
Abstract
In polycystic kidney disease (PKD), a most common human genetic diseases, fluid-filled cysts displace normal renal tubules and cause end-stage renal failure. PKD is a serious and costly disorder. There is no available therapy that prevents or slows down the cystogenesis and cyst expansion in PKD. Numerous efforts have been made to find drug targets and the candidate drugs to treat PKD. Recent studies have defined the mechanisms underlying PKD and new therapies directed toward them. In this review article, we summarize the pathogenesis of PKD, possible drug targets, available PKD models for screening and evaluating new drugs as well as candidate drugs that are being developed.
Similar content being viewed by others

Introduction
Polycystic kidney disease (PKD), an inherited human renal disease, is characterized by massive enlargement of fluid-filled renal tubular and/or collecting duct cysts1. Progressively enlarging cysts compromise normal renal parenchyma, often leading to renal failure. The occurrence of autosomal dominant polycystic kidney disease (ADPKD) is estimated to be between 1 in 1000 and 1 in 400 individuals by a study in Olmsted Country, MN2. ADPKD is caused by mutations in one of two genes (Pkd1 and Pkd2) expressing the interacting polycystic proteins polycystin-1 (PC1) and polycystin-2 (PC2) in renal tubular epithelia3, 4. Mutation of Pkd1 accounts for approximately 85% cases in clinically identified patients5. PC1 is a membrane receptor capable of binding and interacting with many proteins, including carbohydrates and lipids, and eliciting intracellular responses through phosphorylation pathways6, 7. PC2 is thought to act as a calcium permeable channel8, 9. PC1 and PC2 form a complex that localizes to primary cilia10, 11. The polycystin complex has a role in the regulation of the proliferation, differentiation and morphogenesis of renal tubular cells through interactions with protein complexes linked to the actin cytoskeleton, intracellular signaling cascades, and the regulation of gene transcription12, 13 (Figure 1). In ADPKD, the thousands of large, spherical cysts of various sizes throughout the cortex and medulla are derived from the segments of the nephron. Autosomal recessive polycystic kidney disease (ARPKD) results primarily from the mutations in a single gene, Pkhd114. Its frequency is estimated to be one per 20000 individuals. The PKHD1 protein, fibrocystin, has been found to be localized to primary cilia and the basal bodies. The exact function of fibrocystin has not been demonstrated. In ARPKD, smaller, elongated cysts arise as ecstatic expansions of collecting ducts. Patients with PKD often require dialysis and kidney transplantation, which are exceedingly costly. There are currently no approved drug or preventive strategies for PKD.

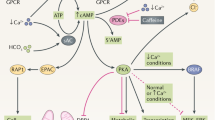
A diagram depicting the effects that PC1 and PC2 exert on signaling pathways. Multiple direct and indirect interactions allow the polycystin proteins to be inhibited or stimulated. Pathways was invoved in cell proliferation and liquid secretion.
Mechanisms of renal cyst formation and enlargement in PKD
The development and growth of PKD cysts involve the abnormal proliferation and apoptosis of immature epithelial cells, accumulation of fluid within the cyst cavity, abnormal cell-cell/cell-matrix interactions and abnormal cilia function.
Role of epithelial cell proliferation and apoptosis in cyst development in PKD
Increased apoptosis and proliferative capacity in renal epithelial cells are essential processes in PKD. While the proliferation of renal tubular epithelial cells halts before birth in normal individuals, cystic epithelia proliferate throughout life in patients with ADPKD15. Several genetic manipulations that increase the proliferation of tubular epithelial cells in mice results in PKD16, 17, 18, 19.
Epidermal growth factor (EGF), transforming growth factor alpha (TGF-α) and EGF receptor (EGFR) promote cystic epithelial proliferation and expand renal cysts. EGFR is overexpressed and mislocalized to the apical membranes of cystic epithelial cells, which leads to a sustained stimulation of cell proliferation in the cysts20. Increased intracellular cAMP level also plays a crucial role in cystogenesis. The reduced calcium caused by mutation of Pkd1 or Pkd2 can inhibit adenylyl cyclase 6 leading to increased cAMP. Studies have demonstrated that cAMP inhibits the proliferation of normal renal epithelial cells. In contrast, cAMP promotes the proliferation of cells derived from PKD patients21. The switch is caused by decreased intracellular calcium levels in a polycystic kidney leading to cAMP-mediated stimulation of the B-Raf/MEK/ERK pathway instead of inhibiting the Ras/Raf/MEK/ERK pathway like in the normal kidney22. B-Raf is inhibited by Akt in normal cells, while it is activated because of decreased activation of Akt in calcium-restricted cells. Inhibitors of Akt and PI3K can reproduce the effects of calcium reduction. However, activation of Akt has been found in animal models of PKD, such as Pkd−/− mice, Han:SPRD rats and jck mice. Additional growth factors, cytokines, lipid factors, and adenosine triphosphate (ATP) also participate in regulating the proliferation of renal epithelial cells23, 24, 25. Cell apoptosis is also a key factor in the development of PKD. Knocking out the anti-apoptotic Bcl-2 and AP-2 genes or overexpression of the pro-apoptotic gene c-myc in mice results in renal cystogenesis26.
Role of fluid secretion in cyst development in PKD
Fluid secretion is a critical pathogenic mechanism associated with cyst formation and growth in PKD. Fluid secretion, coupled with epithelial hyperplasia, is necessary and sufficient to account for the dynamics of cyst growth. In PKD, a large number of cystic lesions lack afferent and efferent tubule connections, suggesting that cysts, which arise from tubular segments, become disconnected from the glomerular filtrate. The development and expansion of cystic lesions therefore requires net transepithelial fluid secretion. An extensive body of in vitro data implicates epithelial chloride secretion in the generation and maintenance of fluid-filled cysts27. The fluid secretion is driven by mechanisms that are similar to those found in other secretory epithelia. Chloride movement drives fluid into the cyst lumen. Fluid accumulation causes cyst enlargement directly by swelling cysts and indirectly by stretching cells to promote cell division28.
Cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-regulated chloride channel, is present on the apical membranes of many secretory epithelia. Chloride secretion through the CFTR has been implicated in the pathway of fluid secretion in PKD. In vitro experiments have suggested that increased cAMP-mediated chloride secretion provides the electrochemical driving force for fluid secretion in cystic epithelia29. CFTR is expressed in the apical membrane of intact cysts dissected from PKD kidneys30. An important role of CFTR in PKD fluid secretion is also supported by the observation that interference with CFTR protein production (by treatment of ADPKD monolayers with antisense oligonucleotide against human CFTR) dramatically reduced fluid secretion by these cells27. Additional evidence supporting a role of CFTR in chloride secretion was obtained from immortalized cystic murine collecting duct cell lines isolated from CFTR mutant and CFTR wild-type mice. The wild-type cell lines formed numerous fluid-filled cysts in response to EGF and forskolin when cultured in three-dimensional collagen gels, whereas the CFTR mutant cell lines failed to form cysts under identical conditions31. These results demonstrate that CFTR is required for in vitro cyst formation. In a single family affected with both ADPKD and cystic fibrosis (CF), individual members with both ADPKD and CF had less severe renal disease than those family members with only ADPKD32, 33. These studies suggest that in vivo, defective CFTR function provides partial protection against renal cyst development and enlargement and suggests that modulation of epithelial chloride secretion may have therapeutic benefit in PKD. Using type I MDCK cells as a cell culture model of cyst development and growth, Sheppard's group found that the CFTR inhibitor CFTRinh-172 (see below) retarded cyst growth. In contrast, blockers of other types of apical membrane Cl− channels, which do not inhibit CFTR, failed to inhibit cyst growth28. Inhibition of cyst growth by CFTR inhibitors correlated with inhibition of cAMP-stimulated Cl− current, but not cell proliferation28. Their studies strongly suggest that CFTR inhibitors might retard cyst growth predominantly by inhibiting fluid accumulation within the cyst lumen. In two ARPKD animal models, PCK rats and the pcy mice, renal cAMP levels were significantly higher compared to that in wild-type animals. Expression of the water channel AQP2 and vasopressin V2 receptor (VPV2R) was also increased. Administration of the VPV2R antagonist OPC-31260 lowered renal cAMP levels and halted progression or caused regression of established cysts33, 34.
Aquaporin (AQP)-mediated water permeability in cyst epithelia may also be involved in fluid secretion in cyst formation and progression, as fluid consists of salts and water. Normally, several AQPs are expressed in kidney: AQP1 in the proximal tubule, thin descending limb of Henle, and vasa recta; AQP2 in the apical membranes of collecting duct; AQP3 and AQP4 in the basolateral membranes of collecting duct. It has been reported that AQP1 and AQP2 are expressed in cyst epithelia from patients with PKD35. Gattone et al36 found AQP2 and AQP3 expression in cysts in C57BL/6J-cpk/cpk mice with autosomal recessive-infantile polycystic kidney disease. High aquaporin-dependent water permeability in cyst epithelium may be important to facilitate near-isosmolar fluid secretion, particularly in growing cysts that have low surface-to-volume ratios.
Role of cell-cell/cell-matrix interactions in cyst development in PKD
PC1 has been detected in tight junctions, adhesions junctions, desmosomes, focal adhesions, apical vesicles, and primary cilia37, 38. A study has shown that PC1 mediates cell-cell adhesion through the formation of strong homophilic interaction of its Ig-like domains39. A significant downregulation of Pkd1 mRNA is detected in MDCK cysts compared to tubules, which leads to a striking reduction of membrane PC1 and mislocalization to the cytoplasmic pools40. It has been demonstrated that a controlled level of PC1 expressed at cell-cell junction is critical for normal tubular differentiation. In normal renal cells, PC1 forms a complex with the protein E-cadherin and its catenins. However, in primary cells from ADPKD patients, the PC1/E-cadherin/β-catenin complex was disrupted and was accompanied by increased PC1 phosphorylation, reduced E-cadherin and upregulated normal mesenchymal N-cadherin41.
Renal epithelial cells in ADPKD show increased PC1 adhesion to type I collagen compared with normal human epithelia42. The defects reduce the cell migratory capacity required for kidney morphogenesis43. The PC1 protein has been proven to regulate the relationships between the cell and matrix through interacting with α1β2 integrin, vinculin, paxillin, p130-cas, talin and focal adhesion kinase (FAK)42. The basement-membrane composition and expression of matrix metalloproteases and their inhibitors are abnormal in PKD kidneys. It has been demonstrated that inactivation of several matrix adhesion receptors and focal adhesion complex-associated proteins result in cystogenesis44, 45, 46.
Role of cilia in cyst development in PKD
Renal cilia are microtubule-based, membrane-bound projections on the epithelia of the renal tubule and duct. Renal cilia have been reported to be mechanosensors and respond to flow by increasing intracellular calcium47. Several studies support that PC1 and PC2 localize to primary cilia10, 38 and form a subfamily of transient receptor potential channels that are responsible for sensing flow and regulating levels of intracellular calcium48. The bending of cilia causes calcium influx into the cell through the PC2 channel42. The mechanosensory response is lost in cells with mutated PC148. Many cellular functions that are related to PKD, such as gene expression, cell cycle, differentiation and apoptosis, are regulated by intracellular calcium concentration.
The dysfunction of cilia has a close relationship with cell cycle progression49, 50. PC1 upregulates p21 (waf1) through activating the JAK-STAT pathway and results in cell cycle arrest in G0/G123. The IFT88/Polaris protein, which is localized to cilia, has been demonstrated to be tightly associated with the centrosome during cell cycle transition51. Overexpression of IFT88/Polaris prevents G1/S transition and induces cell death. In contrast, deletion of IFT88/Polaris promotes cell cycle progression51. PC2 also can regulate the cell cycle through direct interaction with Id2, a member of the helix-loop-helix (HLH) protein family, which has been proven to regulate cell proliferation and differentiation52.
Experimental models for screening and evaluating new drugs for PKD
Several common experimental models that have been used to screen and evaluate the new PKD drugs at the cell, organ and whole animal levels are described in subsequent sections.
Madin-Darby canine kidney (MDCK) cyst model
MDCK type I cells provide a useful in vitro model of cystogenesis for screening candidate inhibitors of cyst formation and growth (Figure 2). MDCK cells cultured in three-dimensional collagen gels with forskolin produce a polarized, single-layer, thinned epithelium surrounding a fluid-filled space similar to the cysts in PKD53. MDCK cells in cysts undergo proliferation, fluid transport and matrix remodeling, as seen in tubular epithelial cells cultured from PKD kidneys. Cyst formation and growth are cAMP-dependent, which is thought to independently increase cell proliferation and activate CFTR-facilitated transepithelial fluid secretion28. Recognizing its limitations, such as differences between MDCK cells versus renal epithelial cells and cell cultures versus intact kidneys, the MDCK cyst model may be used to identify cyst inhibitors that reduce cyst formation and enlargement without demonstrable cell toxicity or inhibition of cell proliferation.

MDCK cyst growth in collagen gels. Light micrographs were taken at indicated days after cell seeding of MDCK cells exposed continuously to 10 μmol/L forskolin without (top) or with cyst inhibitor (bottom). Each series of photographs shows the same cyst on successive days in culture.
Embryonic kidney cyst model
The embryonic kidney culture model permits organotypic growth and differentiation of renal tissue in defined medium without the confounding effects of circulating hormones and glomerular filtration54. In the absence of 8-Br-cAMP, kidneys cultured on porous cell culture inserts increase in size over 4 d, whereas numerous cystic structures were seen in the presence of 8-Br-cAMP (Figure 3). Although embryonic kidney cultures probably represent a better PKD model than MDCK cells, they are avascular and non-perfused and therefore are not exposed to the same environment as the in vivo kidney.

Embryonic kidney cyst model. Embryonic kidneys at d E13.5 were cultured for 4 d. (A) Kidney appearance by transmitted light microscopy for cultures in the absence (top) or continued presence (bottom) of 100 μmol/L 8-Br-cAMP. Each series of photographs shows the same kidney on successive days in culture. (B) Histology (hematoxylin and eosin staining) of embryonic kidneys.
PKD mouse models
Pkd1flox/−;Ksp-Cre mice, are kidney-selective Pkd1 knockout mice that manifest a fulminant course with the development of large cysts (Figure 4), renal failure in the first 2 weeks of life and death by 20 d. This model is suitable to evaluate the efficacy of cyst inhibitors on retarding the growth of cysts in the distal segments of the nephron, including the medullary thick ascending limbs of the loops of Henle, distal convoluted tubule and collecting ducts. In humans, ADPKD develops slowly and causes renal failure at an average age of over 50 years. For experimental studies, this relatively severe model of ADPKD has been used, rather than mouse models in which disease develops more slowly because of the shorter time required for compound administration and the greater likelihood of observing an immediate benefit. Testing cyst inhibitors in the ADPKD mouse model should be of further utility in predicting efficacy in human ADPKD. The CFTR inhibitors significantly reduced cyst formation and clinical signs of PKD, as assessed by lower kidney weights and serum creatinine and urea concentrations in this mouse model55.

Pkd1flox/−; Ksp-Cre PKD mouse model. (A) Kidney from wildtype (left) and Pkd1flox/−; Ksp-Cre PKD mouse (right) at age 5 d. (B) Histology (hematoxylin and eosin staining) of kidneys from Pkd1flox/−; Ksp-Cre PKD mice at ages 1 to 5 d.
Pkd1flox mice and Ksp-Cre transgenic mice have been generated as described56, 57. Ksp-Cre mice express Cre recombinase in the kidney under the control of the Ksp-cadherin promoter58. Pkd1flox/−; Ksp-Cre mice were generated by crossbreeding Pkd1flox/flox mice with Pkd1+/−:Ksp-Cre mice56. Neonatal mice (age 1 d) were genotyped by genomic PCR. Test compound or saline DMSO vehicle control were administered by subcutaneous injection on the backs of neonatal mice four times a day for 3 or 7 d using a 1 mL insulin syringe beginning at age 2 d. Pkd1flox/+; Ksp-Cre or Pkd1flox/+ mice from the same litter were used as controls. Body weight was measured at d 5. Blood and urine samples were collected to measure the test compound concentration and renal function. The kidneys were removed, weighed, and fixed for histological examination or homogenized to determine the test compound content.
Many other mouse models of PKD have been described in which the mutant phenotypes result from spontaneous mutations or gene-specific targeting in mouse orthologs of human PKD genes. These murine phenotypes closely resemble human PKD with common abnormalities observed in the tubular epithelia, interstitial compartment, and extracellular matrix of cystic kidneys59.
Pkd1 and Pkd2 knockout mouse models, which are homologs of human genes, have been generated by targeted mutagenesis59, 60. In most of these models, heterozygous mice develop renal, biliary, and pancreatic cysts at age 4-19 months. Disease progression is rapid, with embryonic lethality occurring in most homozygous mutants.
In the mouse models arising from spontaneous mutations, PKD is generally transmitted as an autosomal recessive trait. Several of these models with cysts distributed along the entire nephron and slower disease progression closely recapitulate human ADPKD59. One of them is the murine autosomal recessive juvenile cystic kidney (jck)61. The jck locus maps to chromosome 11. The mutant allele has a missense change in Nek8, which encodes NIMA (for 'never in mitosis' A)-related kinase 862. In homozygous mutant mice, focal renal cysts are evident as early as 3 d of life, and the renal cystic disease is slowly progressive but not evident by kidney palpation until age 4 to 5 weeks. Histological analysis of jck mutant kidney tubules showed the defects were specific to the connecting segment and collecting duct cells. The proximal tubule cells appeared morphologically normal. Cell membrane and cytoplasmic disruption could be observed in collecting ducts from mutant mice at 2–3 weeks of age. No histological abnormalities in other organs have been described. The mutant mice are fertile and generally survive for 4 months or more.
Another PKD mouse model arose spontaneously by mutation of the “congenital polycystic kidney” (cpk) gene with locus mapping to mouse chromosome 1263. Cys1, the cpk gene, encodes cystin, which localizes to the primary apical cilia on collecting duct cells. Mutant mice develop massive renal cystic disease and progressive renal insufficiency in a pattern that resembles human ARPKD. Initial cystic changes are evident at approximately embryonic d 16 and localize primarily to the proximal tubule. With progressive postnatal age, the cystic changes predominantly involve the collecting duct. Death occurs by 3–4 weeks of age due to uremia64.
PKD in the kat mouse model is caused by a spontaneous mutation occurring in the Nek1 gene, which encodes NIMA-related kinase 1. In Nek1kat-2J homozygotes, fluid-filled cysts and dilated proximal tubules and Bowman spaces are found as early as 1 month of age. The bilateral renal cystic disease involves all levels of the nephron by 3 months of age. Disease progression, including growth of cysts and an increase in the number of cysts, is similar to that in ADPKD.
As a model of ADPKD, the Han:SPRD-cy rat has been used for research extensively65, 66, 67. The gene locus maps to chromosome 568. The spontaneous mutation occurs in the Sprague-Dawley strain. In male Cy/+ rats, the kidneys was enlarged more rapidly, and interstitial fibrosis is more pronounced69. The Han:SPRD Cy/+ rat can be studied for the efficacy of long-term medical therapy. In this model, the renal cyst exclusively develops in the proximal tubules instead of the whole renal segment. Other mouse models, bpk, jcpk, orpk, inv and pcy, also resemble human PKD with respect to renal cyst pathology and disease progression60. Because the murine models share common pathogenic features with human PKD, it is assumed that there are common molecular pathways involved in PKD progression in humans and mice. The jck, cpk, and kat mouse models are commercially available from the Jackson Laboratories.
The dynamics of cyst growth differ in the various models. These differences provide a unique opportunity to study the mechanism of cyst formation. The Nek8jck mouse model can be used mainly to test the preventive role of cyst inhibitors in the formation of cysts in collecting ducts of young mice. The Cys1cpk mouse model is suitable to test the role of cyst inhibitors on the progression of cysts and to compare the effects of treatments on cysts derived from different cell types in all levels of the nephron. The Nek1kat mouse model has been proposed to study the roles of cyst inhibitors on cysts derived from proximal tubules. Heterozygous Pkd2WS25 mice, an ADPKD model generated by targeted mutagenesis, can be used to test the prevention and the treatment with cyst inhibitors on the development of cysts in the kidney and other organs.
Cyst progression can be evaluated by measuring the size and number of cysts in the kidney. At first, the ratio of kidney weight to body weight can be measured. Development of cysts should increase kidney weight. For light microscopic analysis, transverse tissue sections, including cortex, medulla and papilla, can be stained with H&E to measure cyst size and number. The analysis can be performed by a reviewer who is blinded to the identity of the treatment modality. To quantitatively evaluate cyst growth, cyst size can be recorded on the following scale: 0, <0.05 mm (It is difficult to distinguish the cysts from normal renal tubules); 1, 0.05–0.3 mm; 2, 0.3–0.6 mm; 3, 0.6–0.9 mm; 4, 0.9 mm–1.2 mm; and 5, >1.2 mm. The number of cysts can be counted in the cortex, medulla and papilla. In some experiments, the origin of renal tubule cysts can be determined by segment–specific lectin binding using Dolichos biflorus agglutinin (DBA) as a marker for collecting ducts and Lotus tetragonolobus (LTA) as a marker for proximal tubules as described previously70. The numbers of LTA-positive and DBA-positive cysts can be counted in serial sections of bisected whole-mount kidneys from each animal. Proximal tubule cysts can be identified by LTA binding, and collecting duct cysts can be identified by DBA binding. A minimum of 10 sets of serial sections evenly spaced through the kidney from the cortex to the inner medulla can be used to determine the ratio of proximal tubule to collecting duct cysts.
Candidate drugs under research and development
Based on the mechanism of renal cyst development and the pathogenesis of PKD, some chemical and natural compounds have been discovered to have inhibitory activity on renal cysts and to slow PKD progression. Some classes of candidate PKD drugs have been described according to the drug targets in PKD as follows.
Vasopressin 2 receptor (V2R) antagonist
Studies were conducted to target cAMP pathways and take a step further by demonstrating the upregulation of vasopressin and the inhibition of cytogenesis by V2R antagonists OPC-31260 in cpk mice, ARPKD (PCK rat), ADPKD (Pkd2WS25 mice) and adolescent nephronophthisis (pcy mouse)34, 35, 37. As OPC-31260 is a weak antagonist for human V2R, clinical trials with tolvaptan, which has a higher affinity for human V2R, are underway. Tolvaptan was also effective in animal models of ARPKD, ADPKD, and nephronophthisis71, 72, 73. The Tolvaptan Efficacy and Safety in Management of PKD and Outcomes (TEMPO) program is currently active74, 75. Phase 2a studies to determine the response to increasing doses of tolvaptan (15, 30, 60, and 120 mg) in patients with ADPKD and normal renal function have been completed75, 76. A 3-year phase 3, placebo-controlled, double-blind study in 18-to 50-year-old patients with ADPKD to determine long-term safety and efficacy has been initiated and will be completed in 2011.
Renin angiotensin aldosterone system (RAAS) antagonist
Angiotensin-II (AT-II) has been demonstrated to promote cellular proliferation, apoptosis, and the production of TNF-α and other pro-inflammatory cytokines77. RAAS also plays an important role in hypertension. So, RAAS antagonism can prevent cellular proliferation and inflammation and treat hypertension in PKD. Angiotensin-converting enzyme (ACE) inhibitors, which are RAAS antagonists, have been proven to reduce cyst enlargement and blood pressure and improve renal function in Han:SPRD rats78, 79. A randomized 7-year study showed that ACE inhibitors prevented left ventricular hypertrophy better than calcium channel blockers in 75 hypertensive ADPKD patients80. An earlier longitudinal study has shown slower renal progression in those treated only with ACE inhibitor compared to only diuretics. Two HALT PKD trials that are randomized, double-blind, and placebo-controlled are underway to test the impact of intensive blockade of RAAS in ADPKD patients with ACE inhibitor or angiotensin receptor blocker (ARB)81.
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor
EGF is an important factor in cyst epithelial cell proliferation and cystogenesis. EKI-785, an EGFR tyrosine kinase inhibitor, has been shown to be effective in reducing cyst formation and decreasing mortality in murine ARPKD82. EKI-785 and another EGFR tyrosine kinase inhibitor EKB-569 attenuate the development of PKD in Han:SPRD rats66. Contrary to other murine models of ARPKD, overexpression and mislocalization of EGFR are not found at the apical membrane of cystic cells in PCK rats64. This may be the reason that EKI-758 and EKI-569 have no efficacy in PCK rats64.
Peroxisome proliferator-activated receptor-γ (PPARγ) agonist
Proliferation is recognized as an important factor for cysts development in PKD. PPARγ, a member of the superfamily of nuclear hormone receptor transcription factors, has been demonstrated to suppress cell growth and promote differentiation and apoptosis in various cancer cells83. Thus, it may be effective in treating PKD. A recent study showed that the expression of PPARγ was greater in ADPKD kidneys and cyst-lining epithelial cells compared to normal kidneys and human kidney cortex cells84. Rosiglitazone, a PPARγ agonist, significantly inhibits the proliferation of ADPKD cystic epithelial cells by causing a G0/G1 arrest. Short-term treatment in Han:SPRD rats with rosiglitazone has been shown to attenuate the development of kidney cysts and improve renal function, while long-term administration with rosiglitazone can prolong survival in Han:SPRD rats67.
Somatostatin
Octreotide, a kind of somatostatin, has been shown to inhibit hepatic and renal cystogenesis in PCK rats by decreasing cAMP accumulation85. A clinical trial has shown that octreotide safely slows renal volume expansion in 6-month therapy for 13 ADPKD patients86. Recently, octreotide has been tested as long-term treatment for polycystic kidney and polycystic liver disease in a clinical trial.
Phosphodiesterase (PDE) activator
In PKD, cAMP has been proven to be a critical intracellular second messenger involved in cytogenesis. The level of cAMP is largely regulated by the PDE superfamily through hydroxylation. In mixed cortical tubules and microdissected tubular segments, 50%–70% of PDE activity is inhibited by an inhibitor of the calcium-calmodulin-sensitive PDE187. PDE1 is responsible for cAMP and cGMP activity. The reduction of intracellular calcium in PKD may increase cAMP by dysregulating PDE1. PDE3 inhibited by increased cGMP are cAMP-specific PDEs. PDE3 may also be involved in cAMP accumulation in renal cells of PKD kidneys. In mesangial cells, PDE3 inhibitors increase cAMP levels and activate PKA, block phosphorylation of Raf-1 on serine 338 and suppress Raf-1 kinase activity88. PDE inhibitors stimulate MDCK cell proliferation. A recent study showed that the protein levels of PDE1, PDE3, and PDE4 are significantly reduced in the cysts of PCK and Pkd2−/WS25 kidneys compared with wild-type kidneys89, which indicates that a PDE activator may inhibit cystogenesis.
Src inhibitor
Src has been confirmed to be an important intermediary in cAMP pathways that promote epithelial proliferation in PKD and also a critical mediator in the activation of the EGFR axis. Src activity has a relationship with PKD progression in BPK mice and PCK rats90. SKI-606 can inhibit Src activity in a highly specific manner. SKI-606, which is also in clinical trials for breast cancer and malignant tumors, significantly improves renal and biliary lesions in BPK and PCK rodent models of ARPKD90. Thus, Src can be a prospective therapeutic target in PKD.
Raf inhibitor
Sorafenib, a small molecule Raf inhibitor, has been demonstrated to inhibit the proliferation of cells derived from the cysts of human ADPKD kidneys91. Sorafenib has also been proven to treat renal cell carcinomas and prolong survival time92. Cyst growth in human ADPKD cystic cells cultured within three dimensional collagen is completely inhibited by sorafenib91. This study suggests that the inhibition of the Raf kinases may be an effective therapy for PKD.
Mitogen extracellular kinase (MEK) inhibitor
MEK is an important mediator in EGFR and cAMP signaling. PD98059, an inhibitor of MEK, has been shown to completely prevent ADPKD cellular proliferation in response to cAMP agonists21. Another MEK inhibitor, PD184352, improved renal function and reduced the expression of P-ERK and ERK in pcy mice93. However, U0126, an inhibitor of MEK1/2 that blocks phosphorylation of ERK, did not change the rate of cyst growth in Pkd1flox/−:ksp-Cre mice94. More studies on MEK inhibitor efficiency in PKD are needed.
Mammalian target of rapamycin (mTOR) inhibitor
In human ADPKD patients and mouse models, the mTOR pathway is abnormally activated in cyst-lining epithelial cells. It has been shown that the cytoplasmic tail of PC1 interacts with tuberin95. Recently, another experiment96 directly showed that PC1 was able to inhibit the mTORC1(mTOR complex-1) cascade that regulates cell growth and proliferation, ribosome biogenesis and translation of a subset of mRNAs, cellular energy responses and autophagy97, 98. Mutations in PC1 therefore lead to persistent activation of mTOR, which promotes cell growth and proliferation and cyst expansion in PKD. Also, mTOR is activated by increased ERKs through inhibiting tuberin in the renal cells of ADPKD. Rapamycin, an inhibitor of mTOR, was shown to be highly effective in reducing renal cystogenesis in the bpk and orpk-rescue mouse models95. In another study, long-term rapamycin treatment in Han:SPRD rats resulted in a normalization of kidney volume, renal function, blood pressure and heart weight65. Treatment of human ADPKD transplant recipient patients with rapamycin showed a significant reduction in polycystic kidney volumes95. A two-year, placebo-controlled trial of another mTOR inhibitor, everolimus, involving 433 patients with ADPKD has been finished. Everolimus slowed the increase in total kidney volume, but the estimated GFR was not improved99.
Cystic fibrosis transmembrane conductance regulator (CFTR) inhibitor
CFTRinh-17259, a thiazolidinone, has been shown to slow cyst growth in a MDCK cell culture model of PKD28 and in metanephric kidney organ cultures55. CFTRinh-172 maintains the channel closed state, probably by binding to a cytoplasmic domain of CFTR according to patch-clamp analysis100. The other kind of CFTR inhibitors is the glycine hydrazides, which directly bind to the CFTR pore at a site near its external entrance101. In an experiment screening CFTR inhibitors for PKD56, tetrazolo-CFTRinh-172, a tetrazolo-derived thiazolidinone, and Ph-GlyH-101, a phenyl-derived glycine hydrazide, were found to almost completely suppress cyst growth without affecting cell proliferation. These compounds also showed a remarkable inhibition of cyst number and growth in an embryonic kidney cyst model. Subcutaneous delivery of tetrazolo-CFTRinh-172 and Ph-GlyH-101 to neonatal kidney-specific Pkd1 knockout mice for 7 d prominently slowed kidney enlargement, cyst expansion and renal function impairment.
Cyclin-dependent kinase (CDK) inhibitor
As we discussed previously, cilia has a close link with the cell cycle progression. The CDK inhibitor roscovitine effectively inhibited cyst formation through cell cycle arrest in jck and cpk mouse models of PKD102. Roscovitine has also been detected to be active against cysts originating from different parts of the nephron102. Roscovitine significantly downregulates cAMP and aquaporin 2103 and increases p21104, leading to decreased renal tubular epithelial cell proliferation.
TNF-α interventions
A recent study has demonstrated that TNF-α promotes the progression of PKD105. Treating inner medullary collecting duct (IMCD) cells with TNF-α increases the expression of FIP-2 and shows a striking loss of PC2 from its normal location. FIP-2 plays a role in transporting protein from the apical membrane and regulating epithelial cell polarity. TNF-α causes cystogenesis in the wild-type murine embryonic kidney, which is exacerbated in the Pkd2+/− embryonic kidney. Pkd2+/− mice injected with TNF-α have increased cyst development (the frequency was 6/14), while 50 Pkd2+/− mice treated with the TNF-α inhibitor ethanercept did not develop cysts. Another study has found that TNF-α can activate the mTOR pathway106, which plays an important role in PKD development. These studies suggest that inhibition of TNF-α can slow cyst formation in PKD.
Glucosylceramide synthase inhibitor
Glucosphingolipids have been proven to play an important role in regulating cell proliferation and apoptosis107. Recently, a study demonstrated that the glucosylceramide (GlcCer) synthase inhibitor Genz-123346 effectively inhibited cystogenesis in Pkd1−/−, jck and pcy mice108. GlcCer and ganglioside GM3 levels are higher in human and mouse PKD kidneys compared to normal kidneys. Molecular analysis of jck mice and jck cells shows that Genz-123346 prevents cyst growth by dysregulating Akt-mTOR signaling108.
Matrix metalloproteinases (MMPs) inhibitor
MMPs are a large family of proteinases that play an important role in remodeling extracellular matrix components and cleaving a number of cell surface proteins. Kidney tubules derived from the C57BL/6J-cpk mouse contain higher levels of MMP-2 and -9 than normal mice109. Serum MMP-1, -9, and tissue inhibitor of metalloproteinases-1 concentrations in patients with PKD were significantly higher compared to healthy controls 110. MMP-14 mRNA has a higher expression in cyst-lining epithelia and distal tubules in Han:SPRD rats111. Treating Han:SPRD-cy/+ rats with the MMP inhibitor, batimastat, for 8 weeks caused a prominent reduction in cyst number and kidney weight111, which suggests that MMP inhibitor could be potential therapy for PKD.
HMG-CoA reductase inhibitor
Statins, which are HMF-CoA reductase inhibitors, are widely applied to decrease cholesterol in clinical settings. They can be used for improving renal function in PKD. Lovastatin significantly decreased cystic kidney size and improved function in heterozygous male Han:SPRD rats112. It may be related with lovastatin reducing farnesyl pyrophosphate, which is important in cell proliferation112, and lovastatin can also cause actin filament disruption, which can induce apoptosis113. In a double-blind cross-over study, 10 normocholesterolemic ADPKD patients treated with 40 mg/d simvastatin or placebo for 4 weeks showed that simvastatin significantly improved both glomerular filtration rate (GFR) and effective renal plasma flow47. Another study of 16 ADPKD patients with well-preserved renal function treated with 40 mg/d simvastatin for six months proved that simvastatin ameliorated endothelial dysfunction in ADPKD patients using high resolution vascular ultrasound114. A randomized open-label clinical trial was performed with 49 ADPKD patients who were treated with 20 mg/d pravastatin or no treatment for 2 years115. There were no significant changes in the markers of kidney function or urinary protein excretion between the two groups.
Triptolide
Triptolide is a natural product isolated from the “Thunder God Vine”. It has been demonstrated to promote an increase in PC2-mediated calcium release and cytosolic calcium in the murine kidney epithelial Pkd2−/− cells and to inhibit cyst formation in Pkd1−/− embryonic mice116. Triptolide is an inhibitor of NF-κB- and NF-AT-mediated transcription, which results in reduced gene products and cell growth arrest117, 118. It has been proven to inhibit cell growth and increase p21 expression in Pkd1−/− kidney cells. In another study, triptolide significantly inhibited the early phases of cyst expansion and improved renal function at postnatal d 8 in a kidney-specific Pkd1flox−/−; Ksp-cre mouse model of ADPKD119. Recently, a study showed triptolide has a pronounced effect in reducing cyst formation in a Pkd1flox/flox; Mx1Cre mouse model of ADPKD120.
Curcumin
Curcumin is a natural polyphenol derived from the plant Curcuma longa. Numerous studies have indicated that curcumin is a highly pleiotropic molecule capable of treating various cancers. Our studies have proven that curcumin also has a significant inhibitory effect on renal cyst development121. Curcumin slowed cyst formation in both a MDCK cyst model and an embryonic kidney cyst model with a dose-dependent response. Curcumin inhibited forskolin-induced cell proliferation and promoted tubule formation in MDCK cells, which indicates that curcumin promotes MDCK cell differentiation. Curcumin affected intracellular signaling in the MDCK cells exposed to forskolin. Curcumin reduced signaling proteins Ras, B-raf, p-MEK, p-ERK, and c-fos and increased Raf-1 and NAB2 in MDCK cells.
Summary
PKD is a progressive disease with a decline in renal function. The cost of treatment, dialysis, and kidney transplantation related to PKD exceeds $1 billion in USA each year according to the Polycystic Kidney Research Foundation. Up to now, the treatment options for PKD have been limited to renal replacement therapy by dialysis or transplantation. Based on the understanding of the pathogenesis of PKD, the inhibition of cyst epithelia and cyst fluid secretion may provide a new therapeutic option in PKD. Dual or triple therapies may be highly effective in slowing PKD progression. In addition to advancing the understanding of the mechanism in which PKD develops, the functional and morphological improvement in PKD, as seen with chemical compounds, could provide a proof-of-concept for the application of new drugs in treating PKD.
References
Chapin HC, Caplan MJ . The cell biology of polycystic kidney disease. J Cell Biol 2010; 191: 701–10.
Torres VE . Treatment strategies and clinical trial design in ADPKD. Adv Chronic Kidney Dis 2010; 17: 190–204.
Qian F, Watnick TJ, Onuchic LF, Germino GG . The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 1996; 87: 979–87.
Wilson P . Molecular and cellular aspects of polycystic kidney disease. N Engl J Med 2004; 350: 151–64.
Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18: 2143–60.
Geng L, Segal Y, Peissel B, Deng N, Pei Y, Carone F, et al. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest 1996; 98: 2674–82.
International Polycystic Kidney Disease Consortium. Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell 1995; 81: 289–98.
Cai Y, Maeda Y, Cedzich A, Torres VE, Wu GQ, Hayashi T, et al. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem 1999; 274: 28557–65.
Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 2002; 4: 191–7.
Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB . Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol 2002; 12: R378–80.
Yoder BK, Hou X, Guay-Woodford LM . The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are colocalized in renal cilia. J Am Soc Nephrol 2002; 13: 2508–16.
Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, et al. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000; 408: 990–4.
Wu G, Tian X, Nishimura S, Markowitz GS, D'Agati V, Park JH, et al. Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum Mol Genet 2002; 11: 1845–54.
Harris PC, Rossetti S . Molecular genetics of autosomal recessive polycystic kidney disease. Mol Genet Metab 2004; 81: 75–85.
Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG . Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol 1995; 5: 1462–8.
Trudel M, D'Agati V, Costantini F . C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int 1991; 39: 665–71.
MacKay K, Striker LJ, Pinkert CA, Brinster RL, Striker GE . Glomerulosclerosis and renal cysts in mice transgenic for the early region of SV40. Kidney Int 1987; 32: 827–37.
Schaffner DL, Barrios R, Massey C, Banez EI, Ou CN, Rajagopalan S, et al. Targeting of the rasT24 oncogene to the proximal convoluted tubules in transgenic mice results in hyperplasia and polycystic kidneys. Am J Pathol 1993; 142: 1051–60.
Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS . Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc Natl Acad Sci U S A 2002; 99: 943–8.
Du J, Wilson PD . Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol 1995; 269: C487–95.
Yamaguchi T, Pelling JC, Ramaswamy NT . cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 2000; 57: 1460–71.
Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP . Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279: 40419–30.
Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, et al. PKD1 induces p21 (waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002; 109: 157–68.
Grantham JJ, Ye M, Davidow C, Holub BJ, Sharma M . Evidence for a potent lipid secretagogue in the cyst fluids of patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 6: 1242–9.
Chatterjee S, Shi WY, Wilson P, Mazumdar A . Role of lactosylceramide and MAP kinase kinase in the proliferation of proximal tubular cells in human polycystic kidney disease. J Lipid Res 1996; 37: 1334–44.
Torres, VE . Apoptosis in cystogenesis: hands on or hands off? Kidney Int 1999; 55: 334–5.
Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ . The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int 1996; 50: 208–18.
Li H, Findlay IA, Sheppard DN . The relationship between cell proliferation, Cl− secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int 2004; 66: 1926–38.
Sullivan LP, Wallace DP, Grantham JJ . Epithelial transport in polycystic kidney disease. Physiol Rev 1998; 78: 1165–91.
Brill SR, Ross KE, Davidow CJ, Ye M, Grantham JJ, Caplan MJ . Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc Natl Acad Sci U S A 1996; 93: 10206–11.
Sweeney WE, Avner ED, Elmer HL, Cotton CU . CFTR is required for cAMP-dependent: In vitro renal cyst formation. J Am Soc Nephrol 1998; 9: 38A.
O'Sullivan DA, Torres VE, Gabow PA, Thibodeau SN, King BF, Bergstralh EJ . Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis 1998; 32: 976–83.
Gattone VH, Wang X, Harris PC, Torres VE . Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9: 1323–6.
Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH 2nd . Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 2004; 10: 363–4.
Devuyst O, Burrow CR, Smith BL, Agre P, Knepper MA, Wilson PD . Expression of aquaporins-1 and -2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am J Physiol 1996; 271: F169–83.
Gattone VH 2nd, Maser RL, Tian C, Rosenberg JM, Branden MG . Developmental expression of urine concentration-associated genes and their altered expression in murine infantile-type polycystic kidney disease. Dev Genet 1999; 24: 309–18.
Ong AC, Harris PC . Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int 2005; 67: 1234–47.
Yoder BK, Hou X, Guay-Woodford LM . The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are colocalized in renal cilia. J Am Soc Nephrol 2002; 13: 2508–16.
Ibraghimov-Beskrovnaya O, Bukanov NO, Donohue LC, Dackowski WR, Klinger KW, Landes GM . Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum Mol Genet 2000; 9: 1641–9.
Bukanov NO, Husson H, Dackowski WR, Lawrence BD, Clow PA, Roberts BL, et al. Functional polycystin-1 expression is developmentally regulated during epithelial morphogenesis in vitro: downregulation and loss of membrane localization during cystogenesis. Hum Mol Genet 2002; 11: 923–36.
Roitbak T, Ward CJ, Harris PC, Bacallao R, Ness SA, Wandinger-Ness A . A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol Biol Cell 2004; 15: 1334–46.
Wilson PD, Geng L, Li X, Burrow CR . The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest 1999; 79: 1311–23.
Hyink DP, Burrow CR, Bloswick B, Wilson PD . Reversal of the ADPKD epithelial cell decreased motility phenotype by lithium. J Am Soc Nephrol 2000; 11: 392A.
Lo SH, Yu QC, Degenstein L, Chen LB, Fuchs E . Progressive kidney degeneration in mice lacking tensin. J Cell Biol 1997; 136: 1349–61.
Shannon MB, Miner JH . Insertional mutation in laminin alpha 5: a new mouse model for polycystic kidney disease. J Am Soc Nephrol 2004; 15: 652A.
van Dijk MA, Kamper AM, van Veen S, Souverijn JH, Blauw GJ . Effect of simvastatin on renal function in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2001; 16: 2152–7.
Praetorius HA, Spring KR . Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 2001; 184: 71–9.
Nauli SM, Alenghat FJ, Luo Y, Caniams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33: 129–37.
Pazour GJ . Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol 2004; 15: 2528–36.
Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Ruf RG, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of the primary cilia and left-right axis determination. Nat Genet 2003; 34: 413–20.
Robert A, Margall-Ducos G, Guidotti JE, Bregerie O, Celati C, Brechot C, et al. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J Cell Sci 2007; 120: 628–37.
Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J . Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol 2005; 7: 1202–12.
Balkovetz DF . Hepatocyte growth factor and Madin-Darby canine kidney cells: in vitro models of epithelial cell movement and morphogenesis. Microsc Res Tech 1998; 43: 456–63.
Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, et al. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na+, K+, 2Cl− co-transporter-dependent cystic dilation. J Am Soc Nephrol 2006; 17: 3424–37.
Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS . Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19: 1300–10.
Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 2008; 17: 1505–16.
Shao X, Somlo S, Igarashi P . Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 2002; 13: 1837–46.
Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 2002; 110: 1651–8.
Guay-Woodford LM . Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 2003; 285: F1034–49.
Pritchard L, Sloane-Stanley JA, Sharpe JA, Aspinwall R, Buckle V, Strmecki L, et al. A human PKD1 transgene generates functional polycystin-1 mice and is associated with a cystic phenotype. Human Mol Genet 2000; 9: 2617–27.
Atala A, Freeman MR, Mandell J, Beier DR . Juvenile cystic kidneys (jck): a new mouse mutation which causes polycystic kidneys. Kidney Int 1993; 43: 1081–5.
Liu S, Lu W, Obara T, Kuida S, Lehoczky J, Dewar K, et al. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002; 129: 5839–46.
Sommardahl C, Woychik R, Sweeney W, Avner E, Wilkinson J . Efficacy of taxol in the orpk mouse model of polycystic kidney disease. Pediatr Nephrol 1997; 11: 728–33.
Torres VE, Sweeney WE Jr, Wang X, Qian Q, Harris PC, Frost P, et al. Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int 2004; 66: 1766–73.
Zafar I, Belibi FA, He Z, Edelstein CL . Edelstein long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease. Nephrol Dial transplant 2009; 24: 2349–53.
Torres VE, Sweeney WE Jr, Wang X, Qian Q, Harris PC, Frost P, et al. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int 2003; 64: 1573–9.
Dai B, Liu Y, Mei C, Fu L, Xiong X, Zhang Y, et al. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin Sci 2010; 119: 323–33.
Bihoreau M, Ceccherini I, Browne J, Kranzlin B, Romeo G, Lathrop GM, et al. Location of the first genetic locus, PKDr1, controlling autosomal dominant polycystic kidney disease in Han:SPRD cy/- rat. Hum Mol Gen 1997; 6: 609–13.
Gretz N, Ceccherini I, Kranzlin B, Kloting I, Devoto M, Rohmeiss P, et al. Gender-dependent disease severity in autosomal polycystic kidney disease of rats. Kidney Int 1995; 48: 496–500.
Nakanishi K, Sweeney WE Jr, Macrae Dell K, Cotton CU, Avner ED . Role of CFTR in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 2001; 12: 719–25.
Wang X, Gattone VH 2nd, Harris PC, Torres VE . Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16: 846–51.
Gattone VH, Kinne Q, Torres VE . Efficacy of OPC-41061 in the treatment of murine nephronophthisis. J Am Soc Nephrol 2005; 16: 138A.
Wang S, Gattone VH, Somlo S, Harris PC, Torres VE . Effectiveness of OPC-41061 on polycystic kidney disease development in Pkd2WS25/–. J Am Soc Nephrol 2005; 16: 361A.
Grantham JJ, Chapman AB, Torres VE, Ouyang J, Shoaf SE, Czerwiec FS . Acute and chronic osmostasis after vasopressin V2 receptor inhibition with tolvaptan in ADPKD. J Am Soc Nephrol 2005; 16: 361A.
Torres VE, Wang X, Ward CJ, Grantham JJ, Chapman AB, Ouyang J, et al. Urine aquaporin 2 and cyclic AMP responses to tolvaptan administration in autosomal dominant kidney disease. J Am Soc Nephrol 2005; 16: 361A.
Chapman AB, Torres VE, Grantham JJ, Shoaf SS, Ouyang JJ, Czerwiec FS . A phase IIB pilot study of the safety and efficacy of tolvaptan, a vasopressin V2 receptor antagonist (V2RA) in patients with ADPKD. J Am Soc Nephrol 2005; 16: 68A.
Cao Z, Cooper ME . Role of angiotensin II in tubulointerstitial injury. Semin Nephrol 2001; 21: 554–62.
Zafar I, Tao Y, Falk S, McFann K, Schrier RW, Edelstein CL . Effect of statin and angiotensin-converting enzyme inhibition on structural and hemodynamic alterations in autosomal dominant polycystic kidney disease model. Am J Physiol Renal Physiol 2007; 293: F854–9.
Kennefick TM, Al-Nimri MA, Oyama TT, Thompson MM, Kelly FJ, Chapman JG, et al. Hypertension and renal injury in experimental polycystic kidney disease. Kidney Int 1999; 56: 2181–90.
Schrier R, McFann K, Johnson A, Chapman A, Edelstein C, Brosnahan G, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13: 1733–9.
Chapman AB, Torres VE, Perrone RD, Steinman TI, Bae KT, Miller JP, et al. The HALT polycystic kidney disease trials: design and implementation. Cin J Am Soc Nephrol 2010; 5: 102–9.
Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED . Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int 2000; 57: 33–40.
Grommes C, Landreth GE, Heneka MT . Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol 2004; 5: 419–29.
Liu Y, Dai B, Fu L, Jia J, Mei C . Rosiglitazone inhibits cell proliferation by inducing G1 cell cycle arrest and apoptosis in ADPKD cyst-lining epithelia cells. Basic Clin Pharmacol Toxicol 2010; 106: 523–30.
Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF . Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology 2007; 132: 1104–16.
Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene-Iordache B, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int 2005; 68: 206–16.
Kusano E, Yoshida I, Takeda S, Homma S, Yusufi AN, Dousa TP, et al. Nephron distribution of total low Km cyclic AMP phosphodiesterase in mouse, rat and rabbit kidney. Tohoku J Exp Med 2001; 193: 207–20.
Cheng J, Thompson MA, Walker HJ, Homma S, Yusufi AN, Dousa TP, et al. Differential regulation of mesangial cell mitogenesis by cAMP phosphodiesterase isozymes 3 and 4. Am J Physiol Renal Physiol 2004; 287: F940–53.
Wang X, Ward CJ, Harris PC, Torres VE . Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int 2010; 77: 129–40.
Sweeney WE Jr, von Vigier RO, Frost P, Avner ED . Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol 2008; 19: 1331–41.
Yamaguchi T, Reif GA, Calvet JP, Wallace DP . Sorafenib inhibits cAMP-dependent ERK activation, cell proliferation, and in vitro cyst growth of human ADPKD cyst epithelial cells. Am J Physiol Renal Physiol 2010; 299: F944–51.
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007; 356: 125–34.
Omori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, et al. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol 2006; 17: 1604–14.
Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 2008; 17: 1505–16.
Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103: 5466–71.
Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol 2009; 29: 2359–71.
Wullschleger S, Loewith R, Hall MN . TOR signaling in growth and metabolism. Cell 2006; 124: 471–84.
Huang J, Manning BD . The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008; 412: 179–90.
Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363: 830–40.
Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ . Altered channel gating mechanism for CFTR inhibition by a high affinity thiazolidinone blocker. FEBS Lett 2004; 558: 52–6.
Sonawane ND, Muanprasat C, Nagatani R Jr, Song Y, Verkman AS . In vivo pharmacology and antidiarrheal efficacy of a thiazolidinone CFTR inhibitor in rodents. J Pharm Sci 2004; 94: 134–43.
Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O . Long lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 2006; 444: 949–52.
Genzyme Corporation, Framingham, Massachusetts. Targeting dysregulated cell cycle and apoptosis for polycystic kidney disease therapy. Cell Cycle 2007; 6: 776–9.
Park JY, Schutzer WE, Lindsley JN, Bagby SP, Oyama TT, Anderson S, et al. p21 is decreased in polycystic kidney disease and leads to increased epithelial cell cycle progression: roscovitine augments p21 levels. BMC Nephrol 2007; 8: 12.
Li X, Magenheimer BS, Xia S, Johnson T, Wallace DP, Calvet JP, et al. A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat Med 2008; 14: 863–8.
Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007; 130: 440–55.
Bieberich E . Integration of glycosphingolipid metabolism and cell-fate decisions in cancer and stem cells: review and hypothesis. Glycoconj J 2004; 21: 315–27.
Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med 2010; 16: 788–92.
Rankin CA, Suzuki K, Itoh Y, Ziemer DM, Grantham JJ, Calvet JP, et al. Matrix metalloproteinases and TIMPS in cultured C57BL/6Jcpk kidney tubules. Kidney Int 1996; 50: 835–44.
Nakamura T, Ushiyama C, Suzuki S, Ebihara I, Shimada N, Koide H . Elevation of serum levels of metalloproteinase-1, tissue inhibitor of metalloproteinase-1 and type IV collagen, and plasma levels of metalloproteinase-9 in polycystic kidney disease. Am J Nephrol 2000; 20: 32–6.
Obermuller N, Morente N, Kranzlin B, Gretz N, Witzgall R . A possible role for metalloproteinases in renal cyst development. Am J Physiol Renal Physiol 2001; 280: F540–50.
Gile RD, Cowley BD Jr, Gattone VH 2nd, O'Donnell MP, Swan SK, Grantham JJ . Effect of lovastatin on the development of polycystic kidney disease in the Han:SPRD rat. Am J kidney Dis 1995; 26: 501–7.
Limura O, Vrtovsnik F, Terzi F, Friedlander G . HMG-CoA reductase inhibitors induce apoptosis in mouse proximal tubular cells in primary culture. Kidney int 1997; 52: 962–72.
Namli S, Oflaz H, Turgut F, Alisir S, Tufan F, Ucar A, et al. Improvement of endothelial dysfunction with simvastatin in patients with autosomal dominant polycystic kidney disease. Ren Fail 2007; 29: 55–9.
Fassett RG, Coombes JS, Packham D, Fairley KF, Kincaid-Smith P . Effect of pravastatin on kidney function and urinary protein excretion in autosomal dominant polycystic kidney disease. Scand J Urol Nephrol 2010; 44: 56–61.
Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, et al. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci U S A 2007; 104: 4389–94.
Lee KY, Chang W, Qiu D, Kao PN, Rosen GD . PG490 (triptolide) cooperates with tumor necrosis factor-alpha to induce apoptosis in tumor cells. J Biol Chem 1999; 274: 13451–5.
Liu H, Liu ZH, Chen ZH, Yang JW, Li LS . Triptolide: a potent inhibitor of NF-kappa B in T-lymphocytes. Acta Pharmacol Sin 2000; 21: 782–6.
Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM . Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol 2008; 19: 1659–62.
Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM . Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transplant 2010; 25: 2187–94.
Gao J, Zhou H, Lei T, Zhou L, Li W, Li X, et al. Curcumin inhibits renal cyst formation and enlargement in vitro by regulating intracellular signaling pathways. Eur J Pharmacol 2011; 654: 92–9.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sun, Y., Zhou, H. & Yang, Bx. Drug discovery for polycystic kidney disease. Acta Pharmacol Sin 32, 805–816 (2011). https://doi.org/10.1038/aps.2011.29
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.29
Keywords
This article is cited by
-
Autosomal dominant polycystic kidney disease and pioglitazone for its therapy: a comprehensive review with an emphasis on the molecular pathogenesis and pharmacological aspects
Molecular Medicine (2020)
-
Ganoderic acid A is the effective ingredient of Ganoderma triterpenes in retarding renal cyst development in polycystic kidney disease
Acta Pharmacologica Sinica (2020)
-
Analysis of catechol-O-methyltransferase gene mutation and identification of new pathogenic gene for paroxysmal kinesigenic dyskinesia
Neurological Sciences (2016)
-
Identification of people with autosomal dominant polycystic kidney disease using routine data: a cross sectional study
BMC Nephrology (2014)
-
An approach to cystic kidney diseases: the clinician's view
Nature Reviews Nephrology (2014)
