
Abstract
Purpose of Review
Tripartite motif (TRIM) proteins are a large group of E3 ubiquitin ligases involved in different cellular functions. Of special interest are their roles in innate immunity, inflammation, and virus replication. We discuss novel roles of TRIM proteins during virus infections that lead to increased pathogenicity.
Recent Findings
TRIM proteins regulate different antiviral and inflammatory signaling pathways, mostly by promoting ubiquitination of important factors including pattern recognition receptors, adaptor proteins, kinases, and transcription factors that are involved in type I interferon and NF-κB pathways. Therefore, viruses have developed mechanisms to target TRIMs for immune evasion. New evidence is emerging indicating that viruses have the ability to directly use TRIMs and the ubiquitination process to enhance the viral replication cycle and cause increased pathogenesis. A new report on TRIM7 also highlights the potential pro-viral role of TRIMs via ubiquitination of viral proteins and suggests a novel mechanism by which ubiquitination of virus envelope protein may provide determinants of tissue and species tropism.
Summary
TRIM proteins have important functions in promoting host defense against virus infection; however, viruses have adapted to evade TRIM-mediated immune responses and can hijack TRIMs to ultimately increase virus pathogenesis. Only by understanding specific TRIM-virus interactions and by using more in vivo approaches can we learn how to harness TRIM function to develop therapeutic approaches to reduce virus pathogenesis.
Similar content being viewed by others


Introduction
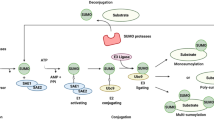
The tripartite motif (TRIM) is a superfamily of proteins conserved throughout the animal kingdom and has spread during vertebrate evolution; there are more than 80 known TRIMs encoded by the human and mouse genomes [1]. TRIM proteins are involved in many different cellular functions by acting as E3 ubiquitin (E3-Ub) ligases [2,3,4]. The consensus N-terminal region of TRIM proteins contains a RING finger domain followed by one or two B-box domains and a coiled-coil domain (CC) [4]. The RING domain comprises conserved cysteine and histidine residues that bind to two zinc atoms in a cross-brace arrangement and is essential for recruiting the E2-conjugated enzyme loaded with ubiquitin (E2 ~ Ub). The CC domain in combination with the B-box domain has been proposed to mediate protein-protein interactions, particularly homomeric and heteromeric interactions and by promoting the formation of interlocking helices between the TRIM family and other protein [5, 6]. Each TRIM protein has a specific C-terminal domain, which confers substrate specificity via protein-protein interactions [3, 7]. Most commonly found are the PRY and SPRY domains (B30.2), either in combination (PRY-SPRY) or individually. SPRY domains are found in some human protein families as well and are evolutionarily conserved in mammals, plants, and fungi [3]. Many TRIM proteins are induced by type I and type II interferons (IFN-I or IFN-II) or pathogen stimulations, in different cell types including human and mouse primary immune cells [8, 9]. Almost half of human TRIMs can positively regulate induction of IFN-I and/or NF-kB-mediated signaling [10, 11]. Since IFN signaling leads to induction of multiple IFN-stimulated genes (ISGs) that are known to have direct antiviral effector functions [12], together these studies suggest that TRIMs may have evolved as defense mechanisms that aid in resistance to pathogens [13,14,15]. In addition to their immune functions, as expected for enzymes involved in the ubiquitination process, TRIMs are also known to play important roles in a wide range of biological processes, including cell proliferation, differentiation, development, apoptosis, oncogenesis, innate immunity, and DNA repair [2, 3]. Intriguingly, some viruses have the ability to use TRIMs to improve several steps of the replication cycle and cause pathogenesis [4, 16••, 17••].
In this review, we discuss the novel mechanisms that are used by TRIM proteins in the context of their different functions related to host defense, which ultimately may affect virus pathogenesis. We focused our discussion on recent developments and on TRIMs that have been thoroughly studied. Some excellent recent reviews highlight the roles of other TRIMs [3,4,5, 13,14,15, 18,19,20,21,22,23,24,25,26,27,28,29].
Antiviral Activity of TRIM Proteins
There are host factors that have the capability of blocking virus replication at almost every step of viral life cycle. TRIMs can act as intrinsic restriction factors with the ability to interact directly with viral proteins, or they can act indirectly by inducing antiviral cytokines thereby regulating the activity of other antiviral effectors. TRIMs can use different mechanisms to inhibit viral entry, uncoating, replication, or viral release, ultimately resulting in reduction of viral pathogenesis (Fig. 1). TRIMs that act as restriction factors are usually expressed at sufficient levels to inhibit virus replication, although expression of many TRIMs can be further enhanced by diverse stimuli, including IFNs. A few recent reviews have highlighted the roles of TRIMs in restricting replication [3, 30, 31••]. TRIM5α is one of the best characterized TRIMs acting as a restriction factor against HIV-1 and other retroviruses [32]. Multiple mechanisms have been proposed for TRIM5α inhibition of retrovirus replication. TRIM5α interacts with the viral capsid through multivalent interactions and inactivates the virus by promoting premature uncoating [20, 31••, 33]. Potential mechanisms of viral inhibition that are still under investigation include degradation of the viral capsid by the proteasome, reduced reverse transcription due to premature uncoating, and potentially via induction of innate immunity [20, 31••, 34, 35]. Recent reviews explain the detailed molecular mechanisms of TRIM5α antiviral activity [20, 36]. In addition to HIV-1 restriction, recent reports indicate that TRIM5α also has antiviral activity against specific flaviviruses, including tick-borne encephalitis virus (TBEV), Kyasanur Forest disease virus (KFDV), and Langat virus (LGTV) but not West Nile virus (WNV), dengue virus (DENV), Zika virus (ZIKV), or yellow fever virus (YFV) [31••]. TRIM5α inhibits RNA replication by promoting proteasomal degradation of the flaviviral NS2B/3 protease and also contributes to the IFN-I mediated antiviral response [31••]. TRIM11 is another TRIM that has also been reported to restrict HIV-1 reverse transcription by interacting with the capsid protein and promoting premature uncoating [37]. TRIM33 inhibits HIV-1 infection by decreasing HIV-1 integrase function, thus, preventing viral cDNA integration into the host cell genome [38].

The TRIM effect: the forked road of host fitness or susceptibility. The TRIM family of proteins influence how hosts respond to foreign organisms leading to beneficial (host survival) or detrimental (host pathology) outcomes. This duality is linked to whether a particular TRIM retains desirable functions (antiviral or pro-viral participation) and if the negative consequences accompanying TRIM involvement outweigh their positive contributions (failure to tolerate subsequent inflammatory responses)
The targeting of important viral components for destruction by the host has been a defining feature of TRIM-mediated antiviral activity. Ubiquitination and subsequent proteasomal degradation affords the host a means for interrupting the virus life cycle by employing a cell’s own garbage disposal system against the invading pathogen. This is the case for hepatitis C virus (HCV), encephalomyocarditis virus (EMCV), hepatitis B virus (HBV), and influenza virus A (IAV). TRIM22 restricts HCV replication by interacting with the viral NS5A protein and targeting it to the proteasome [39, 40]. Other viral non-structural proteins like NS2A of JEV are critical for replication, making them desirable targets for TRIMs like TRIM52 which redirects JEV NS2A to the proteasome [41]. TRIM22 can also inhibit EMCV by targeting viral 3C protease for ubiquitination and degradation [42] and inhibits HBV by suppressing the core promoter responsible for viral pre-genomic RNA synthesis [43]. TRIM14 and TRIM22 have been reported to promote IAV nucleoprotein (NP) ubiquitination, and degradation in a proteasome-dependent manner [44, 45]. Viral NPs are attractive candidates as TRIM targets as TRIM41 also recognizes vesicular stomatitis virus (VSV) NP suggesting this system of TRIM redundancy or “cross-talk” may be applicable to other viruses [46]. The strategy of degrading viral RNA-binding proteins in order to reveal its genome for host detection is a common theme amongst several TRIMs. TRIM21 promotes the destruction of viral capsids via the proteasome and exposes the viral DNA or RNA to cytosolic nucleic acid sensors like cGAS and RIG-I [47]. In addition to NP, the polymerase complexes of viruses are subject to this means of disposal as TRIM32 targets the influenza polymerase subunit PB1 of several IAV strains to the proteasome via ubiquitination, while TRIM21 interacts with the hepatitis B virus (HBD) DNA polymerase to achieve the same effect for ubiquitination and proteasomal degradation [48, 49].
Aside from utilizing the proteasome, TRIMs have been found to obstruct viruses through various means. In addition to its aforementioned involvement with the proteasome, TRIM21 may act as an intracellular receptor through high-affinity binding with the Fc portion of immunoglobulin (Ig) molecules allowing for restriction of adenoviruses and rhinoviruses [50]. TRIM56 was previously shown to inhibit the replication of several viruses in the family Flaviviridae, including DENV serotype 2, YFV, bovine viral diarrhea virus, ZIKV, coronavirus OC43, IAV, and HIV-1 [51, 52, 53•, 54, 55]. In the case of IAV, TRIM56 blocks IAV replication possibly through interactions with viral RNA [55]. The proposed method of interaction between TRIM56 and IAV was observed during ZIKV infection where the TRIM56 C-terminal region and E3 ligase activity mediated an association with ZIKV RNA in infected cells [53•]. Parallels between this study and others can be seen as TRIM41 inhibition of HBV transcription activity also depended on its E3 ligase activity and C-terminal domain [56]. Furthermore, TRIM interference in vRNA events has proven to be a reliable antiviral method as TRIM22 inhibits HBV by suppressing the core promoter responsible for viral pre-genomic RNA synthesis [43]. Additional recent studies identifying inhibition of viral proteins by TRIMs included TRIM28 and TRIM59. TRIM28, a nuclear protein that is known to have transcriptional regulatory activity [57] and is a known repressor of endogenous retroviruses [58], has been recently reported to restrict viral integration of HIV-1 by binding and inhibiting the active, acetylated form of the viral integrase host [29, 59], while TRIM59 interacts with the porcine reproductive and respiratory syndrome virus (PRRSV) nsp11 to inhibit infection [60]. Finally, TRIM69 inhibits viral transcription and the formation of VSV replication compartments, reducing the synthesis of viral RNA and, therefore, the inhibition of viral replication [61]. Further, a recent report showed that TRIM69 interacts directly with DENV nonstructural protein 3 (NS3) and drives its polyubiquitination and degradation [62].
An interesting recent example of a novel TRIM antiviral function independent of its RING domain is TRIM2, which is highly expressed in the brain. TRIM2 binds neurofilament light chain (NEFL) subunit through its RBCC and FIL domain. Using Trim2−/− mice, it was recently shown that TRIM2 suppresses New World arenaviruses (NWA) such as the Junín virus (JUNV) and Tacaribe virus but not Old World arenaviruses such as Lassa or Lymphocytic choriomeningitis virus (LCMV) [63••]. Consistent with this, fibroblasts from a patient encoding a missense mutation on the CC region of TRIM2 are also more susceptible to this virus infection [63••]. TRIM2 limits NWA endocytosis into cells and operates at a post-receptor binding step in the viral life cycle. This antiviral activity is dependent on its FIL domain and not TRIM2 E3-Ub ligase activity. A regulatory protein α (SIRPA) was identified by interactome profiling as a TRIM2-interacting protein and also inhibited virus replication. SIRPA’s role in preventing phagocytosis is harnessed by TRIM2, resulting in the blockade of JUNV internalization [63••].
Indirect Antiviral Activity of TRIM Proteins
TRIM proteins can also have antiviral activity via indirect mechanisms including induction of antiviral cytokines or regulation of other antiviral effectors. An interesting recent example is TRIM43, which was recently shown to inhibit herpesviruses by promoting ubiquitination and proteasomal degradation of the centrosomal protein pericentrin, in turn resulting in nuclear lamina propria-dependent repression of active viral chromatin states [64•]. However, the majority of studies on TRIM antiviral functions continue to be focused on their potential roles as regulators of innate immune signaling and antiviral cytokine production.
TRIMs in Innate Immunity
The innate immune system is the first line of defense against pathogens as it detects virus invasion and subsequently limits virus replication. Innate immune cytokines released upon virus recognition are responsible for directing a proper adaptive immune response that is involved in elimination of pathogens in the later phase of infection and also shapes immunological memory [65].
The first step in innate immune activation occurs when pattern recognition receptors (PRRs), including endosomal Toll-like receptors (TLRs) and cytoplasmic RIG-I-like receptors (RLRs), recognize microbial components encoded in microorganism that are known as pathogen-associated molecular patterns (PAMPs) [66]. PRRs then stimulate a series of downstream signaling cascades that result in the activation and nuclear translocation of transcription factors, such as IRF3, IRF7, and NF-κB, which induce transcriptional upregulation of IFN-I and pro-inflammatory cytokines [66]. A large number of TRIMs have been found to play important regulatory roles at almost every step in PRR-activated signaling pathways [5, 13, 14]. In addition, a large number of TRIMs have been found to enhance cytokine signaling pathways [5, 13].
RIG-I has been thoroughly investigated for its role in virus RNA recognition and its essential role in the antiviral IFN-I response. The structure of RIG-I includes a central helicase domain and a C-terminal domain (CTD), required for RNA binding, and also contains two N-terminal caspase activation and recruitment domains (CARDs) that are essential for downstream signaling. Upon RNA binding, the CARDs undergo conformational changes allowing K63-linked ubiquitination by TRIM25 [67], which allows assembly of a signaling complex with mitochondrial antiviral signaling protein (MAVS) at the mitochondria [68, 69]. TRIM25 has also been reported to have RIG-I-independent antiviral activity to Sindbis virus via the zinc finger antiviral protein (ZAP) [70, 71]. The ubiquitin ligase activity of TRIM25 may be regulated by direct interactions with endogenous RNA [72•, 73••], which also promotes binding to ZAP [72•]. In addition to TRIM25, other TRIMs and additional E3-Ub ligases have also been reported to ubiquitinate and regulate functions of RIG-I-like receptors. TRIM4 mediates K63-linked polyubiquitination of RIG-I to positively regulate RIG-I-mediated IFN induction [74, 75]. TRIM8 has recently been identified as a modulator of innate signaling in plasmacytoid dendritic cells (pDCs), by protecting IRF7 from proteasomal degradation in an E3-Ub ligase-independent manner [76]. On the other hand, TRIM65 has a role in MDA5 K63-linked polyubiquitination by promoting MDA5 activation and oligomerization. Consequently, Trim65−/− mice are more susceptible to EMCV infection due to reduced IFN-I induction [77, 78].
Downstream of RIG-I and MDA5, other TRIMs have also been found to regulate this signaling pathway. TRIM31 catalyzes K63-linked polyubiquitination of K10, K311, and K461 on MAVS and promotes its aggregation in the mitochondria promoting downstream signaling [79]. MAVS signaling then leads to activation of different downstream signaling effectors, facilitating the induction of both NF-kB-mediated inflammatory cytokines and IFN-Is. On the NF-kB branch of the pathway, TRIM5α has been reported to activate TAK1 kinase via synthesis of unanchored K63-linked polyubiquitin chains, leading to NF-kB and AP-1 activation and inflammatory cytokine induction [80, 81]. TRIM23 also mediates activation of NF-kB during human cytomegalovirus (HCMV) infection [82] and can positively regulate NEMO activity, which is a crucial regulator of NF-κB activation, by mediating K27-linked polyubiquitination [83].
Another important PRR subject of intense recent studies is the cytoplasmic DNA sensor cyclic GMP-AMP synthase (cGAS), which can recognize a variety of replicating DNA viruses [84, 85], although studies have also shown that mitochondrial damage during RNA virus infection can result in cytoplasmic DNA leakage [86••]. The cGAS enzyme catalyzes a reaction to form the second messenger cyclic GMP-AMP (cGAMP), which then binds the adaptor protein STING on the endoplasmic reticulum (ER) and triggers downstream activation of TBK1-IRF3 and NF-kB for subsequent IFN-I and cytokine production [85]. TRIM proteins can also modulate cGAS-STING signaling. For example, TRIM14 functions as an adaptor to recruit the deubiquitinating enzyme USP14 and regulate cGAS, improving its stability and enhancing the antiviral response [87]. TRIM38 targets cGAS for sumoylation during the early phase of viral infection, preventing its K48-linked polyubiquitination and proteasomal degradation. TRIM38 also sumoylates STING during the early phase of viral infection, promoting both STING activation and protein stability which prevents STING degradation by the chaperone-mediated autophagy pathway [88]. TRIM41 has also been proposed to be involved in immune responses induced by DNA viruses and cytosolic DNA, via monoubiquitination of cGAS [89]. In addition, TRIM56 can also induce monoubiquitination of cGAS, thereby increasing its ability to interact and sense DNA [90••] and potentially providing a redundant mechanims of cGAS activation. Interestingly, TRIM56 can also ubiquitinate STING [91]. The fact that multiple TRIMs have been proposed to modulate both cGAS and STING functions raises the question whether a complex between cGAS-STING and multiple TRIMs (TRIM38, TRIM41, TRIM56) may provide a feedback activation loop that can sustain downtream signaling and may also be functonaly redundant. In addition, the Ub regulatory X domain protein UBXN3B regulates TRIM56-mediated K63-linked polyubiquitination of STING, which is necessary for STING oligomerization and activation of downstream TBK1-mediated antiviral signaling [92].
Another important immune regulator is TRIM28, which, consistent with its known function as a negative regulator of transcription, inhibits expression of pro-inflammatory cytokines and IFN-I; however, infections with highly pathogenic avian influenza viruses (HPAIV), such as H5N1 or H7N7, trigger a PKR-dependent signaling cascade that culminates with phosphorylation of TRIM28 and enhanced cytokine levels during infection [93]. Interestingly, this anti-inflammatory role of TRIM28 may be associated with its function as a repressor of endogenous retroviruses. For instance, another study identified a loss of sumoylated TRIM28 during IAV infection, which resulted in increased expression of endogenous retroviral elements that can be recognized as a source of “self” dsRNA by the RIG-I pathway [94•]. These studies highlight the complexity of the pathways that are regulated by TRIMs and the potential indirect effects that TRIMs can have on immune and non-immune pathways.
IFN-I produced during virus infection is then released to the extracellular space and triggers its own signaling cascade in an autocrine or paracrine manner via the IFN receptor (IFNAR). TRIMs have also been implicated in regulation of IFNAR signal transduction. The IKKε kinase has been shown to play a non-redundant role in optimal IFNAR signaling and ISG induction by phosphorylating STAT1 on S708 [95]. The JAK-STAT signaling pathway is important for defense against viral infection [96]. Receptor ligation activates the kinases JAK1 and TYK2 and induces phosphorylation of signal transducer and activator of transcription (STAT)1 and STAT2 which together with IRF9 form the interferon-stimulated gene factor 3 (ISGF3) complex, which translocates into the nucleus and induces transcription of an extensive set of antiviral ISGs. TRIM6 promotes the synthesis of unanchored K48-linked polyubiquitin chains that positively regulate IKKε activity resulting in enhanced IFN-I induction and signaling for optimal ISG induction [97].
TRIM Proteins in Pathogenesis
Although the vast majority of studies have associated TRIM activity with antiviral or innate immune inflammatory functions in response to viral infections, new evidence indicates that TRIMs can also be involved in directly promoting virus replication. This novel function could be a consequence of viruses taking advantage or hijacking TRIMs as a “side-effect” of TRIM-viral protein interaction during the antiviral process, or a direct utilization of the host ubiquitin machinery by the virus to enhance its replication. In either case, these effects exemplify complex mechanism of virus adaption to the host as well as constant interaction between TRIMs and viral proteins during evolution. Recent evidence indicates that some viruses have the ability to hijack TRIMs to improve several steps of the replication cycle, and increased replication would lead to increased pathogenesis. On other hand, increased virus pathogenesis could also be a result of virus antagonizing TRIM-mediated antiviral activity or indirectly affected when dysregulation of specific TRIMs involved in cytokines production results in uncontrolled inflammation and tissue damage (Fig. 1).
Increased Viral Pathogenesis via Antagonism of TRIM Antiviral Activity
TRIM21 Antagonism during JEV and SFTSV Infection
TRIM21 can be activated by diverse pathogens like viruses and intracellular bacteria [19, 98]. After virus detection, TRIM21 synthesizes K63-linked polyubiquitin chains and activates the innate immune pathways NF-κB, AP-1, IRF-3, IRF-7, and IRF-8 [19, 98, 99], leading to IFN-I production. JEV infection induces the expression of TRIM21 in human microglial cells, which results in attenuation of JEV-mediated effects in terms of activation of IRF-3 and production of IFNβ [99]. On the other hand, the non-structural (NSs) protein of severe fever with thrombocytopenia syndrome virus (SFTSV) interacts with TRIM21 and inhibits the activation of nuclear factor erythroid 2-related factor 2 (Nrf2) which is responsible for the expression of a series of antioxidant proteins and detoxifying enzymes [19, 100, 101]. Nrf2 is regulated by interactions with Kelch-like ECH-associated protein 1 (Keap1) and the proteasome system [102]. In normal conditions, Keap1 targets Nrf2 for degradation which suppresses intracellular antioxidant responses. In context of SFTSV infection, the viral proteins bind to the C-terminal SPRY subdomain of TRIM21, enhancing p62 stability and oligomerization. This allows p62-mediated Keap1 sequestration and activates the Nrf2-mediated antioxidant response, promoting viral replication and pathogenesis [103].
Antagonism of TRIM25-Mediated IFN Induction by DENV, MERS-CoV, EBV, and IAV TRIM25
A published study identified mutations on the DENV strain PR-2B that emerged during an epidemic in Puerto Rico in the 1990s. These mutations appear to increase production of sub-genomic flavivirus non-coding RNAs (sfRNAs). The PR-2B sfRNAs can bind to TRIM25 and prevent its deubiquitination, which is crucial for TRIM25-mediated activation of RIG-I. These findings suggested that adaptive mutations on DENV sfRNAs have the ability to differentially bind to host antiviral proteins to promote viral evasion of innate immunity and increase viral fitness [104]. In addition, Middle East respiratory syndrome coronavirus (MERS-CoV) infection suppresses RIG-I ubiquitination and downstream IFN-I and IFN-III induction, via interactions between MERS-CoV N protein and TRIM25 [105•].
Epstein-Barr virus (EBV) encodes a large tegument protein BPLF1, a viral deubiquitinase (DUB) that facilitates TRIM25’s interaction with the 14-3-3 scaffold to promote TRIM25 autoubiquitination and sequestration to inhibit IFN-I responses [106].
The IAV-NS1 protein also has the ability to antagonize the immune response by blocking TRIM25-mediated RIG-I ubiquitination [107]. IAV isolates from different species may have varying abilities to inhibit IFN-I induction partly due to differential NS1-TRIM25 and NS1-Riplet interactions that may contribute to differences in pathogenicity between avian, swine, and human IAV isolates [108].
Viruses Targeting TRIM23
An intriguing example of a TRIM being targeted specifically by a virus is TRIM23, which has been shown to ubiquitinate the NS5 protein of YFV [109]. This ubiquitination provides YFV-NS5 the ability to interact with STAT2, which ultimately results in inhibition of IFN-I signaling and increased virus replication. Interestingly, other studies have shown that TRIM23 has antiviral activity and this is dependent on NEMO leading to IRF3 and NF-kB activation and IFN-I induction [83]. Since YFV NS5 binds STAT2 only after IFN-I treatment [109], it suggests that TRIM23-mediated ubiquitination of YFV NS5 versus NEMO are regulated by different mechanisms. Reports indicate that YFV and DENV NS5 protein have 10 residues in their N-terminal regions, which are essential for the IFN antagonism function [109, 110]. TRIM23 has also been reported to have a function in autophagy-mediated antiviral defense mediated by TANK-binding kinase 1 (TBK1) [111]. Autophagy is an evolutionarily conserved process that restricts certain intracellular pathogens [112]. However, herpes virus simplex-1 (HSV-1) inhibits autophagy to enhance its replication in the mucosal epithelium and establish latency in neurons of the peripheral nervous system [113]. This inhibition is caused by the HSV-1 US11 protein which interacts with TRIM23 and blocks the formation of the functional TBK1. The TRIM23 complex is required for autophagy induction [114]. These data provide a new insight into viral escape from autophagy-mediated host restriction mechanisms.
TRIM6 Function in IFN-I Induction and Signaling during Nipah Virus and West Nile Virus Infections
TRIM6 has been shown to play multiple roles during infections with different viruses, leading to protection or pathogenicity depending on the conditions. TRIM6 can play a role in optimal IFN-I-mediated antiviral function against different RNA viruses, including IAV, EMCV and Sendai virus (SeV) [97]. In addition, the Nipah virus (NiV) matrix (M) protein has been described to interact with and degrade TRIM6 [115]. In NiV-infected cells, the endogenous level of TRIM6 is decreased significantly compared with mock cells or cells infected with a recombinant NiV lacking M [115]. IFN promoter luciferase reporter assays demonstrated that NiV-M can inhibit TRIM6’s function in IFN-I induction and signaling, but the functional importance of this antagonism was not tested in the context of NiV infection [115] (Fig. 2b). Several other NiV proteins act as potent antagonists of the IFN-I induction and signaling pathways [116,117,118], including NiV accessory protein V which interacts with human TRIM25 to prevent activation of RIG-I and downstream IFN-I induction [119]. NiV-M’s interaction with and degradation of TRIM6 may play a redundant role in IFN-I antagonism, but it cannot be excluded that the NiV-M-TRIM6 interaction plays an uncharacterized pro-viral function or antagonizes a distinct TRIM6-regulated pathway. In the context of West Nile virus (WNV), TRIM6 is required for efficient IFN-I induction and signaling to dampen replication (Fig. 2b) [97, 120]. TRIM6 was found to be required for the phosphorylation of STAT1 at S708 and the induction of several ISGs known to be involved in WNV antagonism [120]. Additionally, TRIM6 regulates the expression of VAMP8, a vesicle-associated membrane protein we found to be required for optimal JAK1 phosphorylation downstream of IFN-I stimulation [120]. Therefore, TRIM6 is emerging as an important antiviral factor via the IFN-I system.

Pro-viral and antiviral roles of TRIM6. a TRIM6 facilities the ubiquitination (white circles with Ub) of Ebola virus (EBOV) VP35 at lysine residue 309 (K309). This ubiquitination at K309 augments the EBOV VP35’s polymerase co-factor activity in the presence of TRIM6. b Upon virus infection, viral RNA in the cytoplasm triggers the activation of RIG-I-like receptors, including RIG-I and MDA-5, to trigger downstream type-I interferon (IFN-I) induction. TRIMs 6 and 25 both participate in the IFN-I pathway. TRIM6 promotes the synthesis of unanchored K48-linked polyubiquitin chains which act as a scaffold for IKKɛ oligomerization promoting its kinase activity and downstream functions in IFN-I induction and signaling, and TRIM6 regulates that expression of a vesicle-associated protein, VAMP8, which has been shown to promote JAK1 phosphorylation downstream of IFN-I signaling. TRIM25 facilities the covalent conjugation of K63-linked polyubiquitin to RIG-I to promote RIG-I’s activity. The viral antagonism (red arrows) of these TRIMs’ function has been described for EBOV VP35 and Nipah virus (NiV) accessory protein V (V) and matrix protein (M). In addition to the antagonism of TRIMs, these viral proteins also target additional steps of the IFN-I pathways, including NiV-V antagonism of MDA5 and STAT1 and EBOV antagonism of IKKɛ- and TBK1-mediated phosphorylation of IRF3
Increased Viral Pathogenesis by TRIMs Direct Pro-viral Activity
TRIM6 Pro-viral Function
TRIM6 has been identified as an important host factor targeted to enhance Ebola virus (EBOV) replication. EBOV is a highly pathogenic virus that causes severe hemorrhagic fever in humans [121]. VP35 is the viral polymerase co-factor [122, 123] and an IFN-I inhibitory protein [122, 124,125,126] critical for EBOV replication and pathogenesis. We found that TRIM6 ubiquitinates EBOV VP35 to promote optimal viral replication [16••] (Fig. 2a). Using co-immunoprecipitation assays and IFN-I reporter assays, we found that VP35 antagonizes TRIM6-mediated enhancement of IFN-I induction and TRIM6 facilitates ubiquitination of VP35 at K309. Using an EBOV minigenome system [122], it was shown that TRIM6 enhances the minigenome activity when expressed with wild-type (WT) VP35 but not a K309A mutant [16••]. Further, a TRIM6 ubiquitin ligase mutant (C15A) is unable to enhance VP35 polymerase co-factor activity [16••]. Despite EBOV VP35’s capability to antagonize TRIM6-mediated IFN-I activation, EBOV replication was attenuated significantly in TRIM6-KO cells compared with WT cells [16••]. Although the VP35 may interact with TRIM6 to antagonize IFN-I induction, VP35 exploits TRIM6 as pro-viral factor to enhance viral replication. However, TRIM6 knockout cells infected with EBOV express higher levels of the pro-inflammatory cytokine IL-6 as compared with WT cells [16••], indicating that TRIM6 may also play a role in regulating inflammation and could lead to the immune dysregulation observed during EBOV infection. The mechanism underlying TRIM6’s pro-viral activity versus its immune regulatory role is under on-going investigation. In addition to VP35 residues, other VP35 lysine residues are also ubiquitinated [16••], but their identity, function, and dependence on TRIM6 are yet to be determined.
TRIM7 in Pathogenesis
TRIM7 is another E3-Ub ligase that can promote virus pathogenesis or protect against infection depending on the context of virus infection. We recently reported that the envelope (E) protein of ZIKV is K63-linked polyubiquitinated by TRIM7, promoting enhanced replication in brain and reproductive tissues and leading to enhanced pathogenesis in vivo [17••]. Since a proportion of ubiquitinated E is present in infectious viruses and ubiquitinated E-containing viruses infect cells more efficiently, this appears to create a more permissive environment for its replication within specific target tissues (Fig. 3a–b). Indeed, a recombinant infectious ZIKV mutant that lacks ubiquitination on the K38 residue (ZIKV-E K38R) has reduced ability to attach to host cells (Fig. 3b and c#1), leading to reduced virus-endosome membrane fusion (Fig. 3c#2), and lower replication as compared with WT ZIKV, causing less pathology. TRIM7 may play a role in determining ZIKV tissue tropism in vivo, because ZIKV replicated to lower titers in brain and reproductive tissues (uterus and testis) as compared with other tissues of Trim7−/− mice and as compared with WT littermate controls [17••]. Cell fractionation experiments suggested that TRIM7 and its E2-Ub conjugase UbcH5a [127, 128] co-localize in the endoplasmic reticulum (ER) compartment in ZIKV infected cells, although TRIM7 can also re-localize to the Golgi [17••, 129], suggesting that TRIM7 may be hijacked by ZIKV-E during maturation in the Golgi or during replication in the ER (Fig. 3c#3).

a A portion of released Zika virions possess ubiquitinated envelope proteins. Zika virus grown in both human and mosquito cells are ubiquitinated to varying degrees with human grown viruses having longer poly-Ub chains while mosquito-grown viruses retain shorter poly-Ub chains. b Envelope ubiquitination by TRIM7 enhances Zika virus entry in mammalian, but not mosquito, hosts. Ubiquitination of the Zika envelope protein at site K38 is made possible by the E3 ubiquitin ligase TRIM7 allowing for enhanced Zika tissue tropism where levels of TRIM7 are high (brain, uterus, and testis). c Ubiquitination of Zika envelope promotes binding to host receptors and enhances viral entry. The K63-poly-Ub chains of Zika envelope afford for stronger interactions with host receptors (#1), virus-endosome membrane fusion (#2), and higher replication titers (#3). Upon Zika infection, TRIM7 re-localizes to the Golgi and co-localizes with Zika envelope in distinct puncta where ubiquitination presumably occurs. Infectious Zika virions with ubiquitinated envelope can be neutralized with K63-regulate innate immune responses in both a positive and negative manner. TRIM7 promotes herpes virus infection by targeting STING for K48-poly-Ub and proteasome-mediated degradation (#5) while hindering norovirus replication (#6). Additionally, TRIM7 can also enhance TLR4 signaling in macrophages during LPS challenge (#7)
Once ZIKV virions are ubiquitinated by TRIM7, they are released from infected cells (Fig. 3c#4) and such ubiquitination provides an advantage for viral replication by promoting more efficient attachment to cellular receptors (Fig. 3c#1). Additional evidence that ZIKV infectious particles contain ubiquitinated E comes from experiments showing that an anti-K63-Ub antibody can neutralize ZIKV replication in cells and in vivo [17••] (Fig. 3c#4). Although multiple receptors are proposed to be involved in ZIKV attachment and cell entry, including DC-SIGN, AXL, Tyro3, and TIM-1 [130], we demonstrated that at least in the case of TIM-1, efficient ZIKV attachment depends on the presence of K63-linked polyubiquitinated E in the infectious ZIKV particles. This is also supported by data showing that infection of Havcr-1−/− mice (Havcr-1 is the gene that encodes TIM-1 protein), with ZIKV-E WT, exhibited a reduction in viral titer in the brain as compared with WT littermate controls, whereas no difference was observed with the ZIKV-E K38R mutant virus. This suggests that although Tim-1 is not the only receptor that mediates ZIKV entry, it may play a role in specific cell types/tissue when ZIKV contains ubiquitinated E (Fig. 3b and c#1) [17••].
Interestingly, although mosquitoes also express components of the ubiquitin system, including a small number of TRIM orthologues, ZIKV grown in mosquito cells appears to contain reduced and shorter forms of polyubiquitinated E and this does not affect virus replication in live mosquitoes [17••] (Fig. 3a–b). However, previous reports indicate that the mosquito ubiquitin Ub3881 protein may be involved in DENV E protein degradation [131] although other studies have also proposed that virus replication in mosquitoes may be dependent on a functioning ubiquitin proteasome system [132]. Nonetheless, the role of the ubiquitin system and the function of individual E3-ubiquitin ligases during infection and in mosquito transmission are still unclear.
TRIM7 in known to be involved in some important biological processes including tumor cell proliferation and glycogen metabolism [129, 133]. TRIM7 has also been shown to act as an E3 ligase mediating K63-linked polyubiquitination of the AP-1 coactivator RACO-1, leading to RACO-1 protein stabilization [127]. Other studies have also proposed that TRIM7 may play antiviral roles against norovirus [134•] (Fig. 3c#6) and in IFN induction [135••], which is also in line with our own findings that TRIM7 KO cells have reduced IFNβ induction upon ZIKV infection or PRR stimulation, the data from in vivo infections in Trim7−/− mice suggest that the pro-viral roles of TRIM7 are dominant over its potential IFN-mediated antiviral roles in specific cell types and in vivo [17••]. In addition, TRIM7 was also recently described to negatively regulate responses to DNA viruses by targeting STING for degradation [136•] (Fig. 3c#5), so TRIM7 could also play a role in alternative innate immune signaling pathways during ZIKV infection, especially if DNA damage occurs during virus infection that could lead to activation of the cGAS-STING pathway.
Since ubiquitination was observed on residue K38, which is conserved in members of the Flaviviridae family, and we also found that DENV particles also contained ubiquitinated E [17••], it will be interesting to see if other enveloped viruses may use similar mechanisms of virus entry via interactions between potentially ubiquitinated envelope resident protein and host receptors.
Conclusions
This review describes the roles of TRIMs in virus-host interactions and TRIM involvement in immune signaling and direct virus restriction. In addition, viral antagonism of TRIMs exemplifies the importance of this protein family in antiviral responses. Despite these advancements, many TRIMs have yet to be characterized. Additionally, viruses hijack similar innate immune host factors to enhance their replication. More needs to be learned regarding the role of TRIMs in adaptive immunity. A new area of investigation is whether additional antiviral TRIMs may be redirected by others pathogens to improve replication through protein ubiquitination. This information could help design novel broad-spectrum antiviral strategies, including targeting TRIM function that may cause hyper-inflammation. To this end, more studies using in vivo animal models will be required to differentiate between antiviral and pro-viral roles of TRIMs and ultimately elucidate whether virus-induced pathology could be treated using pharmacological approaches to inhibit specific TRIM activity.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sardiello M, Cairo S, Fontanella B, Ballabio A, Meroni G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol Biol. 2008;8:225. https://doi.org/10.1186/1471-2148-8-225.
Vunjak M, Versteeg GA. TRIM proteins. Curr Biol. 2019;29(2):R42–R4. https://doi.org/10.1016/j.cub.2018.11.026.
van Gent M, Sparrer KMJ, Gack MU. TRIM proteins and their roles in antiviral host defenses. Annu Rev Virol. 2018;5(1):385–405. https://doi.org/10.1146/annurev-virology-092917-043323.
Hage A, Rajsbaum R. To TRIM or not to TRIM: the balance of host-virus interactions mediated by the ubiquitin system. J Gen Virol. 2019;100(12):1641–62. https://doi.org/10.1099/jgv.0.001341.
van Tol S, Hage A, Giraldo MI, Bharaj P, Rajsbaum R. The TRIMendous role of TRIMs in virus-host interactions. Vaccines (Basel). 2017;5(3). https://doi.org/10.3390/vaccines5030023.
Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol. 2017;52(4):425–60. https://doi.org/10.1080/10409238.2017.1325829.
Munir M. TRIM proteins: another class of viral victims. Sci Signal. 2010;3(118):jc2. https://doi.org/10.1126/scisignal.3118jc2.
Rajsbaum R, Stoye JP, O'Garra A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol. 2008;38(3):619–30. https://doi.org/10.1002/eji.200737916.
Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, et al. Human TRIM gene expression in response to interferons. PLoS One. 2009;4(3):e4894. https://doi.org/10.1371/journal.pone.0004894.
Versteeg GA, Rajsbaum R, Sanchez-Aparicio MT, Maestre AM, Valdiviezo J, Shi M, et al. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity. 2013;38(2):384–98. https://doi.org/10.1016/j.immuni.2012.11.013.
Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, et al. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol. 2013;87(1):257–72. https://doi.org/10.1128/JVI.01804-12.
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472(7344):481–5. https://doi.org/10.1038/nature09907.
Rajsbaum R, Garcia-Sastre A, Versteeg GA. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol. 2014;426(6):1265–84. https://doi.org/10.1016/j.jmb.2013.12.005.
McNab FW, Rajsbaum R, Stoye JP, O'Garra A. Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol. 2011;23(1):46–56. https://doi.org/10.1016/j.coi.2010.10.021.
Ozato K, Shin DM, Chang TH, Morse HC 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8(11):849–60. https://doi.org/10.1038/nri2413.
•• Bharaj P, Atkins C, Luthra P, Giraldo MI, Dawes BE, Miorin L, et al. The host E3-ubiquitin ligase TRIM6 ubiquitinates the Ebola virus VP35 protein and promotes virus replication. J Virol. 2017;91(18). https://doi.org/10.1128/JVI.00833-17. This study reports a new pro-viral role for the antiviral TRIM6, by promoting efficient EBOV replication.
•• Giraldo MI, Xia H, Aguilera-Aguirre L, Hage A, van Tol S, Shan C, et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature. 2020. https://doi.org/10.1038/s41586-020-2457-8. TRIM7 was identified as the E3-Ub ligase that ubiquitinates envelope and is a determinant of tissue tropism.
Choudhury NR, Heikel G, Michlewski G. TRIM25 and its emerging RNA-binding roles in antiviral defense. Wiley Interdiscip Rev RNA. 2020:e1588. https://doi.org/10.1002/wrna.1588.
Foss S, Bottermann M, Jonsson A, Sandlie I, James LC, Andersen JT. TRIM21-from intracellular immunity to therapy. Front Immunol. 2019;10:2049. https://doi.org/10.3389/fimmu.2019.02049.
Ganser-Pornillos BK, Pornillos O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat Rev Microbiol. 2019;17(9):546–56. https://doi.org/10.1038/s41579-019-0225-2.
Khan R, Khan A, Ali A, Idrees M. The interplay between viruses and TRIM family proteins. Rev Med Virol. 2019;29(2):e2028. https://doi.org/10.1002/rmv.2028.
Wei Y, Chen S, Wang M, Cheng A. Tripartite motif-containing proteins precisely and positively affect host antiviral immune response. Scand J Immunol. 2018;87(6):e12669. https://doi.org/10.1111/sji.12669.
Vicenzi E, Poli G. The interferon-stimulated gene TRIM22: a double-edged sword in HIV-1 infection. Cytokine Growth Factor Rev. 2018;40:40–7. https://doi.org/10.1016/j.cytogfr.2018.02.001.
Esposito D, Koliopoulos MG, Rittinger K. Structural determinants of TRIM protein function. Biochem Soc Trans. 2017;45(1):183–91. https://doi.org/10.1042/BST20160325.
Hu MM, Shu HB. Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cell Mol Immunol. 2017;14(4):331–8. https://doi.org/10.1038/cmi.2016.66.
Kimura T, Jain A, Choi SW, Mandell MA, Johansen T, Deretic V. TRIM-directed selective autophagy regulates immune activation. Autophagy. 2017;13(5):989–90. https://doi.org/10.1080/15548627.2016.1154254.
Rajsbaum R, Garcia-Sastre A. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol. 2013;21(8):421–9. https://doi.org/10.1016/j.tim.2013.06.006.
Versteeg GA, Benke S, Garcia-Sastre A, Rajsbaum R. InTRIMsic immunity: positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 2014;25(5):563–76. https://doi.org/10.1016/j.cytogfr.2014.08.001.
Patil G, Li S. Tripartite motif proteins: an emerging antiviral protein family. Futur Virol. 2019;14(2):107–22. https://doi.org/10.2217/fvl-2018-0161.
Berthoux L. The Restrictome of Flaviviruses. Virol Sin. 2020. https://doi.org/10.1007/s12250-020-00208-3.
•• Chiramel AI, Meyerson NR, McNally KL, Broeckel RM, Montoya VR, Mendez-Solis O, et al. TRIM5alpha restricts flavivirus replication by targeting the viral protease for proteasomal degradation. Cell Rep. 2019;27(11):3269–83 e6. https://doi.org/10.1016/j.celrep.2019.05.040. This study identified a novel role of TRIM5α as an anti-flavivirus restriction factor that differs from its well-know function as restriction factor of retroviruses.
Skorupka KA, Roganowicz MD, Christensen DE, Wan Y, Pornillos O, Ganser-Pornillos BK. Hierarchical assembly governs TRIM5alpha recognition of HIV-1 and retroviral capsids. Sci Adv. 2019;5(11):eaaw3631. https://doi.org/10.1126/sciadv.aaw3631.
Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, et al. Rhesus TRIM5alpha disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 2011;7(3):e1002009. https://doi.org/10.1371/journal.ppat.1002009.
Mandell MA, Jain A, Arko-Mensah J, Chauhan S, Kimura T, Dinkins C, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30(4):394–409. https://doi.org/10.1016/j.devcel.2014.06.013.
Campbell EM, Weingart J, Sette P, Opp S, Sastri J, O'Connor SK, et al. TRIM5alpha-mediated ubiquitin chain conjugation is required for inhibition of HIV-1 reverse transcription and capsid destabilization. J Virol. 2016;90(4):1849–57. https://doi.org/10.1128/JVI.01948-15.
Nakayama EE, Shioda T. Impact of TRIM5alpha in vivo. AIDS. 2015;29(14):1733–43. https://doi.org/10.1097/QAD.0000000000000812.
Yuan T, Yao W, Tokunaga K, Yang R, Sun B. An HIV-1 capsid binding protein TRIM11 accelerates viral uncoating. Retrovirology. 2016;13(1):72. https://doi.org/10.1186/s12977-016-0306-5.
Ali H, Mano M, Braga L, Naseem A, Marini B, Vu DM, et al. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat Commun. 2019;10(1):926. https://doi.org/10.1038/s41467-019-08810-0.
Yang C, Zhao X, Sun D, Yang L, Chong C, Pan Y, et al. Interferon alpha (IFNalpha)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5A. Cell Mol Immunol. 2016;13(1):94–102. https://doi.org/10.1038/cmi.2014.131.
Wang S, Chen Y, Li C, Wu Y, Guo L, Peng C, et al. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Sci Rep. 2016;6:32336. https://doi.org/10.1038/srep32336.
Fan W, Wu M, Qian S, Zhou Y, Chen H, Li X, et al. TRIM52 inhibits Japanese encephalitis virus replication by degrading the viral NS2A. Sci Rep. 2016;6:33698. https://doi.org/10.1038/srep33698.
Mancia G, Omboni S, Parati G. Assessment of antihypertensive treatment by ambulatory blood pressure. J Hypertens Suppl. 1997;15(2):S43–50. https://doi.org/10.1097/00004872-199715022-00002.
Gao B, Duan Z, Xu W, Xiong S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology. 2009;50(2):424–33. https://doi.org/10.1002/hep.23011.
Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, et al. TRIM22 inhibits influenza a virus infection by targeting the viral nucleoprotein for degradation. J Virol. 2013;87(8):4523–33. https://doi.org/10.1128/JVI.02548-12.
Wu X, Wang J, Wang S, Wu F, Chen Z, Li C, et al. Inhibition of influenza a virus replication by TRIM14 via its multifaceted protein-protein interaction with NP. Front Microbiol. 2019;10:344. https://doi.org/10.3389/fmicb.2019.00344.
Patil G, Xu L, Wu Y, Song K, Hao W, Hua F et al. TRIM41-mediated ubiquitination of nucleoprotein limits vesicular stomatitis virus infection. Viruses. 2020;12(2). https://doi.org/10.3390/v12020131.
Watkinson RE, McEwan WA, Tam JC, Vaysburd M, James LC. TRIM21 promotes cGAS and RIG-I sensing of viral genomes during infection by antibody-opsonized virus. PLoS Pathog. 2015;11(10):e1005253. https://doi.org/10.1371/journal.ppat.1005253.
Mu T, Zhao X, Zhu Y, Fan H, Tang H. The E3 Ubiquitin ligase TRIM21 promotes HBV DNA polymerase degradation. Viruses. 2020;12(3). https://doi.org/10.3390/v12030346.
Fu B, Wang L, Ding H, Schwamborn JC, Li S, Dorf ME. TRIM32 senses and restricts influenza a virus by ubiquitination of PB1 polymerase. PLoS Pathog. 2015;11(6):e1004960. https://doi.org/10.1371/journal.ppat.1004960.
Vaysburd M, Watkinson RE, Cooper H, Reed M, O'Connell K, Smith J, et al. Intracellular antibody receptor TRIM21 prevents fatal viral infection. Proc Natl Acad Sci U S A. 2013;110(30):12397–401. https://doi.org/10.1073/pnas.1301918110.
Liu B, Li NL, Wang J, Shi PY, Wang T, Miller MA, et al. Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J Virol. 2014;88(23):13821–35. https://doi.org/10.1128/JVI.02505-14.
Wang J, Liu B, Wang N, Lee YM, Liu C, Li K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J Virol. 2011;85(8):3733–45. https://doi.org/10.1128/JVI.02546-10.
• Yang D, Li NL, Wei D, Liu B, Guo F, Elbahesh H et al. The E3 ligase TRIM56 is a host restriction factor of Zika virus and depends on its RNA-binding activity but not miRNA regulation, for antiviral function. PLoS Negl Trop Dis 2019;13(6):e0007537. doi:https://doi.org/10.1371/journal.pntd.0007537. This study identified for the first time TRIM56 as an intrinsic restriction host factor against ZIKV.
Kane M, Zang TM, Rihn SJ, Zhang F, Kueck T, Alim M, et al. Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe. 2016;20(3):392–405. https://doi.org/10.1016/j.chom.2016.08.005.
Liu B, Li NL, Shen Y, Bao X, Fabrizio T, Elbahesh H, et al. The C-terminal tail of TRIM56 dictates antiviral restriction of influenza a and B viruses by impeding viral RNA synthesis. J Virol. 2016;90(9):4369–82. https://doi.org/10.1128/JVI.03172-15.
Zhang S, Guo JT, Wu JZ, Yang G. Identification and characterization of multiple TRIM proteins that inhibit hepatitis B virus transcription. PLoS One. 2013;8(8):e70001. https://doi.org/10.1371/journal.pone.0070001.
Bunch H, Calderwood SK. TRIM28 as a novel transcriptional elongation factor. BMC Mol Biol. 2015;16:14. https://doi.org/10.1186/s12867-015-0040-x.
Groh S, Schotta G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol Life Sci. 2017;74(11):2055–65. https://doi.org/10.1007/s00018-017-2454-8.
Allouch A, Di Primio C, Alpi E, Lusic M, Arosio D, Giacca M, et al. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe. 2011;9(6):484–95. https://doi.org/10.1016/j.chom.2011.05.004.
Jing H, Ke W, Tao R, Li Y, Zhao Y, Cao S, et al. TRIM59 inhibits porcine reproductive and respiratory syndrome virus (PRRSV)-2 replication in vitro. Res Vet Sci. 2019;127:105–12. https://doi.org/10.1016/j.rvsc.2019.10.004.
Kueck T, Bloyet LM, Cassella E, Zang T, Schmidt F, Brusic V et al. Vesicular stomatitis virus transcription is inhibited by TRIM69 in the interferon-induced antiviral state. J Virol. 2019;93(24). https://doi.org/10.1128/JVI.01372-19.
Wang K, Zou C, Wang X, Huang C, Feng T, Pan W, et al. Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLoS Pathog. 2018;14(8):e1007287. https://doi.org/10.1371/journal.ppat.1007287.
•• Sarute N, Ibrahim N, Medegan Fagla B, Lavanya M, Cuevas C, Stavrou S, et al. TRIM2, a novel member of the antiviral family, limits New World arenavirus entry. PLoS Biol. 2019;17(2):e3000137. https://doi.org/10.1371/journal.pbio.3000137. This study reported for the first time that TRIM2 has antiviral function against new word arenaviruses in vivo and in human cells encoding a TRIM2 mutation.
• Full F, van Gent M, Sparrer KMJ, Chiang C, Zurenski MA, Scherer M, et al. Centrosomal protein TRIM43 restricts herpesvirus infection by regulating nuclear lamina integrity. Nat Microbiol. 2019;4(1):164–76. https://doi.org/10.1038/s41564-018-0285-5. This study shows a novel indirect antiviral mechanism of TRIM43 against herpesvirus.
Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy, Asthma Clin Immunol. 2018;14(Suppl 2):49. https://doi.org/10.1186/s13223-018-0278-1.
Hoffmann J, Akira S. Innate immunity. Curr Opin Immunol. 2013;25(1):1–3. https://doi.org/10.1016/j.coi.2013.01.008.
Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446(7138):916–20. https://doi.org/10.1038/nature05732.
Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–61. https://doi.org/10.1016/j.cell.2011.06.041.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20. https://doi.org/10.1016/j.cell.2010.01.022.
Li MM, Lau Z, Cheung P, Aguilar EG, Schneider WM, Bozzacco L, et al. TRIM25 enhances the antiviral action of zinc-finger antiviral protein (ZAP). PLoS Pathog. 2017;13(1):e1006145. https://doi.org/10.1371/journal.ppat.1006145.
Zheng X, Wang X, Tu F, Wang Q, Fan Z, Gao G. TRIM25 is required for the antiviral activity of zinc finger antiviral protein. J Virol 2017;91(9). https://doi.org/10.1128/JVI.00088-17.
• Choudhury NR, Heikel G, Trubitsyna M, Kubik P, Nowak JS, Webb S, et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017;15(1):105. https://doi.org/10.1186/s12915-017-0444-9. This study identified RNA interactions with TRIM25 as a mechanism to regulate its ubiquitin ligase activity.
•• Sanchez JG, Sparrer KMJ, Chiang C, Reis RA, Chiang JJ, Zurenski MA, et al. TRIM25 Binds RNA to Modulate Cellular Anti-viral Defense. J Mol Biol. 2018;430(24):5280–93. https://doi.org/10.1016/j.jmb.2018.10.003. This study shows a novel mechanim of regulation of TRIM25 activity via intrecations with RNA.
Yan J, Li Q, Mao AP, Hu MM, Shu HB. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J Mol Cell Biol. 2014;6(2):154–63. https://doi.org/10.1093/jmcb/mju005.
Okamoto M, Kouwaki T, Fukushima Y, Oshiumi H. Regulation of RIG-I activation by K63-linked polyubiquitination. Front Immunol. 2017;8:1942. https://doi.org/10.3389/fimmu.2017.01942.
Maarifi G, Smith N, Maillet S, Moncorge O, Chamontin C, Edouard J, et al. TRIM8 is required for virus-induced IFN response in human plasmacytoid dendritic cells. Sci Adv. 2019;5(11):eaax3511. https://doi.org/10.1126/sciadv.aax3511.
Lang X, Tang T, Jin T, Ding C, Zhou R, Jiang W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J Exp Med. 2017;214(2):459–73. https://doi.org/10.1084/jem.20160592.
Kamanova J, Sun H, Lara-Tejero M, Galan JE. The Salmonella effector protein SopA modulates innate immune responses by targeting TRIM E3 ligase family members. PLoS Pathog. 2016;12(4):e1005552. https://doi.org/10.1371/journal.ppat.1005552.
Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2017;18(2):214–24. https://doi.org/10.1038/ni.3641.
Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461(7260):114–9. https://doi.org/10.1038/nature08247.
Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472(7343):361–5. https://doi.org/10.1038/nature09976.
Poole E, Groves I, MacDonald A, Pang Y, Alcami A, Sinclair J. Identification of TRIM23 as a cofactor involved in the regulation of NF-kappaB by human cytomegalovirus. J Virol. 2009;83(8):3581–90. https://doi.org/10.1128/JVI.02072-08.
Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, et al. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc Natl Acad Sci U S A. 2010;107(36):15856–61. https://doi.org/10.1073/pnas.1004621107.
Chanut R, Petrilli V. [Cytosolic DNA sensing by the cGAS-STING pathway in cancer]. Med Sci (Paris). 2019;35(6–7):527–34. https://doi.org/10.1051/medsci/2019095.
Ma Z, Ni G, Damania B. Innate sensing of DNA virus genomes. Annu Rev Virol. 2018;5(1):341–62. https://doi.org/10.1146/annurev-virology-092917-043244.
•• Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. 2017;2:17037. https://doi.org/10.1038/nmicrobiol.2017.37. This study identified a novel mechansm of DNA-mediated cGAS activation upon infection with an RNA virus.
Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell. 2016;64(1):105–19. https://doi.org/10.1016/j.molcel.2016.08.025.
Hu MM, Yang Q, Xie XQ, Liao CY, Lin H, Liu TT, et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity. 2016;45(3):555–69. https://doi.org/10.1016/j.immuni.2016.08.014.
Liu ZS, Zhang ZY, Cai H, Zhao M, Mao J, Dai J, et al. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018;8:35. https://doi.org/10.1186/s13578-018-0233-3.
•• Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, Jung JU. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat Commun. 2018;9(1):613. https://doi.org/10.1038/s41467-018-02936-3. This study used in vitro and in vivo approaches that indicate that the TRIM56-induced monoubiquitination of cGAS is need for efficient cytosolic DNA detection in response to DNA virus infection.
Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33(5):765–76. https://doi.org/10.1016/j.immuni.2010.10.013.
Yang L, Wang L, Ketkar H, Ma J, Yang G, Cui S, et al. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat Commun. 2018;9(1):2329. https://doi.org/10.1038/s41467-018-04759-8.
Krischuns T, Gunl F, Henschel L, Binder M, Willemsen J, Schloer S, et al. Phosphorylation of TRIM28 enhances the expression of IFN-beta and proinflammatory cytokines during HPAIV infection of human lung epithelial cells. Front Immunol. 2018;9:2229. https://doi.org/10.3389/fimmu.2018.02229.
• Schmidt N, Domingues P, Golebiowski F, Patzina C, Tatham MH, Hay RT, et al. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc Natl Acad Sci U S A. 2019;116(35):17399–408. https://doi.org/10.1073/pnas.1907031116. This study shows a novel role of TRIM28 in inhibiting endogenous retrovirus RNA sequences that can trigger innate immune response to “self’ RNA during IAV infection.
Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315(5816):1274–8. https://doi.org/10.1126/science.1136567.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. https://doi.org/10.1038/nri3581.
Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha-Villanueva A, Martinez-Romero C, et al. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKepsilon kinase-mediated antiviral response. Immunity. 2014;40(6):880–95. https://doi.org/10.1016/j.immuni.2014.04.018.
McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL, James LC. Intracellular antibody-bound pathogens stimulate immune signaling via the fc receptor TRIM21. Nat Immunol. 2013;14(4):327–36. https://doi.org/10.1038/ni.2548.
Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK. Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J Neuroinflammation. 2014;11:24. https://doi.org/10.1186/1742-2094-11-24.
Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91(21):9926–30. https://doi.org/10.1073/pnas.91.21.9926.
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–22. https://doi.org/10.1006/bbrc.1997.6943.
Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9. https://doi.org/10.1128/MCB.24.16.7130-7139.2004.
Choi Y, Jiang Z, Shin WJ, Jung JU. Severe fever with thrombocytopenia syndrome virus NSs interacts with TRIM21 to activate the p62-Keap1-Nrf2 Pathway. J Virol. 2020;94(6). https://doi.org/10.1128/JVI.01684-19.
Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science. 2015;350(6257):217–21. https://doi.org/10.1126/science.aab3369.
• Chang CY, Liu HM, Chang MF, Chang SC. Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses type I and type III interferon induction by targeting RIG-I signaling. J Virol. 2020. https://doi.org/10.1128/JVI.00099-20. TRIM25 was identified an important factor in MERS-CoV infection, its sequestration suppress production type I and type III IFNs. This study reveal how MERS-CoV evades innate immunity.
Gupta S, Yla-Anttila P, Sandalova T, Sun R, Achour A, Masucci MG. 14-3-3 scaffold proteins mediate the inactivation of trim25 and inhibition of the type I interferon response by herpesvirus deconjugases. PLoS Pathog. 2019;15(11):e1008146. https://doi.org/10.1371/journal.ppat.1008146.
Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, et al. Influenza a virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5(5):439–49. https://doi.org/10.1016/j.chom.2009.04.006.
Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, Nistal-Villan E, et al. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza a virus NS1 protein. PLoS Pathog. 2012;8(11):e1003059. https://doi.org/10.1371/journal.ppat.1003059.
Laurent-Rolle M, Morrison J, Rajsbaum R, Macleod JML, Pisanelli G, Pham A, et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe. 2014;16(3):314–27. https://doi.org/10.1016/j.chom.2014.07.015.
Ashour J, Laurent-Rolle M, Shi PY, Garcia-Sastre A. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol. 2009;83(11):5408–18. https://doi.org/10.1128/JVI.02188-08.
Sparrer KMJ, Gableske S, Zurenski MA, Parker ZM, Full F, Baumgart GJ, et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat Microbiol. 2017;2(11):1543–57. https://doi.org/10.1038/s41564-017-0017-2.
Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–51. https://doi.org/10.1080/15548627.2017.1402992.
Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, et al. The cycle of human herpes simplex virus infection: virus transport and immune control. J Infect Dis. 2006;194(Suppl 1):S11–8. https://doi.org/10.1086/505359.
Liu X, Matrenec R, Gack MU, He B. Disassembly of the TRIM23-TBK1 complex by the Us11 protein of herpes simplex virus 1 impairs autophagy. J Virol. 2019;93(17). https://doi.org/10.1128/JVI.00497-19.
Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, et al. The matrix protein of Nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKepsilon kinase-mediated type-I IFN antiviral response. PLoS Pathog. 2016;12(9):e1005880. https://doi.org/10.1371/journal.ppat.1005880.
Rodriguez JJ, Parisien JP, Horvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol. 2002;76(22):11476–83. https://doi.org/10.1128/jvi.76.22.11476-11483.2002.
Shaw ML, Garcia-Sastre A, Palese P, Basler CF. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol. 2004;78(11):5633–41. https://doi.org/10.1128/JVI.78.11.5633-5641.2004.
Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J Virol. 2009;83(16):7828–41. https://doi.org/10.1128/JVI.02610-08.
Sanchez-Aparicio MT, Feinman LJ, Garcia-Sastre A, Shaw ML, Paramyxovirus V. Proteins interact with the RIG-I/TRIM25 regulatory complex and inhibit RIG-I signaling. J Virol. 2018;92(6). https://doi.org/10.1128/JVI.01960-17.
van Tol S, Atkins C, Bharaj P, Johnson KN, Hage A, Freiberg AN, et al. VAMP8 contributes to the TRIM6-mediated type I interferon antiviral response during West Nile virus infection. J Virol. 2020;94(2). https://doi.org/10.1128/JVI.01454-19.
Jacob ST, Crozier I, Fischer WA 2nd, Hewlett A, Kraft CS, Vega MA, et al. Ebola virus disease. Nat Rev Dis Primers. 2020;6(1):13. https://doi.org/10.1038/s41572-020-0147-3.
Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol. 1999;73(3):2333–42.
Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol. 2010;84(20):10581–91. https://doi.org/10.1128/JVI.00925-10.
Basler CF, Wang X, Muhlberger E, Volchkov V, Paragas J, Klenk HD, et al. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci U S A. 2000;97(22):12289–94. https://doi.org/10.1073/pnas.220398297.
Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77(14):7945–56. https://doi.org/10.1128/jvi.77.14.7945-7956.2003.
Prins KC, Cardenas WB, Basler CF. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83(7):3069–77. https://doi.org/10.1128/JVI.01875-08.
Chakraborty A, Diefenbacher ME, Mylona A, Kassel O, Behrens A. The E3 ubiquitin ligase Trim7 mediates c-Jun/AP-1 activation by Ras signalling. Nat Commun. 2015;6:6782. https://doi.org/10.1038/ncomms7782.
Napolitano LM, Jaffray EG, Hay RT, Meroni G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem J. 2011;434(2):309–19. https://doi.org/10.1042/BJ20101487.
Montori-Grau M, Pedreira-Casahuga R, Boyer-Diaz Z, Lassot I, Garcia-Martinez C, Orozco A, et al. GNIP1 E3 ubiquitin ligase is a novel player in regulating glycogen metabolism in skeletal muscle. Metabolism. 2018;83:177–87. https://doi.org/10.1016/j.metabol.2018.02.005.
Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, et al. Biology of Zika virus infection in human skin cells. J Virol. 2015;89(17):8880–96. https://doi.org/10.1128/JVI.00354-15.
Troupin A, Londono-Renteria B, Conway MJ, Cloherty E, Jameson S, Higgs S, et al. A novel mosquito ubiquitin targets viral envelope protein for degradation and reduces virion production during dengue virus infection. Biochim Biophys Acta. 2016;1860(9):1898–909. https://doi.org/10.1016/j.bbagen.2016.05.033.
Choy MM, Sessions OM, Gubler DJ, Ooi EE. Production of infectious dengue virus in Aedes aegypti is dependent on the ubiquitin proteasome pathway. PLoS Negl Trop Dis. 2015;9(11):e0004227. https://doi.org/10.1371/journal.pntd.0004227.
Zhan W, Han T, Zhang C, Xie C, Gan M, Deng K, et al. TRIM59 promotes the proliferation and migration of non-small cell lung Cancer cells by Upregulating cell cycle related proteins. PLoS One. 2015;10(11):e0142596. https://doi.org/10.1371/journal.pone.0142596.
• Orchard RC, Sullender ME, Dunlap BF, Balce DR, Doench JG, Virgin HW. Identification of antinorovirus genes in human cells using genome-wide CRISPR activation screening. J Virol. 2019;93(1). https://doi.org/10.1128/JVI.01324-18. This study shows TRIM7 has antivirial activity against norovirus.
•• Lu M, Zhu X, Yang Z, Zhang W, Sun Z, Ji Q, et al. E3 ubiquitin ligase tripartite motif 7 positively regulates the TLR4-mediated immune response via its E3 ligase domain in macrophages. Mol Immunol. 2019;109:126–33. https://doi.org/10.1016/j.molimm.2019.01.015. This study shows the importance of TRIM7 in innate immunity by regulation of the TLR4-mediated response, promoting IFN-I and cytokine production in macrophages.
• Yang B, Liu Y, Cui Y, Song D, Zhang G, Ma S, et al. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog. 2020;16(3):e1008387. https://doi.org/10.1371/journal.ppat.1008387. This study uses a TRIM7−/− mice to demonstrate a negative regulation of STING in innate immune response to a DNA virus.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest. The Rajsbaum lab is supported by grants R01AI134907, R21AI126012, and R21AI132479 from the National Institute of Health/National Institute of Allergy and Infectious Diseases (NIH/NIAID) and funds from UTMB Institute for Human Infections & Immunity (IHII). A.H. is supported by T32 AI007526 and S.v.T is supported by T32 AI060549 from NIH/NIAID.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Virology
Rights and permissions
About this article

Cite this article
Giraldo, M.I., Hage, A., van Tol, S. et al. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr Clin Micro Rpt 7, 101–114 (2020). https://doi.org/10.1007/s40588-020-00150-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40588-020-00150-8