG, suggesting that ribosomes might initiate translation via leaky scanning. This region in the S-protein shows extremely rapid evolution20, but in the SARS-CoV-2 isolates that have been sequenced, its coding capacity is maintained23. Future work is needed to delineate whether this ORF, which is highly expressed (Fig. 4b), represents a functional transmembrane protein. Translated ORFs that do not act as functional polypeptides could still be an important part of the immunological repertoire of the virus, as MHC class I bound peptides are generated at higher efficiency from rapidly degraded polypeptides24.
Finally, although we identified two internal out-of-frame ORFs within ORF3a, we did not detect translation of the SARS-CoV ORF3b homologue, which contains a premature stop codon in SARS-CoV-2 (Extended Data Fig. 10h, i). We also did not find evidence of translation of ORF14, which appears in some SARS-CoV-2 annotations15 (Extended Data Fig. 10c, d).
Translation of viral proteins relies on the cellular translation machinery, and coronaviruses, like many other viruses, are known to cause host shut-off25. To quantitatively evaluate whether SARS-CoV-2 skews the translation machinery to preferentially translate viral transcripts, we compared the ratio of footprints to mRNAs for virus and host CDSs at 5 hpi and 24 hpi in Vero E6 cells and at 7 hpi in Calu3 cells. Because ribosome densities were masked by a contaminant signal at 24 hpi, for samples from this time point we used the footprints that were mapped to subgenomic RNA junctions (and therefore reflect bona fide transcripts) to estimate ribosome densities. In all samples, the virus translation efficiencies fall within the low range of most of the host genes (Fig. 4d–f), indicating that viral transcripts are not preferentially translated in infected cells. Instead, viral transcripts take over the mRNA pool, probably through massive transcription coupled to host-induced RNA degradation26,27.
In summary, we have delineated the translation landscape of SARS-CoV-2. Comprehensive mapping of the expressed ORFs is a prerequisite for the functional investigation of viral proteins and for deciphering virus–host interactions. An in-depth analysis of the ribosome-profiling experiments demonstrated a highly complex landscape of translation products, including translation of 23 novel viral ORFs and revealed the relative production of canonical viral proteins. The ORFs that we have identified may serve as accessory proteins or as regulatory units controlling the balanced production of different viral proteins. Studies on the functional importance and antigenic potential of these ORFs will increase our understanding of SARS-CoV-2 and coronaviruses in general.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Cells and viruses
Vero C1008 (Vero E6) cells (ATCC CRL-1586) were cultured in T-75 flasks with DMEM supplemented with 10% fetal bovine serum (FBS), MEM non-essential amino acids, 2mM l-glutamine, 100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin, 12.5 U ml−1 nystatin (Biological Industries). Calu3 cells (ATCC HTB-55) were cultured in 10-cm plates with DMEM supplemented with 10% FBS, MEM non-essential amino acids, 2 mM l-glutamine, 100 U ml−1 penicillin, 1% non-essential amino acid and 1% sodium pyruvate. Caco-2 cells (ATCC HTB-37) were cultured in 10-cm plates with DMEM supplemented with 20% BS, 1% GlutaMAX, 100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin and 1% sodium pyruvate. A549 cells (ATCC CCL-185) were cultured in 10-cm plates with DMEM supplemented with 10% FBS, 100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin and 2 mM l-glutamine. All cell lines were purchased from and authenticated by ATCC and tested negative for mycoplasma. Monolayers were washed once with DMEM (for Vero E6) or RPMI (for Calu3, A549 and Caco-2) without FBS and infected with SARS-CoV-2 virus, at a multiplicity of infection (MOI) of 0.2, For Calu3 infection, 20 μg ml−1 TPCK trypsin (Thermo scientific) was added unless otherwise stated. After 1 h of infection, cells were cultured in their respective medium supplemented with 2% FBS, MEM non-essential amino acids, l-glutamine and penicillin–streptomycin–nystatin at 37 °C with 5% CO2. SARS-CoV-2 (GISAID accession no. EPI_ISL_406862), was provided by Bundeswehr Institute of Microbiology, Munich, Germany. It was propagated (4 passages) and titred on Vero E6 cells and then sequenced (details below) before it was used. SARS-CoV-2 BavPat1/2020 Ref-SKU: 026V-03883 was provided by C. Drosten, Charité–Universitätsmedizin Berlin, Germany. It was propagated (5 passages), titred on Vero E6 and then sequenced before use in experiments. Infected cells were collected at the indicated times as described below. Handling and work with SARS-CoV-2 virus was conducted in a Biosafety Level 3 facility in accordance with the biosafety guidelines of the Israel Institute for Biological Research. The Institutional Biosafety Committee of Weizmann Institute approved the protocol used in these studies.
Preparation of ribosome-profiling and RNA-seq samples
For RNA-seq, cells were washed with PBS and then collected with Tri-Reagent (Sigma-Aldrich), total RNA was extracted, and poly(A) selection was performed using Dynabeads mRNA DIRECT Purification Kit (Invitrogen). mRNA samples were subjected to DNaseI treatment and 3′ dephosphorylation using FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific) and T4 PNK (NEB) followed by 3′ adaptor ligation using T4 ligase (NEB). The ligated products were used for reverse transcription with SSIII (Invitrogen) for first-strand cDNA synthesis. The cDNA products were 3′ ligated with a second adaptor using T4 ligase and amplified with 8 cycles of PCR for final library products of 200–300 bp. For Ribo-seq libraries, cells were treated with 50 μM LTM for 30 min or 2 μg ml−1 Harr for 5 min for translation initiation libraries, or left untreated for the translation-elongation libraries (CHX library). All three samples were subsequently treated with 100 μg ml−1 CHX for 1 min. Cells were then placed on ice, washed twice with PBS containing 100 μg ml−1 CHX, scraped from the T-75 flasks (Vero E6 cells) or 10-cm plates (Calu3 cells), pelleted and lysed with lysis buffer (1% Triton X-100 in 20 mM Tris pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol supplemented with 10 U ml−1 Turbo DNase and 100 μg ml−1 CHX). After lysis, samples stood on ice for 2 h and subsequent Ribo-seq library generation was performed as previously described4. In brief, cell lysate was treated with RNaseI for 45 min at room temperature followed by SUPERase-In quenching. Sample was loaded on sucrose solution (34% sucrose, 20 mM Tris pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol and 100 μg ml−1 CHX) and spun for 1 h at 100,000 rpm in a TLA-110 rotor (Beckman) at 4 °C. Pellet was collected using TRI reagent and the RNA was collected using chloroform phase separation. For size selection, 15 μg total RNA was separated on a 15% TBE-urea gel for 65 min, and 28–34 footprints were excised using 28 and 34 flanking RNA oligos, followed by RNA extraction and Ribo-seq protocol4.
Virus genomic sequencing
RNA from viruses (culture supernatant after removal of cell debris) was extracted using the viral RNA kit (Qiagen). The SMARTer Pico RNA V2 Kit (Clontech) was used for library preparation. Genome sequencing was conducted on the Illumina Miseq platform, in a single read mode 60 bp for BetaCoV/Germany/BavPat1/2020 EPI_ISL_406862 and in a paired-end mode 150 bp × 2 for BavPat1/2020 Ref-SKU: 026V-03883 producing 2,239,263 and 4,332,551 reads, respectively. Reads were aligned to the viral genome using STAR 2.5.3a aligner. Even coverage along the genome was assessed and the relative abundance junctions (which may reflect genomic deletion) were calculated. For EPI_ISL_406862 passage 4 (that was used for Vero E6 cells infection) the junctions that were found in more than 1% of genomes are listed in Supplementary Table 2. For BavPat1/2020 Ref-SKU: 026V-03883 passage 5 (that was used for Calu3 infection) no junctions in abundance of more than 1% of the genomes were detected. All genomic sequencing data was recorded.
Sequence alignment, metagene analysis
Sequencing reads were aligned as previously described28. In brief, linker (CTGTAGGCACCATCAAT) and poly(A) sequences were removed and the remaining reads were aligned to the Chlorocebus sabaeus genome (ENSEMBL release 99) and to the SARS-Cov-2 genome (Genebank NC_045512.2 with 3 changes to match the used strain (BetaCoV/Germany/BavPat1/2020 EPI_ISL_406862): C241T, C3037T, A23403G) for infection of Vero E6 cells, or to the hg19 and NC_045512.2 sequence with the same sequence changes for infection of Calu3. Alignment was performed using Bowtie v.1.1.229 with a maximum of two mismatches per read. Reads that were not aligned to the genome were aligned to the transcriptome of C. sabaeus (ENSEMBL) and to SARS-CoV-2 junctions that were recently annotated12. The aligned position on the genome was determined as the 5′ position of RNA-seq reads, and for Ribo-seq reads, the P site of the ribosome was calculated according to reads length using the offset from the 5′ end of the reads that was calculated from canonical cellular ORFs. The offsets used are +12 for reads that were 28–29 bp and +13 for reads that were 30–33 bp. Reads that were different in length were discarded. In all figures presenting ribosome-density data, all footprint lengths (28–33 bp) are presented.
Novel junctions were mapped using STAR 2.5.3a aligner30, with running flags as suggested12 to overcome filtering of non-canonical junctions. Reads aligned to multiple locations were discarded. Junctions with 5′ break sites mapped to genomic location 55–85 were assigned as leader-dependent junctions. Matching of leader junctions to ORFs and categorization of junctions as canonical or non-canonical, was adapted from supplementary table 3 in ref. 12 or was assigned manually for strong novel junctions that appear only in our data.
For the metagene analysis only genes with more than 50 reads were used. For each gene, normalization was done to its maximum signal and each position was normalized to the number of genes contributing to the position. In the virus 24-h samples, normalization for each gene was done to its maximum signal within the presented region.
Quantification of gene expression
The deconvolution of RNA expression was done by subtracting the reads per kilobase of transcript, per million mapped reads (RPKM) of an ORF from the RPKM of the ORF located just upstream of it in the genome. The junction counts were based on STAR alignment number of uniquely mapped reads crossing the junction. For comparing transcript and footprint expression level, RNA and footprint counts from bowtie alignments were normalized to units of RPKM to normalize for gene length and for sequencing depth. On the basis of the correlation between the deconvoluted RPKM and junction abundance of the subgenomic RNAs, the genomic RNA abundance was estimated and was used to estimate ORF1a and ORF1b RNA levels compared with footprint levels.
The estimation of the viral footprint densities from the 24 hpi samples was performed by calculating the ratio of the RPKM of ORF1a to the total number of leader canonical junctions at 5 hpi. This ratio was used as a factor to calculate a proxy for the ‘true’ viral footprint densities from the number of footprints that were mapped to leader canonical junctions at 24 hpi.
To quantify the translation levels of novel viral ORFs at 5 hpi and 7 hpi, many of which are overlapping, three types of calculations were used, based on ORF type. For ORFs that have a unique region, with no overlap to any other ORF, bowtie-aligned read density was calculated in that region. For out-of-frame iORFs, the read density of the iORF region was calculated by estimating the expected 3-bp periodicity distribution of footprints based on non-overlapping translated regions in the main ORF. Using linear regression, we calculated the relative contribution of the frames of the main and of the iORF to the reads covering the region of the iORF. The relative contribution of the iORF was then multiplied by the read density in that region to obtain the estimated translation level of the internal out-of-frame ORF. For in-frame iORFs the read density of the main overlapping ORF was calculated from a non-overlapping region and then subtracted from the read density in the overlapping iORF region to get an estimate of translation levels of the iORF. In cases where the unique region used to calculate read density contained the start codon of the ORF, the first 20% of the codons in the region were excluded from the calculation to avoid bias from initiation peaks, unless the region was very short and trimming it would harm the ability to estimate coverage (ORF 8 and extended ORF M). The exact regions that were used for calculation can be found in Supplementary Table 5. Finally, read density was normalized to the length of the region used for calculation and to the sum of length normalized reads in each sample to get transcripts per kilobase million (TPM) values. P values for the relative contribution levels of out-of-frame ORFs were calculated from both replicates using a mixed-effects linear model using the three-base periodicity distribution as the fixed effect and the replicates as random effect. In parallel, ORF-RATER was used to quantify the translation levels of the viral ORFs (using regression), giving largely similar values (Spearman’s R = 0.92 and 0.87 in Vero E6 and Calu3 cells, respectively).
Prediction of translation initiation sites
Translation initiation sites were predicted using PRICE16 and ORF-RATER17. To estimate the codons generating the sequencing reads with maximum likelihood, PRICE requires a predefined set of annotated coding sequences from the same experiment. Thus, it does not perform well on reference sequences with a small number of annotated ORFs such as SARS-CoV-2. As our experiment generated ribosome footprints from both SARS-CoV-2 and host mRNAs, which were exposed to the exact same conditions in the protocol, we used annotated CDSs from the host cells to evaluate the parameters of the experiment. For libraries of infected Vero E6 cells, sequencing reads were aligned using Bowtie to a fasta file containing chromosome 20 of C. sabaeus (1,240 annotated start codons, downloaded from ensembl: ftp://ftp.ensembl.org/pub/release99/fasta/chlorocebus_sabaeus/dna/) and the genomic sequence of SARS-CoV-2 (Refseq NC_045512.2). A gtf file with the annotations of C. sabaeus and SARS-CoV-2 genomes was constructed and provided as the annotations file when running PRICE. For technical reasons, the annotation of the first CDS of the two CDSs in the ORF1ab gene was deleted because having two CDSs encoded from a single gene was not permitted by PRICE. For libraries of infected Calu3 cells sequencing reads were mapped to a fasta file containing chromosome 1 of hg19 (2,843 annotated start codons) and the genomic sequence of SARS-CoV-2 (Refseq NC_045512.2). A gtf file with the annotations of hg19 and SARS-CoV-2 genomes was constructed and provided as the annotations file when running PRICE. For the data that were generated from infected Vero E6 cells at 5 hpi, training and ORF prediction by PRICE were done once using the CHX data from both replicates, and again using all Ribo-seq libraries from both replicates, and the resulting predictions were combined. To test reproducibility, the same predictions were performed on each replicate separately. For the data that were generated from infected Calu3 cells at 7 hpi training and ORF prediction by PRICE were done using all Ribo-seq libraries from both replicates. The predictions were further filtered to include only ORFs with at least 100 reads at the initiation site in the LTM samples of at least one replicate. ORFs were then defined by extending each initiating codon to the next in-frame stop codon.
ORF-RATER was used with the default values besides allowing all start codons with at most one mismatch to ATG. For each cell type, two runs of ORF-RATER were used: one in which ORF-RATER was trained on cellular annotations (chr 20 for the Vero E6 cells, and chr 1 for the Calu cells) and SARS-CoV-2 canonical ORFs (similar to the procedure that was used for running PRICE); and a second in which SARS-CoV-2 canonical ORFs were used for training. In both cases, ORF1b and ORF10 were omitted from the training set. BAM files from STAR alignment were used as input. The CHX data from both replicates was used in the first prune step to omit low-coverage ORFs. The calculations of the P-site offsets and the regression were performed for each type of Ribo-Seq library separately. The final score was calculated on the basis of all three types of libraries. A score of 0.5 was used as cut-off for the final predictions; these were further manually curated. Additional ORFs that were not recognized by the trained models (probably owing to differences in the features of viral genome compared with cellular genomes) but presented reproducible translation profile in the two cell lines were added manually to the final ORF list (Supplementary Table 4). ORFs were manually identified as such if they had reproducible initiation peaks in the CHX libraries that were enhanced in the LTM and Harr libraries, and exhibited increased CHX signal in the correct reading frame along the coding region.
Mapping reads to CUG initiation upstream of the TRS leader
Reads from ribosome-profiling libraries were aligned using bowtie to a single reference that contained the transcripts and the genome allowing no mismatches or gaps. Reads with P-site mapped to position 59 of the viral genome were collected and divided to four groups according to the nucleotide in position +17 of the read (position 76 of the genome). The first group contains reads that are short (28 nucleotides) and do not have any nucleotide at position +17. The other three groups, referred to as T, A and G, correspond to combinations of genomic and subgenomic RNAs based on their sequence, as shown in Extended Data Fig. 6d. Group T is attributed to the genome or to ORF E and ORF M subgenomic RNAs, group A to the subgenomic RNAs of ORF S, ORF7a, ORF8 and ORF N, and group G to the subgenomic RNA of ORF 6. Reads that mapped uniquely to the subgenomic RNA of ORF3a were excluded from calculation, and the number of reads in each group was summed. Group T, containing genomic reads, was further divided on the basis of the nucleotide at position +18, where reads with A at that position can originate from the subgenomic RNA of ORF M and reads with T at that position can originate from the genome or from the subgenomic RNA of ORF E. Final division of the genomic group was done based on position +19 where T corresponds to genomic reads and A corresponds to ORF E subgenomic reads. RNA values as calculated from junction densities (as described in ‘Quantification of gene expression’) were summed for the subgenomic and genomic RNAs in each group. The analysis was performed for each ribosome-profiling library separately.
Mining of proteomics data and transmembrane predictions
Data from ref. 8 was searched using the Byonic search engine with 10 ppm tolerance for MS1 and 20 ppm tolerance for MS2, against the concatenated database containing our 26 novel ORFs as well as the human proteome DB (SwissProt, November 2019), and the SARS-CoV-2 proteome. Modifications allowed were fixed carbamidomethylation on C, fixed TMT6 on K and peptide N terminus, variable K8 and R10 SILAC labelling, variable M oxidation and Variable NQ deamidation. Data downloaded from ref. 9 was searched with the Byonic search engine using 10 ppm tolerance for MS1 and 0.6 Da tolerance for MS2, against the concatenated database containing our 26 novel ORFs as well as the human proteome DB (SwissProt, November 2019), and the SARS-CoV-2 proteome. Modifications allowed were fixed carbamidomethylation on C, variable N-terminal protein acetylation, M oxidation and NQ deamidation. Transmembrane and signal peptide predictions were performed using Phobius31.
Immunofluorescence
Cells were plated on Ibidi slides, fixed in 3% paraformaldehyde for 20 min, permeabilized with 0.5% Triton X-100 in PBS for 2 min, and then blocked with 2% FBS in PBS for 30 min. Immunostaining was performed with rabbit anti-SARS-CoV-2 serum32 at a 1:200 dilution. Cells were washed and labelled with anti-rabbit FITC antibody and with DAPI at a 1:200 dilution. Imaging was performed on a Zeiss AxioObserver Z1 wide-field microscope using a ×40 objective and Axiocam 506 mono camera.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All next-generation sequencing data files were deposited in Gene Expression Omnibus under accession number GSE149973. All the RNA-seq and ribosome-profiling data generated in this study can be accessed through a UCSC browser session (http://genome.ucsc.edu/s/aharonn/CoV2%2DTranslation). The proteomics data analysed in this study are available from the PRIDE repository with the identifiers PXD0177108 and PXD0182419.
References
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Stern-Ginossar, N. et al. Decoding human cytomegalovirus. Science 338, 1088–1093 (2012).
Irigoyen, N. et al. High-resolution analysis of coronavirus gene expression by RNA sequencing and ribosome profiling. PLoS Pathog. 12, e1005473 (2016).
Finkel, Y. et al. Comprehensive annotations of human herpesvirus 6A and 6B genomes reveal novel and conserved genomic features. eLife 9, e50960 (2020).
Sola, I., Almazán, F., Zúñiga, S. & Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2, 265–288 (2015).
Lai, M. M. & Stohlman, S. A. Comparative analysis of RNA genomes of mouse hepatitis viruses. J. Virol. 38, 661–670 (1981).
Yogo, Y., Hirano, N., Hino, S., Shibuta, H. & Matumoto, M. Polyadenylate in the virion RNA of mouse hepatitis virus. J. Biochem. 82, 1103–1108 (1977).
Bojkova, D. et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583, 469–472 (2020).
Davidson, A. D. et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 12, 68 (2020).
Ingolia, N. T. et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 8, 1365–1379 (2014).
Dinan, A. M. et al. Comparative analysis of gene expression in virulent and attenuated strains of infectious bronchitis virus at subcodon resolution. J. Virol. 93, 714–733 (2019).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914–921 (2020).
Schaecher, S. R., Mackenzie, J. M. & Pekosz, A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. J. Virol. 81, 718–731 (2007).
Davidson, A. D. et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 12, 68 (2020).
Wu, A. et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27, 325–328 (2020).
Erhard, F. et al. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 15, 363–366 (2018).
Fields, A. P. et al. A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol. Cell 60, 816–827 (2015).
Cagliani, R., Forni, D., Clerici, M. & Sironi, M. Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses. Infect. Genet. Evol. 83, 104353 (2020).
Firth, A. E. A putative new SARS-CoV protein, 3c, encoded in an ORF overlapping ORF3a. J. Gen. Virol. https://doi.org/10.1099/jgv.0.001469 (2020).
Jungreis, I., Sealfon, R. & Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Preprint at https://doi.org/10.1101/2020.06.02.130955 (2020).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Hachim, A. et al. Beyond the spike: identification of viral targets of the antibody response to SARS-CoV-2 in COVID-19 patients. Preprint at https://doi.org/10.1101/2020.04.30.20085670 (2020).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Yewdell, J. W. DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 32, 548–558 (2011).
Abernathy, E. & Glaunsinger, B. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 479-480, 600–608 (2015).
Huang, C. et al. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. 7, e1002433 (2011).
Kamitani, W., Huang, C., Narayanan, K., Lokugamage, K. G. & Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 16, 1134–1140 (2009).
Tirosh, O. et al. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host–pathogen interactions. PLoS Pathog. 11, e1005288 (2015).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Käll, L., Krogh, A. & Sonnhammer, E. L. L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 (2004).
Yahalom-Ronen, Y. et al. A single dose of recombinant VSV-ΔG-spike vaccine provides protection against SARS-CoV-2 challenge. Preprint at https://doi.org/10.1101/2020.06.18.160655 (2020).
Acknowledgements
We thank Stern-Ginossar laboratory members, I. Ulitsky and S. Schwartz for providing valuable feedback; E. Zehavi, M. Shnayder, I. Ulitsky and N. Gil for technical assistance; E. Wyler for the Calu3 cells; and I. Cohen-Gihon for sharing sequencing and bioinformatics data. This study was supported by the Ben B. and Joyce E. Eisenberg Foundation. Work in the Stern-Ginossar lab is supported by ERC-CoG-2019- 864012 and by the ISF grant no. 1526/18. S.W.-G. is the recipient of the HSFP fellowship, EMBO non-stipendiary Long-Term Fellowship, the Gruss-Lipper Postdoctoral Fellowship, the Zuckerman STEM Leadership Program and the Rothschild Postdoctoral Fellowship. N.S.-G. is an incumbent of the Skirball Career Development Chair in New Scientists and is a member of EMBO Young Investigator Program.
Author information
Authors and Affiliations
Contributions
Y.F., O.M., N.P and N.S.-G. conceptualized the study; O.M. performed experiments; Y.F., A.N. and S.W.-G. performed data analysis; Y.Y.-R., H.T., H.A., S.M., S.W., D.S., O.I., A.B.-D., T.I. and N.P. performed work with SARS-CoV-2. D.M. mined published proteomic data; Y.F., O.M., A.N., M.S. and N.S.-G. interpreted data; and M.S. and N.S.-G. wrote the manuscript with contributions from all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Volker Thiel, Petra Van Damme and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 24 h infection with SARS-CoV-2 of Vero E6 cells.
Vero E6 cells were infected with SARS-CoV-2 at an MOI = 0.2 and 24 hpi the cells were fixed and stained with antisera against SARS-CoV-2 (green) and D API (blue). The experiment was performed once and representative microscopy images are presented. Scale bars are 200μm.
Extended Data Fig. 2 Footprint and RNA-seq profiles of cellular and viral genes from SARS-CoV-2 infected Vero E6 cells.
a, b, Metagene analysis of read densities at the 5′ and the 3′ regions of cellular (a) and viral (b) protein coding genes as measured by the different Ribo-seq approaches and RNA-seq at 5 hpi and 24 hpi, from two biological replicates. The x axis shows the nucleotide position relative to the start or the stop codons. The read densities are shown with different colours indicating the three frames relative to the main ORF (red, frame 0; black, frame +1; grey, frame +2). NA reflect samples in which we did not obtain enough cellular genes that contain 50 reads at the 5′ or 3′ regions to generate metagene profile. c, Scatter plots depicting the number of reads in every position along the SARS-CoV-2 genome in two independent biological replicates, demonstrating reproducibility between our replicates at single nucleotide resolution. Pearson’s R of log-transformed values is presented.
Extended Data Fig. 3 Footprint profiles reveal potential ribosome pausing sites within ORF1a and ORF1b.
Read densities are presented for ORF1a and ORF1b at 5 hpi from two biological replicates. The read densities are shown with different colours indicating the three frames relative to the translated frame of ORF1a (red, frame 0; black, frame +1; grey, frame +2). Black arrows mark potential ribosome pausing sites and their genomic positions.
Extended Data Fig. 4 The abundance of subgenomic RNAs.
a, Measurement of subgenomic RNA abundance using deconvolution of RNA densities versus using relative abundance of RNA reads spanning leader-body junctions, for seven canonical viral ORFs. ORF6, ORF7b and ORF10 obtained negative values in the RNA deconvolution, probably due to their short length and relative weaker expression. For ORF10 also no reads spanning leader-body junctions were detected. Spearman’s R is presented. b, c, Scatter plots presenting the abundance of junction-spanning RNA-seq reads from two biological replicates from (b) 5 hpi and (c) 24 hpi. d, Scatter plots presenting the average abundance of junction-spanning RNA-seq reads from 24 hpi versus data from Kim et al.12 Viral reads that span canonical leader-dependent junctions are marked in red, non-canonical leader-dependent junctions are marked in green, non-canonical leader-independent junctions are marked in purple and non-canonical leader-dependent junctions originating from genomic deletions are marked in cyan. Pearson’s R of log-transformed values is presented. e, Ribo-seq (CHX) and RNA densities over the 7 aa deletion in the S protein. Lower panels present the amino acid sequence in the region and the translation of the WT and of the 7-aa-deleted version of the S protein.
Extended Data Fig. 5 Footprint profiles reveal novel viral coding regions.
a–i, Read densities over SARS-CoV-2 unannotated translated ORFs as measured by the different Ribo-seq approaches from both replicates at 5 hpi. The read densities are shown with different colours indicating the three frames relative to the main ORF in each figure (red, frame 0; black, frame +1; grey, frame +2). Filled and open rectangles indicate the canonical and novel ORFs, respectively. ORFs starting in near cognate start codon are labelled with stripes. a, In frame internal initiation within ORF6 generating truncated product. b, In frame internal initiation within ORF7a. c, Out of frame internal initiations within ORF3a. d, Out of frame internal initiations within ORF-S. e, Out of frame internal initiation within ORF-M. f, An extended version of ORF-M. g, uORF that overlaps ORF10 initiation and in frame internal initiation generating truncated ORF10 product. h, Two uORFs embedded in ORF1ab 5′ UTR. i, Non-canonical CUG initiation upstream of the TRS leader.
Extended Data Fig. 6 The CUG initiation is enriched on the genomic RNA.
a, Comparison of the ribosome footprint read length distributions of reads that align to the CUG upstream of the TRS-L (red) and reads that align to ORF-N AUG (green). b, c, Read densities over the TRS-L CUG on the genomic RNA (b) or ORF-N transcript (c) as measured by the different Ribo-seq approaches at 5 hpi. The read densities are shown with different colours indicating the three frames relative to the CUG (red, frame 0; black, frame +1; grey, frame +2). Rectangles indicate adjacent ORFs and the striped rectangles indicate the ORFs initiating at the TRS-L CUG. d, The sequences of the genome and the most abundant subgenomic transcripts divided to three groups based on the base in position 76. The CUG position is labelled in grey and the location of the junction is labelled by a vertical line. e, The relative number of footprint reads that their P-site was mapped to the CUG (dark red) for each of the transcript groups (as defined in c) and the relative RNA abundance of these transcript groups (green). Data are presented for CHX, Harr and LTM libraries. f, The relative number of footprint reads that their P-site was mapped to the CUG (dark red) in ORF-M transcript or in ORF-E and the Genome RNA and the relative RNA abundance (green). Only footprint sizes 31–33 bp that allow unique alignment were used. Data are presented for CHX, Harr and LTM libraries. g, The number of footprint reads that their P-site was mapped to the CUG in the genome or in ORF-E transcript out of all reads and the relative abundance of these RNAs. Only footprint sizes 32–33 bp that allow unique alignment were used. Read numbers is presented for CHX, Harr and LTM libraries and the relative RNA abundance is presented as percentage of total RNA included in the comparison.
Extended Data Fig. 7 SARS-CoV-2 infection of Calu3 cells.
a, Calu3 cells were either left uninfected, infected with SARS-CoV-2, or infected with SARS-CoV-2 in the presence of trypsin. 12 hpi the cells were fixed and stained with antisera against SARS-CoV-2 (green) and DAPI (blue). The experiment was performed once and representative microscopy images are presented. Scale bars are 200 μm. b, Metagene analysis of read densities at the 5′ and the 3′ regions of viral protein coding genes as measured by the different Ribo-seq approaches and RNA-seq at 7 hpi from two biological replicates. The x axis shows the nucleotide position relative to the start or the stop codons. The read densities are shown with different colours indicating the three frames relative to the main ORF (red, frame 0; black, frame +1; grey, frame +2).
Extended Data Fig. 8 Footprint profiles reveal novel viral coding regions are also translated in infected Calu3 cells.
a–i, Read densities over SARS-CoV-2 unannotated translated ORFs as measured by the different Ribo-seq approaches from both replicates at 7 hpi. The read densities are shown with different colours indicating the three frames relative to the main ORF in each figure (red, frame 0; black, frame +1; grey, frame +2). Filled and open rectangles indicate the canonical and novel ORFs, respectively. ORFs starting in near cognate start codon are labelled with stripes. a, In frame internal initiation within ORF6 generating truncated product. b, In frame internal initiation within ORF7a. c, Out of frame internal initiation within ORF3a. d, Out of frame internal initiation within ORF-S. e, Out of frame internal initiation within ORF-M. f, An extended version of ORF-M. g, uORF that overlap ORF10 initiation and in frame internal initiation generating truncated ORF10 product. h, Two uORFs embedded in ORF1ab 5′ UTR. i, Non-canonical CUG initiation upstream of the TRS-leader.
Extended Data Fig. 9 Reproduciblity of viral ORFs translation.
a–c, Scatter plot presenting the correlation of footprint densities at each initiation site relative to a 8bp window 3 nucleotides downstream of the initiation site in LTM treated samples between two biological replicates of infected Vero E6, 5 hpi (a), between two biological replicates of infected Calu3 cells, 7 hpi (b) and between infected Vero E6 cells at 5 hpi and Calu3 cells at 7 hpi (c). Canonical ORFs are marked in dark green and newly identified ORFs are marked in light green. As a control, the relative occupancy of random positions was calculated in the same way (grey). Dashed lines mark the equal ratio of one. Spearman’s R is presented. d, The position of ribosome footprints relative to the reading frame in all canonical ORFs and novel ORFs excluding in-frame iORFs are presented for our measurements of infected Vero E6 cells at 5 hpi (lower panels) and infected Calu3 cells (upper panels). Filled and open rectangles indicate the canonical and novel ORFs, respectively. ORFs starting in near cognate start codons are labelled with stripes. The frame of the footprints is summed on each of the indicated regions and is presented relative to the frame of the canonical ORF in each of these loci. In all non-overlapping regions, beside ORF10 in Vero E6 cells, clear enrichment to the translated frame is observed indicating active translation. For out-of-frame overlapping ORFs the bar of its frame is labelled by colour and the percentage of the footprints that originate from the overlapping frame as was calculated from linear regression is presented together with the corresponding P value for the contribution of the out-of-frame ORF to the frame distribution of the total reads in this region, using two degrees of freedom that reflect the two replicates. In two out-of-frame overlapping ORFs, ORF3.iORF2 and ORF8.iORF the expression relative to the main ORF was low and did not lead to a significant shift in the translation signal. In all other ORFs there is a significant signal in the alternative frame indicating active translation.
Extended Data Fig. 10 Characteristics of novel predicted ORFs.
a, b, Scatter plots presenting the correlation between translation levels as estimated by our curated quantification and as calculated by ORF-RATER for Vero (a) and Calu3 cells (b). Points representing canonical ORFs are outlined in black. Spearman’s R is presented. c, d, Read densities over ORF-N as measured by the different Ribo-seq approaches in two replicates in Vero cells at 5 hpi (c) and Calu3 cells at 7 hpi (d). The read densities are shown with different colours indicating the three frames relative to the main ORF in each figure (red, frame 0; black, frame +1; grey, frame +2). Filled and open rectangles indicate the canonical and novel ORFs, respectively. ORF14 location is marked based on the homology to SARS-CoV. e, Transmembrane region predicted in S.iORF1 using Phobius. f, Transmembrane region predicted in ORF3c (3a.iORF1) using Phobius. g, Signal peptide prediction in 3a.iORF2 as predicted using Phobius. h, i, Read densities over ORF3a as measured by the different ribosome profiling approaches in two replicates in Vero cells at 5 hpi (h) and Calu3 cells at 7 hpi (i). The read densities are shown with different colours indicating the three frames relative to the main ORF in each figure (red, frame 0; black, frame +1; grey, frame +2). Filled and open rectangles indicate the canonical and novel ORFs, respectively. ORF3b and extended iORF2 are marked based on the homology to SARS-CoV.
Supplementary information
Supplementary Table 1.
Junctions sites detected from junction spanning reads This table lists junction sites that were identified by Kim et al. with more than 100 reads and were also detected in our RNA reads.
Supplementary Table 2.
Junctions sites uniquely detected in our samples This table lists junction sites that were identified in our RNA samples with more than 50 reads but were low or unidentified by Kim et al.
Supplementary Table 3.
Potential junction spanning SARS-CoV-2 ORFs that can be translated from the CUG initiation upstream the TRS-leader This table lists junction spanning SARS-CoV-2 ORFs that can be translated from the CUG initiation upstream the TRS-leader at each of the subgenomic RNAs.
Supplementary Table 4.
Novel SARS-CoV-2 ORFs that have been identified in our study This table lists all the SARS-CoV-2 translated ORFs identified in this study.
Supplementary Table 5.
Translation levels of SARS-CoV-2 ORFs This table lists all translated SARS-CoV-2 ORFs, canonical and newly identified, and their estimated translation levels based on ribosome profiling
Supplementary Table 6.
Multiple sequence alignment for canonical and novel SARS-CoV-2 ORFs in Sarbecoviruses This table includes links for annotated multiple sequence alignment (MSA) views of all SARS-CoV-2, canonical and novel, described in this study, using the CodAlignView tool 38. The MSA includes SARS-CoV-2, SARS-CoV and 42 bat coronavirus genomes 30.
Rights and permissions
About this article

Cite this article
Finkel, Y., Mizrahi, O., Nachshon, A. et al. The coding capacity of SARS-CoV-2. Nature 589, 125–130 (2021). https://doi.org/10.1038/s41586-020-2739-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2739-1
This article is cited by
-
Riboformer: a deep learning framework for predicting context-dependent translation dynamics
Nature Communications (2024)
-
Thunder-DDA-PASEF enables high-coverage immunopeptidomics and is boosted by MS2Rescore with MS2PIP timsTOF fragmentation prediction model
Nature Communications (2024)
-
Coronavirus takeover of host cell translation and intracellular antiviral response: a molecular perspective
The EMBO Journal (2024)
-
SARS-CoV-2 biology and host interactions
Nature Reviews Microbiology (2024)
-
Non-mutational neoantigens in disease
Nature Immunology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
