
Abstract
The recognition of microbial nucleic acids is a major mechanism by which the immune system detects pathogens. Cyclic GMP-AMP (cGAMP) synthase (cGAS) is a cytosolic DNA sensor that activates innate immune responses through production of the second messenger cGAMP, which activates the adaptor STING. The cGAS–STING pathway not only mediates protective immune defense against infection by a large variety of DNA-containing pathogens but also detects tumor-derived DNA and generates intrinsic antitumor immunity. However, aberrant activation of the cGAS pathway by self DNA can also lead to autoimmune and inflammatory disease. Thus, the cGAS pathway must be properly regulated. Here we review the recent advances in understanding of the cGAS–STING pathway, focusing on the regulatory mechanisms and roles of this pathway in heath and disease.
Similar content being viewed by others

Main
As a first line of defense, the innate immune system deploys germline-encoded receptors to detect molecular patterns associated with extracellular or intracellular pathogens. For extracellular pathogens, transmembrane receptors, including TLRs and CLRs, are the main sensors1. The ligand-binding domains of these receptors project extracellularly or into the lumen of the endosome, whereas the signaling domains of these receptors project to the cytosol and initiate signaling cascades that result in the induction of a battery of genes encoding products involved in immune and inflammatory responses. When pathogens gain access to and replicate inside a cell, various classes of cytosolic sensors are engaged, depending on the nature of the pathogen-associated molecular pattern. Sensors of the NLR family, which include the inflammasomes, detect a variety of microbial molecules, toxins and cellular damage and trigger downstream signaling pathways. For inflammasomes, signaling leads to pyroptosis as well as to the maturation of interleukin 1 (IL-1) and IL-18 (ref. 2). Receptors of the RLR family detect viral RNA in the cytosol and then trigger a signaling cascade that leads to the production of type I interferons and inflammatory cytokines3. Cyclic GMP-AMP synthase (cGAS), the focus of this Review, is a cytosolic DNA sensor that activates innate immune responses, including the induction of interferons4.
As the blueprint of life, DNA is best known for carrying genetic information in living organisms that range from viruses to humans. Before DNA was known to be the carrier of genes, it was found to stimulate immune responses, such as the recruitment of phagocytes5. The recognition of microbial DNA by the immune system provides a general mechanism for the detection of a wide variety of pathogens, because with the exception of RNA viruses, all microorganisms contain and need DNA in their life cycle. However, host cells contain abundant self DNA. In eukaryotic cells, DNA is contained in the nucleus and mitochondria, which leaves the cytoplasm largely free of self DNA. Delivery of DNA to the cytoplasm through microbial infection activates the innate immune system. Self DNA can also enter and accumulate in the cytoplasm under certain pathological conditions, which results in autoimmune attack of self tissues. While the recognition of self DNA by the immune system is undesirable in general, the detection of tumor-derived DNA after dead tumor cells are taken up by phagocytes provides a mechanism for the innate sensing of tumors and activation of anti-tumor immunity. In this Review, we will discuss the role of the cGAS pathway in immune defense against DNA-containing pathogens and malignant cells, as well as the role of this pathway in autoimmune diseases.
The cGAS–STING pathway of cytosolic DNA sensing
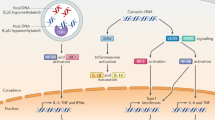
cGAS contains a nucleotidyltransferase domain and two major DNA-binding domains6. In the absence of DNA, cGAS exists in an autoinhibited state7,8,9,10,11. cGAS binds to DNA to form a 2:2 complex7,8, and binding to DNA induces a conformational change in the active site, which catalyzes the synthesis of cyclic GMP-AMP (cGAMP) from ATP and GTP6,7,8,12 (Fig. 1). The cGAMP synthesized by cGAS contain two phosphodiester bonds, one between the 2′-OH of GMP and 5′-phosphate of AMP, and the other between the 3′-OH of AMP and 5′-phosphate of GMP10,13,14,15. cGAS catalyzes the formation of the 2′-5′ linkage, followed by cyclization, which forms the 3′-5′ linkage. This cGAMP isomer, called '2′3′-cGAMP', functions as a second messenger that binds to the endoplasmic-reticulum (ER)-membrane adaptor STING12,13,16,17 and induces a conformational change that presumably results in the activation of STING. STING then traffics from the ER to an ER-Golgi intermediate compartment and the Golgi apparatus18,19,20. During this process, the carboxyl terminus of STING recruits and activates the kinase TBK1, which in turn phosphorylates the transcription factor IRF3 (refs. 21,22,23). The phosphorylated IRF3 dimerizes and then enters the nucleus. STING also activates the kinase IKK, which phosphorylates the IkB family of inhibitors of the transcription factor NF-kB16. Phosphorylated IkB proteins are degraded by the ubiquitin-proteasome pathway; this releases NF-kB, which enters the nucleus, where it functions together with IRF3 and other transcription factors to induce the expression of interferons and inflammatory cytokines such as TNF, IL-1b and IL-6.

Debbie Maizels/Nature PublishingGroup
DNA is a pathogen-associated molecular pattern when it is delivered to the host cytoplasm by microbial infection, and is a danger-associated molecular pattern when enters the cytoplasm from the nucleus (e.g., through DNA damage and reverse transcription of retroelements), mitochondria or dead cells. Cytosolic DNA binds to and activates cGAS, which catalyzes the synthesis of 2′3′-cGAMP from ATP and GTP. 2′3′-cGAMP binds to the ER adaptor STING, which traffics to the ER-Golgi intermediate compartment (ERGIC) and the Golgi apparatus. STING then activates IKK and TBK1. TBK1 phosphorylates STING, which in turn recruits IRF3 for phosphorylation by TBK1. Phosphorylated IRF3 dimerizes and then enters the nucleus, where it functions with NF-kB to turn on the expression of type I interferons and other immunomodulatory molecules.
Regulation of cGAS activation by DNA binding
cGAS is activated by double-stranded DNA (dsDNA) independently of the DNA sequence6. The crystal structure of the cGAS–DNA complex shows that cGAS binds to the sugar-phosphate backbone of dsDNA but not to any of the base, which explains the DNA-sequence-independent activation of cGAS7,8,10,11. Single-stranded DNA that can form internal duplex structures can also activate cGAS. Some single-stranded DNAs that form a Y-shaped structure that includes a duplex and single-stranded overhangs containing a stretch of guanine residues can potently activate cGAS, but the structural basis of this activation remains to be determined24. Oxidation of DNA bases, such as that caused by ultraviolet irradiation, does not impair or enhance the DNA's ability to activate cGAS, but the oxidized DNA is more resistant to cellular nucleases, which leads to greater induction of interferons25. dsRNA can bind to cGAS but cannot activate it. Modeling studies suggest that B-form DNA binds to and 'pushes' an activation loop of cGAS, which results in rearrangement of the active site that causes its activation7. In contrast, A-form dsRNA cannot push the activation loop of cGAS, which might explain why RNA cannot activate cGAS. Short dsDNA (∼15 base pairs in length) is sufficient to bind and activate cGAS in vitro, but longer DNA is needed to activate the cGAS pathway in cells, presumably because of the presence of nucleases and other regulatory factors in cells.
Regulation of cGAS by post-translational modifications
Although the main mechanism for the activation of cGAS is through its binding to DNA, additional mechanisms must exist to regulate the cGAS pathway to modulate a robust and sensitive response to foreign DNA while ensuring the organism remains unresponsive to self DNA. While understanding of cGAS regulation is still in its infancy, studies have suggested that post-translational modifications of cGAS regulate its enzymatic activity. Human cGAS is phosphorylated at Ser305 (Ser291 in mouse cGAS) by the kinase Akt26. This phosphorylation inhibits cGAS activity, suggestive of potential crosstalk between the cGAS pathway and other pathways that regulate Akt. cGAS is glutamylated by the enzymes TTLL4 and TTLL6, and this modification inhibits cGAS activity27. Reversal of this modification by the carboxylpeptidases CCP5 and CCP6 activates cGAS activity. In both cases, only a small fraction of cGAS proteins are modified by inhibitory phosphorylation or glutamylation, which raises the question of how such modifications influence the activity of the bulk of cGAS enzymes that are not modified.
Transcriptional and epigenetic regulation of cGAS
The gene encoding cGAS is induced by interferon; this induction provides a positive feedback mechanism for amplification of activation of the cGAS pathway28,29. Many tumor cell lines have lost the expression of cGAS and are therefore defective in producing interferons and cytokines in response to stimulation by DNA or infection with DNA viruses6,30. In some of these cells, suppression of cGAS expression can be reversed by inhibitors of DNA methylation, which suggests that expression of the gene encoding cGAS is silenced through epigenetic mechanisms31. Similar to cGAS expression, STING expression is also suppressed in many cancer cell lines. The frequent disabling of the cGAS–STING pathway in cancer cells suggests that activation of this pathway might be an impediment to cellular transformation. Alternatively, inactivation of this pathway might assist the evasion of cancer cells from surveillance by the host immune system. In addition to cancer cells, some primary cells, including hepatocytes and T cells, also lack a functional cGAS–STING pathway32,33. The lack of an interferon response to DNA in hepatocytes and T cells might contribute to the infection of these cells by hepatitis B virus and human immunodeficiency virus (HIV), respectively.
Regulation of cGAMP stability and transport
As for other secondary messengers, the intracellular amount of cGAMP is regulated not only by the rate of synthesis but probably also by the rate of decay. Although no intracellular cGAMP phosphodiesterase has been identified so far, the extracellular enzyme ENPP1 degrades 2′3′-cGAMP with high specificity34. Whether ENPP1 regulates the DNA-sensing pathway in vivo requires further investigation. cGAMP can be transferred between cells through gap junctions35, a mechanism that allows virus-infected cells to alert uninfected neighboring cells to activate the interferon pathway to resist infection. This might be particularly important for immune defense against viruses that produce antagonists of immune responses in the virus-infected cells. Some viruses can also incorporate cGAMP into the viral particles and deliver cGAMP into the next infected cell36,37; such a mechanism presumably allows the infected cells to mount a rapid response to viral infection.
Regulation of STING by cGAMP binding
STING contains four transmembrane domains at the amino terminus that anchor the protein to the ER membrane, with the large carboxy-terminal domain projecting into the cytoplasm. STING forms a dimer on the ER membrane in the presence or absence of a ligand13,38,39,40,41,42. One molecule of cGAMP binds to a central crevice of a STING dimer through extensive hydrophobic interactions and hydrogen bonding13,38. This binding induces an extensive conformational change in STING that is postulated to release a carboxy-terminal tail (CTT) that recruits and activates TBK1. However, the CTT of STING is not visible in crystal structures of STING. Through molecular modeling and functional assays, it has been proposed that a 'lid' structure of STING that forms as a result of cGAMP's binding provides an 'anchor' site that binds to the carboxyl terminus of STING; this causes the CTT to form a structure that resembles a TBK1 substrate and thereby allows TBK1 to phosphorylate the CTT of STING, which is important for IRF3 activation (discussed below)43. 2′3′-cGAMP binds to STING with a dissociation constant of approximately 4 nM, which represents much tighter binding than that of other cGAMP isomers or bacterial cyclic dinucleotides (CDNs), including cyclic di-GMP and cyclic di-AMP13. Analyses of free ligand conformations and STING-bound ligand conformations have revealed that 2′3′-cGAMP, but not the other isomers, adopts an organized conformation in solution that is similar to the STING-bound conformation and therefore 'pays' lower enthalpy and entropy costs when it binds to STING44. This model provides an explanation for the higher affinity of endogenous cGAMP for STING than that of other cGAMP isomers or bacterial CDNs. Certain flavonoid compounds, such as DMXAA and CMA, can bind and activate mouse STING but not human STING45,46,47. In these cases, two molecules of DMXAA or CMA bind to the same cGAMP-binding site of a mouse STING dimer, and this binding induces a conformational rearrangement of STING similar to that caused by cGAMP's binding38,47. The observation that DMXAA activates mouse STING but not human STING might explain in part the finding that this compound has antitumor activity in mice but has failed in human clinical trials.
Regulation of STING by post-translational modifications
Shortly after cGAMP binds, STING activates IKK and TBK1, which in turn activate NF-kB and IRF3, respectively. TBK1 phosphorylates STING at several serine and threonine residues, including Ser366. This phosphorylation of STING is critical for the subsequent phosphorylation of IRF3 by TBK1 (ref. 48). Phosphorylated STING binds to a positively charged region of IRF3 and thereby recruits IRF3 for phosphorylation by TBK1. Once IRF3 is phosphorylated, it forms a homodimer that enters the nucleus to induce interferon expression. Notably, several adaptors of innate immunity, including MAVS, TRIF and STING, share a highly conserved motif (p-Leu-X-Ile-Ser, where 'p-' indicates phosphorylation and 'X' indicates any amino acid) that is phosphorylated in response to stimulation with a cognate ligand. Phosphorylation of these adaptors provides a 'licensing' mechanism for the phosphorylation of IRF3 by TBK1. This mechanism ensures that among all agents that can activate TBK1, only a few, such as viral infection, lead to the activation of IRF3 and induction of interferon expression. This is an important mechanism that prevents excessive production of interferons, which can otherwise cause autoimmune diseases. It has also been proposed that phosphorylation of STING at Ser366 leads to the degradation of STING and inactivation of the pathway49. However, this model is inconsistent with the data showing that substitution of that serine at position 366 with alanine abolishes STING activity and that this STING mutant is still degraded in response to stimulation by DNA or cGAMP21,48,49.
Several studies have suggested that STING is also regulated by ubiquitination. At least three distinct ubiquitin E3 ligases that positively regulate STING's activity have been identified. The E3 ligases TRIM56 and TRIM32 have been found to promote the Lys63 (K63) polyubiquitination of STING, which enhances its activation of the downstream pathway50,51. The ER-localized E3 ligase complex consisting of AMFR-GP78 and INSIG1 promotes the K27 polyubiquitination of STING, recruitment of TBK1 and induction of interferons52. In contrast, the E3 ligases RNF5 and TRIM30a promote the K48 polyubiquitination of STING, which targets it for degradation by the proteasome; this results in inhibition of the DNA-sensing pathway53,54. Overall, the roles of ubiquitination in STING activation or inactivation appear to be quite complex, and further work is needed to clarify the roles of different types of ubiquitination and E3 ligases in the STING pathway in vivo.
Regulation of STING trafficking and degradation
After binding to cGAMP, STING rapidly traffics from the ER to perinuclear compartments and forms large punctate structures18. Inhibition of the trafficking of STING by brefeldin A, an inhibitor of ARF GTPase, blocks activation of the downstream pathway49, which suggests that the trafficking of STING is important for its activation. The shigella effector protein IpaJ, which inactivates the ARF family of GTPases and thereby blocks trafficking from the ER-Golgi intermediate compartment to the Golgi apparatus, also inhibits the induction of interferon by STING20. STING is palmitolated at Cys88 and Cys91 at the Golgi apparatus, and inhibition of the palmitolation of STING by a chemical inhibitor or by alteration of the two cysteine residues blocks STING activation55. After STING accomplishes its signaling function, it is rapidly degraded, possibly through autophagy49. The autophagy kinase ULK1-ATG1 is reported to have a role in triggering the degradation of STING, but the mechanism by which STING is targeted to the autophagy pathway remains to be elucidated.
Other regulators of the cytosolic DNA-sensing pathway
Prior to the discovery of cGAS as a cytosolic DNA sensor that activates the interferon pathway, several other proteins were proposed to function as DNA sensors56. These included DAI, DDX41, DNA-PK and IFI16, among others. However, the role of these proteins in the cytosolic DNA-sensing pathway is controversial, in part because none have been confirmed by genetic studies to be involved in this. Mice that lack DAI can mount a normal interferon response to stimulation by DNA or infection with DNA viruses57. Similarly, a mouse strain engineered to lack all cytosolic-receptor-AIM2-like receptors, which include the mouse homologs of IFI16, displays no apparent defect in the induction of interferon by cytosolic DNA or infection with DNA viruses58. Similarly, human cells depleted of IFI16 via CRISPR technology show normal induction of interferon by cytosolic DNA58. In contrast to results obtained for those putative DNA sensors, genetic deletion of cGAS completely abolishes the induction of interferon by transfection of DNA or infection with DNA-containing pathogens, including DNA viruses, retroviruses and bacteria4. Thus, cGAS has a non-redundant role in the cytosolic DNA-sensing pathway that leads to interferon production.
The polyglutamine-binding protein PQBP1 has been reported to facilitate the sensing of HIV-1 DNA by cGAS59. Knockdown of PQBP1 inhibits the induction of interferon by HIV-1 but not the induction achieved by infection with DNA viruses or transfection of DNA. What in the HIV-1 DNA that is specifically recognized by PQBP1 but not by cGAS remains unclear. More genetic studies are needed to clarify the role of PQBP1 in the sensing of HIV-1.
cGAS also binds to the autophagy regulator beclin-1; this interaction induces autophagy through a STING-independent mechanism60. The binding of cGAS to beclin-1 releases RUBICON, a negative regulator of autophagy, from the beclin-1 complex and thereby leads to the induction of autophagy, which facilitates the removal of cytosolic DNA.
Physiological functions of the cGAS–STING pathway
The ability of cGAS to detect and mount a rigorous immune response to almost any dsDNA suggests that cGAS is a general sensor for a large spectrum of microbial pathogens that contain DNA. This has been demonstrated by genetic approaches, especially the use of cGAS-deficient mice. Several DNA viruses, including herpes simplex virus, vaccinia virus, adenovirus, cytomegalovirus and Kaposi's sarcoma-associated herpesvirus (KSHV) induce interferons in a cGAS- and STING-dependent manner61,62,63,64,65,66,67. Mice deficient in cGAS or STING do not produce interferons in response to infection by herpes simplex virus type 1, murine g-herpesvirus virus 68 or vaccinia virus, have high viral titers and quickly succumb to infections. cGAS- and STING-deficient mice are also more sensitive to infection with RNA viruses, despite the fact that cells that lack cGAS or STING have normal interferon production in response to RNA viruses67. It is possible that the cGAS pathway is responsible for producing low or 'tonic' amounts of interferons at steady state and that such amounts of interferons are important for controlling infection with DNA or RNA viruses in vivo. Alternatively, infection with RNA viruses in vivo might cause cellular damage and cell death that results in 'leakage' of cellular DNA, which in turn activates the cGAS pathway to help the immune defense against infection with RNA viruses. Membrane fusion by viral particles is also suggested to activate the STING pathway and might contribute to immune defense against RNA viruses68.
cGAS is a key sensor for retroviruses, including HIV-1 and HIV-2 (refs. 69,70,71). After HIV viral capsids enter macrophages and dendritic cells (DCs), reverse transcription inside the viral capsid converts the viral RNA into cDNA, which is injected directly into the nucleus and is integrated into the host genome. Thus, retroviruses do not normally trigger strong innate immune responses. However, if the integrity of the viral capsid is compromised or if some of the host factors (such as SAMHD1, TREX1 and CPSF6) that normally prevent the accumulation of cytoplasmic DNA lose their function, the reverse-transcribed viral DNA can be detected in the cytoplasm by cGAS, which triggers the induction of interferons and other cytokines.
The role of cGAS in detecting reverse-transcribed cDNA has been extended to endogenous retroviruses and retroelements. Mice that lack cGAS or STING fail to mount T cell–independent B cell responses to multivalent antigens, such as bacterial capsular polysaccharides72. Immunization with such antigens leads to the upregulation of endogenous retroviral RNA in antigen-specific B cells. The RNA is detected by the pathway of the RNA helicase RIG-I and MAVS and is also reverse-transcribed to DNA, which is then detected by the cGAS–STING pathway. Deletion of both MAVS and cGAS leads to more profound defects in the B cell response than does deletion of either alone. Thus, both MAVS and cGAS are important for the detection of endogenous retroviruses, which facilitate antibody production in B cells.
Bacteria contain abundant DNA. Many bacteria can invade mammalian cells and replicate in the phagosomes or even in the cytosol73. Bacteria utilize their secretion apparatus to deliver effector molecules into the cytoplasm. Although it is still not clear how bacterial DNA might gain access to the cytoplasm, many intracellular bacteria induce interferons through the cGAS–STING pathway. The growing list of bacteria that activate the cGAS pathway includes Mycobacteria, Legionella, Listeria, Shigella, Francisella, Chlamydia, Neisseria and group B streptococcus20,74,75,76,77,78,79,80,81. With the exception of Listeria monocytogenes, which induces interferons in mouse macrophages in a manner dependent on STING but not on cGAS, the induction of interferon by most bacteria is largely abolished in the absence of cGAS. This is surprising, given that many bacteria produce CDNs such as cyclic di-GMP and cyclic di-AMP, which can directly activate STING. One possibility is that the bacterial CDNs do not gain easy access to the cytoplasm. Another possibility is that bacterial and host cells contain enzymes that selectively digest the bacterial CDNs but not the 2′3′-cGAMP produced by host cells.
Evasion of the cGAS–STING pathway by microbial pathogens
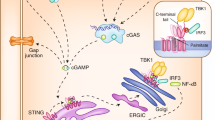
During the constant 'arms race' between pathogens and hosts, most microbes have evolved ways to successfully evade the attack by the immune system of their susceptible host. Thus, viruses and bacteria have numerous strategies with which to antagonize the cGAS–STING pathway (Fig. 2). The g-herpesvirus KSHV encodes ORF52, a tegument protein that inhibits cGAS activity through direct binding to cGAS as well as through binding to DNA64. A cytoplasmic form of the KSHV nuclear antigen LANA also inhibits the cGAS activity63. Further, the viral interferon-regulatory factor vIRF1 of KSHV inhibits STING by preventing its binding to TBK1 (ref. 65). Herpes simplex virus type 1 encodes the regulatory protein ICP27, which interacts with STING and TBK1 and thereby prevents the phosphorylation of IRF3 by TBK1 (ref. 82). The E1A protein of adenovirus and E7 protein of human papilloma virus inhibit STING signaling by binding to STING through a LXCXE motif (Leu-X-Cys-X-Glu, where 'X' is any amino acid) of these viral proteins83. Shigella utilizes the effector IpaJ to inhibit ARF GTPases and thereby prevents the trafficking and activation of STING20.

Debbie Maizels/Nature PublishingGroup
Multiple steps along the cGAS pathway are regulated by host or viral factors. Enzymes (including SAMHD1, TREX1, DNase II and RNase H2) control the synthesis or stability of cytosolic DNA. cGAS expression is induced by interferon signaling and is suppressed by DNA methylation. cGAS activity is also regulated by phosphorylation. The cellular amount of 2′3′-cGAMP is further controlled by its degradation by ENPP1 and perhaps other enzymes. cGAMP can also be transferred to neighboring cells through the gap junction. STING is regulated by its trafficking to the ER-Golgi intermediate compartment and the Golgi apparatus and by its degradation through autophagy. Several viral proteins (ORF52 and LANA of KHSV; E7 of human papilloma virus (HPV); E1A of adenovirus (AdV); and vIRF1 of KSHV) inhibit cGAS and STING in the pathway. TYK and SYK, kinases; STAT, signal transducer and transcription activator; PDE, phosphodiesterase; HSV-1, herpes simplex virus type 1; ISG, interferon-stimulated gene.
For all microbes, the most common and effective way of avoiding triggering the cGAS pathway is by 'hiding' their DNA from the cytoplasm. Retroviruses and many DNA viruses keep their DNA in the viral capsid while they traverse the cytoplasm and then deliver their viral DNA into the nucleus. For HIV-1, the viral capsid in the cytoplasm further recruits the host factors CPSF6 and cyclophilins69. These host factors help coordinate reverse transcription, capsid uncoating and entry of the viral pre-integration complex into the nucleus, such that viral DNA is not exposed to the cytoplasm. Depletion of CPSF6 leads to the induction of interferon in macrophages infected with wild-type HIV-1. Similarly, depletion of the DNase Trex1 leads to the accumulation of HIV-1 DNA in the cytoplasm, which triggers the induction of interferon via cGAS. These results suggest that HIV-1 co-opts host proteins to minimize the exposure and accumulation of viral DNA in the cytoplasm, which would otherwise trigger the cGAS–STING pathway.
The cGAS–STING pathway in autoimmune and inflammatory disease
While the cGAS–STING pathway has evolved as a major defense mechanism for the detection of microbial infection, activation of cGAS by self DNA represents a threat for triggering autoimmunity. Any DNA released or leaked from the nucleus and mitochondria into the cytoplasm might trigger the cGAS pathway. A link between the cGAS pathway and autoimmune diseases has been established in Aicardi-Gourtières syndrome, a collection of monogenic autoimmune diseases characterized by elevated expression of genes induced by type I interferon84. Genetic studies of patients with this syndrome have identified causal mutations in several key genes, including those encoding TREX1, RNase H2, SAMHD1, the adenosine deaminase ADAR1 and the cytosolic receptor MDA5. Among those, in mice lacking functional TREX1 or RNaseH2, activation of the cGAS–STING pathway causes the diseases85,86,87,88,89. TREX1 is an exonuclease that degrades nicked dsDNA and single-stranded DNA90. TREX1-deficient mice die within a few months after birth because of multi-organ inflammation, especially myocarditis91, while deletion of just one allele of the gene encoding cGAS or STING in TREX1-deficient mice largely rescues them from the lethal autoimmunity phenotype85,86,87. RNase H2 degrades RNA in the RNA–DNA hybrid and removes ribose that is mis-incorporated into dsDNA during DNA replication. A knock-in mouse strain expressing a catalytically inactive mutant of RNase H2A dies in the perinatal period, and primary cells from the embryos of these mice exhibit elevated expression of interferon-stimulated genes, which is abolished by deletion of cGAS or STING88. Deletion of STING partially rescues these mice from the perinatal lethality. Deletion of DNase II, a major enzyme that digests DNA in the lysosome, causes embryonic death in mice owing to excessive production of type I interferons in the embryos, which kills erythrocytes92. Deletion of the interferon receptor IFNAR rescues mice from the embryonic lethality, but deletion of TLR9 or the adaptor MyD88 does not93,94. Interestingly, mice lacking both DNase II and IFNAR develop polyarthritis, probably owing to the continued production of inflammatory cytokines such as TNF. Deletion of cGAS or STING in Dnase-II-deficient mice completely rescues them from the embryonic lethality, as well as from autoimmune and inflammatory phenotypes86.
Direct evidence that activation of STING causes human autoinflammatory disease has been provided by the identification of gain-of-function mutations of the gene encoding STING in human patients with early-onset vasculopathy and pulmonary inflammation95. The diseases caused by such mutations, now categorized as SAVI ('STING-associated vasculopathy with onset in infancy'), affect mainly the skin, blood vessels and lungs. In addition to developing cutaneous inflammation, such as ulcers and necrosis of the skin, patients with SAVI can also develop widespread lung damage that leads to pulmonary fibrosis and difficulty in breathing. The gain-of-function mutations in the gene encoding STING render the protein constitutively active or more sensitive to stimulation by cGAMP, which results in elevated production of interferons by a variety of cells, including vascular endothelial cells. Collectively, these genetic studies of human and mice provide proof-of-concept findings for targeting the cGAS–STING pathway to treat certain human autoimmune and inflammatory diseases.
Roles of the cGAS–STING pathway in cancer
Interferons have a critical role in the immunosurveillance of cancer cells96,97. In addition to their direct cytotoxic effects on cancer cells, interferons also promote the maturation and antigen presentation of DCs and thereby link innate immune responses to adaptive immune responses. How the innate immune system detects tumor cells for the production of interferons has been a longstanding mystery. Self DNA from dying tumor cells has been suggested to be an important danger signal that triggers the cGAS–STING pathway to induce interferons98 (Fig. 3). After the transplantation of immunogenic tumors into syngeneic mice, tumors grow more rapidly in STING-deficient mice than in wild-type mice or TRIF-deficient mice99. Priming of CD8+ T cells against tumor-associated antigens is defective in STING-deficient mice but not in those lacking TLRs, MyD88 or MAVS. STING is also required for the antitumor effects of radiation, but MyD88 and TRIF are not. An antibody to CD47 (a phagocytosis-inhibitory protein with high expression in tumor cells, including cancer stem cells) also exerts antitumor effects in a STING-dependent manner100,101. Radiation and treatment with antibody to CD47 kill tumor cells, which are taken up by phagocytes, including DCs. Through an unknown mechanism, the tumor-derived DNA is delivered to the cytoplasm of CD8a+ DCs, which activates the cGAS–STING pathway; this in turn facilitates antigen presentation by major histocompatibility complex class I and the activation of CD8+ T cells. Consistent with that model, intratumor delivery of cGAMP or its analogs into tumor-bearing mice leads to substantial inhibition of tumor growth and improves the survival of the mice102,103. The combination of cGAMP with irradiation or immune-system-checkpoint inhibitors (such as antibodies to PD-1 and PD-L1) produces synergistic antitumor effects, which indicates that the cGAS–STING pathway is important for the sensing of tumors by the innate immune system and has a critical role in intrinsic antitumor immunity. However, activation of the cGAS–STING pathway can also lead to tolerogenic responses through the induction of indolamine 2,3-dioxygenase104. Activation of STING promotes the growth of tumors with low antigenicity but not of those with higher antigenicity105. Brain-metastatic cancer cells produce cGAMP, which is transferred to neighboring astrocytes to activate the production of inflammatory cytokines, which facilitates metastasis106. Such studies warrant caution in ongoing clinical efforts to enhance cancer immunotherapy by stimulating the STING pathway with cGAMP analogs. Ultimately, a good clinical outcome will probably be achieved through an optimal combination of different treatments, including stimulation of the innate immune system, blockade of immune-system checkpoints, radiation, targeted cytotoxic agents and even adoptive T cell transfer.

Debbie Maizels/Nature PublishingGroup
Dying tumor cells are taken up by DCs, and the tumor DNA is delivered to the cytoplasm through an unknown mechanism. The DNA activates the cGAS pathway to induce the expression of interferons, major histocompatibility complex class I (MHCI) and co-stimulatory molecules such as CD86. interferons stimulate the maturation of DCs and facilitates presentation of tumor associated antigens on major histocompatibility complex class I. The DCs then migrate to lymph nodes to activate CD8+ T cells, which seek and attack tumors in target tissues. TCR, T cell antigen receptor; CD28, costimulatory receptor; IFN, interferon.
Summary and future perspectives
Since the discovery of the cGAS–cGAMP–STING pathway of cytosolic DNA sensing, a series of biochemical, structural and genetic studies have established the basic framework and mechanisms of this DNA-sensing pathway. However, much remains to be learned about the regulation of this pathway and details of the signaling mechanism at each step of the pathway. The nature of the DNA that directly activates cGAS in the cytoplasm in the context of microbial infection and autoimmune disease is still largely unknown. Additional regulators that control the activity of cGAS, stability of cGAMP and trafficking and degradation of STING remain to be discovered. Further work is also needed to understand how STING activates downstream kinases and transcription factors. Another interesting area of investigation is the exploration of potential crosstalk between the cGAS–STING pathway and other innate-immune-system pathways.
Genetic and microbiological studies have demonstrated an essential role for the cGAS–STING pathway in immunological defense against a variety of DNA viruses, retroviruses and bacteria. It is expected that the range of pathogens detected by this pathway will continue to expand, and it will probably include fungi and parasites that also contain abundant DNA. Because cGAMP has potent adjuvant activities66, this endogenous small molecule has great potential to aid in the development of new vaccines for the prevention and treatment of infectious diseases such as AIDS, tuberculosis and malaria.
Because all cells and tissues contain DNA that can potentially function as a 'danger' molecule to trigger inflammation, activation of the cGAS pathway might be linked to inflammatory diseases in many vital organs. Thus, the cGAS pathway could have a role in many common diseases that involve inflammation, such as cardiovascular disease, neurodegenerative disease, inflammatory bowel disease, diabetes, fibrosis, lupus, arthritis and psoriasis. An important and exciting task now is to develop potent and specific inhibitors of the cGAS–STING pathway, which will be not only very useful as research tools but also valuable as potential therapeutic agents for the treatment of a variety of human diseases.
The finding that the cGAS–STING pathway has a critical role in intrinsic antitumor immune responses has inspired intense efforts to harness this natural defense pathway to boost antitumor immunity in next-generation cancer immunotherapies. Parallel to the drug-development efforts, basic research must be carried out to delineate how stimulation of the cGAS–STING pathway might lead to stimulatory immune responses versus tolerogenic immune responses under different conditions. Knowledge obtained from this line of research will be important in guiding cancer therapies with better precision.
References
Pandey, S., Kawai, T. & Akira, S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 7, a016246 (2014).
Broz, P. & Dixit, V.M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Yoneyama, M., Onomoto, K., Jogi, M., Akaboshi, T. & Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 32, 48–53 (2015).
Cai, X., Chiu, Y.H. & Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296 (2014).
Land, W.G. Innate Alloimmunity, Part 1: Innate Immunity and Host Defense (Pabst Science Publishers, 2011).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Zhang, X. et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 (2014).
Li, X. et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 (2013).
Kranzusch, P.J., Lee, A.S., Berger, J.M. & Doudna, J.A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Reports 3, 1362–1368 (2013).
Gao, P. et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Zhang, X. et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235 (2013).
Diner, E.J. et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Reports 3, 1355–1361 (2013).
Ablasser, A. et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Ishikawa, H. & Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Zhong, B. et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 (2008).
Saitoh, T. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 106, 20842–20846 (2009).
Ishikawa, H., Ma, Z. & Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Dobbs, N. et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168 (2015).
Tanaka, Y. & Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20 (2012).
Fitzgerald, K.A. et al. IKKe and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003).
Sharma, S. et al. Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 (2003).
Herzner, A.-M. et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 16, 1025–1033 (2015).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 (2013).
Seo, G.J. et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 13, 440–449 (2015).
Xia, P. et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 17, 369–378 (2016).
Schoggins, J.W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
Ma, F. et al. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 194, 1545–1554 (2015).
Chiu, Y.H., Macmillan, J.B. & Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Xia, T., Konno, H., Ahn, J. & Barber, G.N. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282–297 (2016).
Thomsen, M.K. et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 64, 746–759 (2016).
Berg, R.K. et al. T cells detect intracellular DNA but fail to induce type I IFN responses: implications for restriction of HIV replication. PLoS One 9, e84513 (2014).
Li, L. et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 10, 1043–1048 (2014).
Ablasser, A. et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503, 530–534 (2013).
Gentili, M. et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science 349, 1232–1236 (2015).
Bridgeman, A. et al. Viruses transfer the antiviral second messenger cGAMP between cells. Science 349, 1228–1232 (2015).
Gao, P. et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154, 748–762 (2013).
Yin, Q. et al. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46, 735–745 (2012).
Shu, C., Yi, G., Watts, T., Kao, C.C. & Li, P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 19, 722–724 (2012).
Shang, G. et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 19, 725–727 (2012).
Ouyang, S. et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36, 1073–1086 (2012).
Tsuchiya, Y., Jounai, N., Takeshita, F., Ishii, K.J. & Mizuguchi, K. Ligand-induced ordering of the C-terminal tail primes STING for phosphorylation by TBK1. EBioMedicine 9, 87–96 (2016).
Shi, H., Wu, J., Chen, Z.J. & Chen, C. Molecular basis for the specific recognition of the metazoan cyclic GMP-AMP by the innate immune adaptor protein STING. Proc. Natl. Acad. Sci. USA 112, 8947–8952 (2015).
Kim, S. et al. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem. Biol. 8, 1396–1401 (2013).
Conlon, J. et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J. Immunol. 190, 5216–5225 (2013).
Cavlar, T., Deimling, T., Ablasser, A., Hopfner, K.P. & Hornung, V. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J. 32, 1440–1450 (2013).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Konno, H., Konno, K. & Barber, G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698 (2013).
Zhang, J., Hu, M.M., Wang, Y.Y. & Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655 (2012).
Tsuchida, T. et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 (2010).
Wang, Q. et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014).
Zhong, B. et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 (2009).
Wang, Y. et al. TRIM30a Is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog. 11, e1005012 (2015).
Mukai, K. et al. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 7, 11932 (2016).
Paludan, S.R. & Bowie, A.G. Immune sensing of DNA. Immunity 38, 870–880 (2013).
Ishii, K.J. et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 (2008).
Gray, E.E. et al. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45, 255–266 (2016).
Yoh, S.M. et al. PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 161, 1293–1305 (2015).
Liang, Q. et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15, 228–238 (2014).
Paijo, J. et al. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog. 12, e1005546 (2016).
Lio, C.W. et al. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J. Virol. 90, 7789–7797 (2016).
Zhang, G. et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 113, E1034–E1043 (2016).
Wu, J.J. et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18, 333–344 (2015).
Ma, Z. et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 112, E4306–E4315 (2015).
Li, X.D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Schoggins, J.W. et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014).
Holm, C.K. et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 13, 737–743 (2012).
Rasaiyaah, J. et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405 (2013).
Lahaye, X. et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 39, 1132–1142 (2013).
Gao, D. et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 (2013).
Zeng, M. et al. MAVS, cGAS, and endogenous retroviruses in T-independent B cell responses. Science 346, 1486–1492 (2014).
Portnoy, D.A., Auerbuch, V. & Glomski, I.J. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J. Cell Biol. 158, 409–414 (2002).
Watson, R.O. et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17, 811–819 (2015).
Wassermann, R. et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17, 799–810 (2015).
Collins, A.C. et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe 17, 820–828 (2015).
Hansen, K. et al. Listeria monocytogenes induces IFNb expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 33, 1654–1666 (2014).
Storek, K.M., Gertsvolf, N.A., Ohlson, M.B. & Monack, D.M. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 194, 3236–3245 (2015).
Zhang, Y. et al. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-b during Chlamydia trachomatis infection. J. Immunol. 193, 2394–2404 (2014).
Andrade, W.A. et al. Type I interferon induction by Neisseria gonorrhoeae: dual requirement of cyclic GMP-AMP synthase and Toll-like receptor 4. Cell Rep. 15, 2438–2448 (2016).
Andrade, W.A. et al. Group B streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe 20, 49–59 (2016).
Christensen, M.H. et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 35, 1385–1399 (2016).
Lau, L., Gray, E.E., Brunette, R.L. & Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571 (2015).
Crow, Y.J. Type I interferonopathies: mendelian type I interferon up-regulation. Curr. Opin. Immunol. 32, 7–12 (2015).
Gray, E.E., Treuting, P.M., Woodward, J.J. & Stetson, D.B. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutières syndrome. J. Immunol. 195, 1939–1943 (2015).
Gao, D. et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 112, E5699–E5705 (2015).
Gall, A. et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 (2012).
Pokatayev, V. et al. RNase H2 catalytic core Aicardi-Goutières syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J. Exp. Med. 213, 329–336 (2016).
Mackenzie, K.J. et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844 (2016).
Lindahl, T., Barnes, D.E., Yang, Y.G. & Robins, P. Biochemical properties of mammalian TREX1 and its association with DNA replication and inherited inflammatory disease. Biochem. Soc. Trans. 37, 535–538 (2009).
Yang, Y.G., Lindahl, T. & Barnes, D.E. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886 (2007).
Kawane, K. et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292, 1546–1549 (2001).
Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2005).
Okabe, Y., Kawane, K., Akira, S., Taniguchi, T. & Nagata, S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J. Exp. Med. 202, 1333–1339 (2005).
Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014).
Dunn, G.P., Koebel, C.M. & Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 6, 836–848 (2006).
Fuertes, M.B., Woo, S.R., Burnett, B., Fu, Y.X. & Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 34, 67–73 (2013).
Corrales, L. & Gajewski, T.F. Endogenous and pharmacologic targeting of the STING pathway in cancer immunotherapy. Cytokine 77, 245–247 (2016).
Woo, S.R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Deng, L. et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014).
Liu, X. et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 21, 1209–1215 (2015).
Demaria, O. et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 112, 15408–15413 (2015).
Corrales, L. et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018–1030 (2015).
Huang, L. et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J. Immunol. 191, 3509–3513 (2013).
Lemos, H. et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 76, 2076–2081 (2016).
Chen, Q. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Chen, Q., Sun, L. & Chen, Z. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol 17, 1142–1149 (2016). https://doi.org/10.1038/ni.3558
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3558
This article is cited by
-
cGAS-STING, inflammasomes and pyroptosis: an overview of crosstalk mechanism of activation and regulation
Cell Communication and Signaling (2024)
-
Second messenger 2'3'-cyclic GMP-AMP (2'3'-cGAMP): the cell autonomous and non-autonomous roles in cancer progression
Acta Pharmacologica Sinica (2024)
-
Innate immune sensing of lysosomal dysfunction drives multiple lysosomal storage disorders
Nature Cell Biology (2024)
-
Conservation and similarity of bacterial and eukaryotic innate immunity
Nature Reviews Microbiology (2024)
-
Cystoid Macular Oedema in a Patient Treated with STING Agonist and Ezabenlimab for Disseminated Melanoma
Ophthalmology and Therapy (2024)
