
Abstract
Purpose of Review
To review current literature on endothelial dysfunction with previous coronaviruses, and present available data on the role of endothelial dysfunction in coronavirus disease-2019 (COVID-19) infection in terms of pathophysiology and clinical phenotype
Recent Findings
Recent evidence suggests that signs and symptoms of severe COVID-19 infection resemble the clinical phenotype of endothelial dysfunction, implicating mutual pathophysiological pathways. Dysfunction of endothelial cells is believed to mediate a variety of viral infections, including those caused by previous coronaviruses. Experience from previous coronaviruses has triggered hypotheses on the role of endothelial dysfunction in the pathophysiology of SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), which are currently being tested in preclinical and clinical studies.
Summary
Endothelial dysfunction is the common denominator of multiple clinical aspects of severe COVID-19 infection that have been problematic for treating physicians. Given the global impact of this pandemic, better understanding of the pathophysiology could significantly affect management of patients.
Similar content being viewed by others


Introduction
Recent evidence suggests that signs and symptoms of severe coronavirus disease-2019 (COVID-19) infection resemble the clinical phenotype of endothelial dysfunction and share mutual pathophysiological mechanisms [1]. Importantly, endothelial dysfunction has been suggested as a main pathophysiological process in several viral infections, including previous coronaviruses [2, 3]. Experience from previous coronaviruses has triggered studies testing hypotheses on the role of the endothelial dysfunction in patients with SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). As a result, recent lines of evidence implicate endothelial dysfunction in the pathophysiology of this systemic infection. Endothelial dysfunction appears to be the common denominator of multiple clinical aspects of severe COVID-19 that have been problematic for treating physicians. Given the global impact of this pandemic, better understanding of the pathophysiology could significantly affect management of patients. Therefore, we systematically reviewed current literature on the pathophysiology of endothelial dysfunction, evidence of endothelial dysfunction in coronaviruses, and particularly in COVID-19 focusing on the clinical phenotype.
Pathophysiology of Endothelial Dysfunction
Vascular endothelium is a continuous monolayer of endothelial cells that forms the inner cellular lining of arteries, veins, and capillaries. Mechanistically, it constitutes a barrier between tissues and blood with the functional capacity of an endocrine organ. Endothelium is directly involved in a number of pathophysiological processes through its dynamic interaction with blood components and other circulating cells. Under physiological conditions, the complex functions of this large endocrine organ are critical to maintain hemostatic balance. Well-described functions of the endothelium include restoration of vascular integrity upon vascular injury and inhibition of excessive thrombosis and clot formation through multiple anticoagulant pathways, including the protein C/protein S pathway [4]. Endothelial cells interact with platelets and leucocytes, prompting their recruitment, adhesion, and interaction on thrombogenic surfaces, at sites of vascular injury driven by inflammation or infection, or at lesion-prone sites such as the carotid bifurcation [4, 5].
In addition, endothelium plays an important role in the regulation of vascular tone and growth by synthesizing and releasing a variety of vasoactive substances, both vasodilatory, such as nictric oxide (NO), prostaglandins, and endothelium-dependent hyperpolarization (EDH) factors, as well as endothelium-derived contracting factors, such as endothelin and angiotensin II [5, 6]. NO biosynthesis by endothelial cells is the most important for the maintenance of vascular homeostasis. The main source of circulating NO is endothelial nitric oxide synthase-3 (eNOS3), expressed in endothelial cells and platelets [7]. NOS3 activity is regulated by the CA2+/calmodulin (CaM) complex, and can also be activated by phosphorylation, which can be CA2+/CaM-independent as most of the stimuli, such as shear stress applied on the vessel wall, do not require the presence of CA2+/CaM [8, 9]. Increased oxidative stress promotes oxidation of tetrahydrobiopterin (BH4), which is essential for the formation and stability of NOS3 [10]. When the supply of BH4 is reduced, NOS3 becomes uncoupled and generates superoxide instead of NO [11].
Based on the above, it becomes apparent that conditions characterized by increased oxidative stress are associated with diminished biosynthesis and availability of NO. Therefore, an imbalance between excessive formation of reactive oxygen species with inadequate antioxidant defense capacity is considered the hallmark of endothelial dysfunction [12]. Under such conditions, the protective properties of the endothelium are lost, with a shift toward impaired vasodilation and the expression of a pro-inflammatory, pro-atherosclerotic, and pro-thrombotic phenotype in the vasculature which is directly associated with the pathogenesis, the progression, and the complications of cardiovascular diseases (CVDs) [13]. Endothelial dysfunction has been documented early in the course of CVD by use of several vascular markers, such as the gold standard flow-mediated vasodilation (FMD), or by measurement of circulating biomarkers, including asymmetrical dimethylarginine (ADMA), oxidized LDL, and endothelial microvesicles (EMVs) [14, 15].
Endothelial dysfunction triggers coagulation disorders in severe infectious diseases, including viral infections. In such cases, free radicals damage the endothelium and disrupt the endothelial barrier by quenching NO, thus allowing toxins to pass into underlying tissues. Vascular leakage is a key feature of endothelial dysfunction in viral infections and can be caused either directly by the viral attack or indirectly by excessive endothelial activation mediated by maladaptive immunological responses [4].
Endothelial Dysfunction in Previous Coronavirus Infections
Disruption of the endothelium, either directly through signaling effects or indirectly through increased pro-inflammatory mediator production and subsequent deregulation of the coagulation cascade, has been described in the pathophysiology of previous coronavirus infections [2, 3].
SARS-CoV was confirmed to be the causative agent for a SARS epidemic associated with severe respiratory failure two decades ago. SARS-CoV infection primarily targets pneumocytes and enterocytes due to their abundant expression of angiotensin-converting enzyme 2 (ACE2), the main functional SARS-CoV receptor, followed by immune system deregulation. Although endothelial cells express ACE2 in the vasculature of several organs, direct evidence regarding endothelial cell infection and dysfunction in SARS patients is limited [16, 17]. In a study that included 22 SARS patients, Yang et al. showed the development of autoantibodies against human umbilical venous and pulmonary endothelial cells in the convalescent phase that could mediate complement-dependent cytotoxicity, thus suggesting a possible pathogenic mechanism [18]. In addition, vasculitis and evidence of endothelial cell inflammation have been documented in postmortem analyses of individual SARS patients, but their role in the pathogenesis of the disease is unknown [19,20,21].
Thrombotic Predisposition in Previous Coronavirus Infections
Coronavirus-infected patients (SARS-CoV and Middle East Respiratory Syndrome/MERS-CoV) have an enhanced thrombotic tendency that has been documented both in vitro and in vivo [22]. Studies in mice showed that SARS-CoV infection is associated with dysregulation of the urokinase pathway, leading to inflammatory vascular damage and activation of the coagulation cascade, culminating in thrombosis [23, 24]. Similarly, in vitro studies showed that SARS-CoV-infected human cells significantly upregulate the expression of genes implicated in inflammation and coagulation and their related proteins [25,26,27], thus contributing to a pro-coagulant profile that mimics the thrombotic alterations observed in SARS patients.
In this context, data have shown that SARS patients exhibit abnormal coagulation parameters and thromboembolic events. In a retrospective study including 153 SARS patients, 49% of them presented reactive thrombocytosis and 63% presented prolonged activated partial thromboplastin time (aPTT) during the course of their illness with no evidence of thromboembolism or other coagulation abnormalities, whereas 2.5% developed disseminated intravascular coagulation [28]. In another study of 138 SARS patients, almost 50% of the study population had a prolonged aPTT and increased levels of d-dimers, while 71% showed increased levels of lactate dehydrogenase, a significant predictor of intensive care unit admission and death [29]. An abnormal coagulation profile, including increased levels of thrombopoietin, von Willebrand factor (vWF), and plasminogen activator inhibitor-1 (PAI-1), has also been documented in SARS patients [30, 31]. In a study of 46 critically ill SARS patients, an increased frequency of thromboembolic complications, including pulmonary embolism and deep vein thrombosis, was observed [32]. In another study, a uniform pattern of large-vessel ischemic strokes was observed in 5 out of 206 SARS patients with relatively few vascular risk factors, while approximately 30% of those who were critically ill had venous thromboembolism [33]. Of note, increased levels of anticardiolipin antibodies have been found in post-SARS patients [34].
Despite relatively limited reports of confirmed clinical thromboembolic events, evidence of enhanced coagulation in SARS patients has been firmly established in histopathological analyses, with fibrin clots in the pulmonary vasculature being a prominent feature [35]. Further, postmortem findings of individual SARS cases were consistent with microvascular thrombi formation in the pulmonary veins and evidence of systemic vasculitis in multiple organs, associated with endothelial cell inflammation, proliferation, swelling, and apoptosis [19]. In a case-series study of 8 SARS patients by Chong et al., autopsy findings included pulmonary thromboemboli in the main and segmental pulmonary arteries as well as deep vein thrombosis and widespread multi-organ infarctions associated with intravascular thrombi [36]. In addition, a larger study including 20 autopsies from SARS patients by Hwang et al. revealed vascular endothelial damage of both small- and medium-sized pulmonary vessels and multiple intravascular fibrin thrombi and thromboemboli associated with pulmonary infarctions [20]. Another postmortem analysis of an individual SARS patient revealed evidence of endothelial cell inflammation and thromboemboli in the veins and microcirculation of multiple organs, thereby highlighting the thrombogenic potential of SARS-CoV in a wider spectrum, including the systemic vasculature [21].
Endothelial Dysfunction in COVID-19 Infection
- Pathophysiology
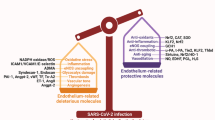
The first step in understanding new complex pathophysiological mechanisms is to combine experimental and translational research and apply a bench-to-bedside approach. Figure 1 summarizes our understanding of the pathophysiology of endothelial dysfunction in COVID-19. Taking into account previous experience from other coronaviruses, several recent studies have implicated complement activation as part of the vicious cycle of endothelial dysfunction in COVID-19 [37]. Complement is a major regulator of endothelial injury syndromes, such as thrombotic microangiopathies [38, 39]. Severe COVID-19 appears to resemble complement-mediated thrombotic microangiopathies in both pathophysiology and clinical phenotype [40]. This resemblance could be also important for management [41], since complement inhibitors have shown safety and efficacy in COVID-19 [42, 43].

Schematic representation of endothelial dysfunction in COVID-19
As mentioned above for other coronaviruses, pericytes with high expression of ACE2 are target cells of COVID-19, resulting in endothelial cell and microvascular dysfunction. Since ACE2 is highly expressed on cardiac myocytes, cardiac injury is expected in COVID-19 [44]. Similarly, a recent study has suggested a coronaviral tropism for the kidney, since ACE2 is highly expressed on podocytes and tubular epithelial cells of the kidney [45]. A puzzling question is the neuroinvasive potential of SARS-CoV-2 that has been suggested by the clinical observation of neurological signs in infected patients. Animal models of structurally similar coronavirus infections point to direct nervous system invasion by coronaviruses. Another hint is that ACE2 is also expressed in in the vasculature of the brain [46]. Potential routes of central nervous system entry, including hematogenic spread, olfactory bulbs, synapse-connected routes, and peripheral nerves, have been suggested. It has been proposed that even before neuronal damage occurs, endothelial injury leads to rupture of cerebral capillaries and eventually to fatal intracerebral hemorrhage in patients with COVID-19 [47]. Further research is needed to explore how such invasiveness could cause direct neurological tissue damage through endothelial dysfunction and the probable variable interaction of the virus with different hosts leading to fluctuating clinical manifestations and severity [48].
The most critical step in unraveling the neuroinvasive propensity of the new coronavirus is to study human neuropathological findings. The first evidence of SARS-CoV-2 presence in brain tissue from postmortem examination strongly supported the hypothesis of endothelial injury and hematogenous dissemination as the primary route of central nervous system (CNS) invasion as viral particles were found in brain capillary endothelium [49]. In a prospective cohort study, SARS-CoV-2 RNA was detected at low titers in autopsied brains of 4 (33%) patients [50]. These data appear to confirm the predominant theory of hematogenous dissemination, leading to endothelial damage. Attempts to isolate SARS-CoV-2 from CSF have been made but were not always successful [51, 52]. The latter study described the first case of meningitis/encephalitis associated with SARS-CoV-2 virus in which RNA was detected in CSF but not in the nasopharyngeal swab [52]. Another case report of acute disseminated encephalomyelitis supported an immune-mediated CNS involvement that could occur after COVID-19 infection [53]. Hence, alternative routes of direct nervous system invasion or an indirect immune-mediated involvement of CNS cannot be ruled out.
- Clinical Phenotype
Thromboembolic Events
The interplay between endothelial injury, complement activation, hypercoagulable state, and thrombin production seems to be a common denominator of COVID-19 clinical features such as deep vein thrombosis (DVT), pulmonary embolism (PE), microvascular thrombosis, cerebrovascular, and cardiac disease [41, 54]. Table 1 summarizes clinical features of endothelial dysfunction and microvascular thrombosis in COVID-19.
Microvascular Thrombosis
Autopsy findings in SARS-CoV-2 patients have demonstrated extensive microvascular injury mediated by complement activation and an associated hypercoagulability [55•]. Another postmortem examination of COVID-19-affected lungs revealed that pulmonary vascular endotheliitis, thrombosis, and angiogenesis are distinctive pulmonary vascular pathophysiologic features of COVID-19 infection not seen in influenza A (H1N1) infection or in uninfected controls [56]. In this context, use of term MicroCLOTS (microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome) has been proposed [57].
Venous Thromboembolism
Venous thromboembolism (VTE) manifested as DVT or PE is encountered commonly in critically ill COVID-19 patients despite prophylactic anticoagulant treatment.
Autopsy studies revealed generalized thrombotic microangiopathy and endothelial dysfunction together with PE and DVT in COVID-19-infected patients [58, 59]. In the first study, PE was found in 4 of 21 specimens while receiving anticoagulation [58••]. In the other autopsy study of 12 consecutive COVID-19 patients, only four of which were receiving anticoagulation; DVT was revealed in 7 (58%), but in no case was DVT suspected clinically [59]. PE was identified and was the primary cause of death in 4 patients. Both studies reported high body mass index and high prevalence of cardiovascular disease, hypertension, and diabetes mellitus history in the afflicted patients.
In parallel with autopsy findings, high rates of VTE (10 to 43%, especially PE) have been clinically diagnosed in COVID-19 patients in intensive care units (ICUs) [60, 61]. A multicenter Dutch study in COVID-19 ICU patients confirmed VTE in 27%. PE was the most frequently encountered thrombotic complication (81%) [60]. A Chinese ICU study identified a 25% incidence of VTE in 81 patients, of which 8 were fatal (10%) [61]. Furthermore, in a series of 184 ICU patients with severe COVID-19, PE was observed in 25 (14%) and DVT in 1 case [62]. PE incidence rate was 21% in another series of 107 ICU patients despite prophylactic or therapeutic anticoagulation [63]. In contrast, in another cohort, the incidence of VTE was 20% overall, but no VTE events were observed in patients receiving therapeutic level anticoagulation on admission [64]. Α high rate of VTE (43%, mainly PE) overall was reported in another series of 150 ICU patients in which patients with COVID-19-associated acute respiratory distress syndrome (ARDS) had higher rates of thrombotic complications compared with non-COVID-19-ARDS [65]. In a small series of 26 individuals with COVID-19, in whom screening leg ultrasounds were performed in the ICU, VTE was found in 18 (69%) cases [66]. All patients were receiving either prophylactic or therapeutic anticoagulation, and some had additional VTE risk factors. Non-ICU COVID-19 patients had lower rates (3% to 21%) of VTE during hospitalization [64, 67, 68]. Data on thrombotic events in patients with COVID-19 infections who were not hospitalized are insufficient for analysis.
The repeated reports of high rates of VTE in COVID-19 have led to the development of a guidance document recommending more aggressive thromboprophylaxis dosing intensities, extended-duration post-discharge thromboprophylaxis, and an individualized approach, taking into account average body mass index, severe thrombocytopenia, and drug-to-drug interactions [69, 70].
Acute Limb Ischemia
Acute upper or lower extremity ischemia has been identified as a COVID-19 arterial thrombotic complication, in some cases requiring surgical embolectomy [71, 72]. In one case series, two young patients who had no comorbidities developed acute limb ischemia despite low molecular weight heparin prophylaxis [73]. A case report linked acute COVID-19-associated limb thrombosis with a probable diagnosis of antiphospholipid syndrome [74]. The largest single-center, observational cohort study assessed 20 COVID-19 patients who presented with and were treated for acute limb ischemia over a 3-month period [75]. An increased incidence rate of acute limb ischemia was observed in 2020 compared with the same months in 2019, and successful revascularization was achieved in 12 of the 17 (70.6%) patients.
Cardiovascular Events
Acute myocardial injury, evidenced by elevated cardiac biomarkers (cardiac troponins, ECG changes, or echocardiographic abnormalities) is the most commonly reported cardiovascular complication of COVID-19 infection [76]. The frequency of myocardial injury varies among hospitalized patients, with reported incidence of 7 to 28% [77•, 78,79,80,81].
Acute cardiac injury occurred in five out of the first 41 COVID-19 patients in Wuhan, four of whom were admitted to ICU, suggesting that cardiac involvement was a predictor of disease severity [82••]. Some studies identified an association of troponin elevation in hospitalized patients with a more severe clinical course and worse outcomes [77, 78, 81]. In one of the aforementioned cohorts of 416 COVID-19 hospitalized patients in Wuhan, one out of five patients had extremely elevated high-sensitivity troponin I (hs-TnI) on admission [77•]. Similarly, in another study from Wuhan, elevation of hs-TnI on admission was identified in half of nonsurvivors compared with 1% of survivors [83]. In contrast, in a study carried out in the USA, troponin levels on ICU admission were elevated in only 2 of 13 critically ill COVID-19 patients, with a 50% mortality rate in the group as a whole [80].
Heart failure has been proposed as another predictor of COVID-19 outcome. In a study from Wuhan of 191 hospitalized patients, heart failure was found in half of the fatal cases and in only 12% of survivors [84]. These observations were confirmed in a second Chinese cohort of 799 hospitalized COVID-19 patients [85].
Apart from myocardial injury and heart failure, evidence of other forms of cardiovascular disease and risk factors in COVID-19 is limited. Both tachyarrhythmia and bradyarrhythmia have been described. The incidence was higher in patients requiring ICU admission (44.4% versus 8.9%) but the exact type of arrhythmia is unknown [83]. According to the report from National Health Commission of China (NHC), cardiovascular symptoms, e.g., palpitations and chest pain, were the first manifestation of COVID-19 infection for some patients [86]. Importantly, pre-existing cardiovascular risk factors and/or development of acute cardiac injury have been consistently found to be associated with significantly poorer prognosis in COVID-19 patients [84]. Furthermore, a significantly increased prevalence of pre-existing cardiovascular disease/cardiac risk factors has been reported in COVID-19 patients with evidence of myocardial injury [77•]. However, there is not yet sufficient evidence to conclude that cardiac involvement is a surrogate biomarker of COVID-19 severity rather than just a confounding factor.
Cerebrovascular Events
Acute ischemic stroke is a recognized extra-pulmonary thromboembolic clinical feature of COVID-19. There are reports of stroke as the presenting clinical feature of polymerase chain reaction (PCR)–confirmed COVID-19 infection, not simply a complication of inhospital stay [87]. In one case series, four elderly patients with cardiovascular disease histories presented with fever and neurologic deficits related to acute infarction in the middle cerebral artery territory but were deemed unsuitable candidates for thrombolysis or any acute neurointervention. Another report of five patients younger than 50 years of age identified large-vessel stroke as a presenting feature of COVID-19 [88]. Two of these had unremarkable medical histories; four out of five received acute treatment; and only one was hospitalized in ICU.
A large study that examined the neurologic signs and symptoms of 214 consecutively hospitalized COVID-19 patients in Wuhan reported six acute cerebrovascular events and found that neurologic symptoms were present in severely affected COVID-19 patients [89]. In a nationwide, multidisciplinary study from the UK, 77 (62%) COVID-19 patients had a cerebrovascular event, 57 of whom had acute ischemic strokes [90]. In contrast, in a Spanish registry, ischemic stroke was recorded in only 11 (1.3%) cases, and in an ICU cohort of 184 patients, only three cases were found [60, 91]. Another three strokes were reported in a similar French ICU cohort of 150 COVID-19 patients [65]. A UK report confirmed the observation of COVID-19 association with large-vessel occlusion, 8–24 days after COVID-19 symptom onset in six cases (aged 53–85 years, with cardiovascular medical history) [92]. Five of six patients had a positive lupus anticoagulant, one with coexisting low-medium titer of antiphospholipid antibodies (aPL). The theory that COVID-19 might stimulate the production of aPL has been put forward [93]. However, aPL are usually transient in the postinfection period and their clinical significance is ambiguous [94].
The most likely mechanism of early cerebrovascular accidents during the course of COVID-19 infection appears to be hypercoagulability and vascular endothelial dysfunction mediated by pro-inflammatory cytokines. An Italian retrospective study reported significantly increased rates of cerebrovascular disease in COVID-19 patients with neurological manifestations compared with hospitalized neurological patients without COVID-19 (76.8% vs 58.1%, respectively) [95]. In that study, COVID-19 patients with cerebrovascular disease presented with a hypercoagulability state characterized by longer prothrombin times, higher fibrinogen levels and inflammatory indices, and statistically significantly worse outcomes (COVID-19 patients with a good outcome 25.6% vs non-COVID-19 70.6%). Yet, further investigation is needed to assess the risk factors, causative relationship, mechanisms, and stroke phenotype, especially in young patients with COVID-19. Increased awareness is needed to recognize the coexistence of COVID-19 with stroke and promptly treat a COVID-19 patient presenting with an acute neurologic symptom with thrombolysis or mechanical thrombectomy when appropriate [96].
The other side of cerebrovascular disease, intracerebral hemorrhage (ICH), has also been also linked with COVID-19, but occurs less frequently. Nine intracerebral hemorrhages were reported in the UK-wide cohort and three in a Spanish registry [90, 91]. In a case report, evaluation of acute loss of consciousness in a febrile 79-year-old patient with an unremarkable medical history revealed COVID-19 infection and a massive intracerebral hemorrhage together with intraventricular and subarachnoid hemorrhage [97]. Moreover, in an ICU cohort of 150 COVID-19 patients receiving anticoagulation, one patient presented with both ICH and ischemic lesions [65].
Cerebral vein and dural sinus thrombosis (CVT) and CNS vasculitis are less common types of cerebrovascular disease. A rare case of CVT was observed in a cohort of 50 COVID-19 ICU patients [98]. Small-vessel CNS vasculitis was present as a result of presumably direct viral infection of endothelial cells based on a radiological finding of gadolinium-enhanced white matter lesions. Similar findings occurred in one case in the UK-wide cohort [90, 99].
Neurologic Invasion and Phenotypes
In addition to cerebrovascular accidents, SARS-CoV-2 can affect the central and peripheral nervous systems, causing a variety of neurologic disease phenotypes [89, 100]. Many neurological symptoms, such as headache, dizziness, myalgia, loss of smell and taste, ophthalmoparesis, pseudoexacerbations of multiple sclerosis and altered level of consciousness have been reported in COVID-19 patients and have been attributed to the coronavirus [101,102,103,104,105,106]. Severe cases may manifest as impaired level of consciousness, encephalopathy, or encephalitis [89, 90, 107]. In this context, the idea of COVID-19 disease classification according to the presence or absence of neurological involvement of COVID-19 neurological complications has been supported.
Reports of COVID-19 neurological complications have described a 6–36% incidence of secondary neurological events [108]. Of note, a large Spanish registry revealed that 57.4% of 841 hospitalized COVID-19 patients developed some type of neurological symptom [91]. The most common was altered level of consciousness. However, neurological complications were considered the primary cause of death only in 4.1% of total deaths. Some patients may present with nonspecific neurological symptoms, as reported in one of the first retrospective cohorts from Wuhan with 99 patients. Muscle ache occurred in 11%,confusion in 9% and headache in 8% of the patients [100]. This large study from Wuhan reported an incidence of 36.4% involving CNS, peripheral, and skeletal muscles [89•]. They observed an association of severe infection with the development of neurologic symptoms. In a French retrospective cohort of 58 inhospital patients, encephalopathy with consistent electroencephalography findings and also prominent agitation and corticospinal tract signs were observed in the majority of ICU patients [109•]. The coronavirus could not be isolated from CSF, but MRI showed lesions that included leptomeningeal enhancement, bilateral frontotemporal hypoperfusion and ischemic strokes. Neurologic sequelae in ICU-survivors of COVID-19 also included critical illness neuropathy or myopathy and neuropsychiatric manifestations [110, 111]. Acute de novo psychotic and neuropsychiatric symptoms, hyperkinetic movements, generalized myoclonus, and status epilepticus have also been reported in association with COVID-19 infection [91, 112,113,114]. A multicenter retrospective study that investigated the risk of seizures or status epilepticus during COVID-19 infection did not record any event, although 108 of 304 patients enrolled had a severe infection [115]. This contrasts with previous reports of frequent neurological complications in COVID-19 patients [89, 116]. Furthermore, an increasing number of case reports support the causative role of COVID-19 in postinfectious neurologic complications such as Miller Fisher and Guillain-Barré syndrome (GBS) [117,118,119,120,121,122]. However, in some cases no SARS-CoV-2 PCR assay was performed to confirm the diagnosis of infection before the hospitalization for GBS [123, 124].
Hence, it is important to note that the evidence of nervous system involvement in COVID-19 disease is mainly based on case reports and retrospective data. In the middle of the pandemic, the research community is trying to understand the new virus, but publication bias lies in wait.
Conclusions and Future Perspectives
Our review summarizes for the first time available evidence of endothelial dysfunction caused by coronaviruses, focusing on COVID-19. Our data confirm previous and recently acquired knowledge that endothelial dysfunction plays a key role in these infections. These data are important in order to understand the multisystemic attack of these viruses, and could also be helpful in patient management. Although there is no direct therapeutic target for endothelial dysfunction, several agents have shown beneficial effects. A promising new strategy for treating severe COVID-19 is complement inhibition. Given the global impact of the pandemic, further well-designed, mechanistic, and clinical studies are urgently needed to translate this knowledge into clinical practice.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8. https://doi.org/10.1016/S0140-6736(20)30937-5.
Steinberg BE, Goldenberg NM, Lee WL. Do viral infections mimic bacterial sepsis? The role of microvascular permeability: a review of mechanisms and methods. Antivir Res. 2012;93(1):2–15. https://doi.org/10.1016/j.antiviral.2011.10.019.
•• Goeijenbier M, van Wissen M, van de Weg C, Jong E, Gerdes VE, Meijers JC, et al. Review: viral infections and mechanisms of thrombosis and bleeding. J Med Virol. 2012;84(10):1680–96. https://doi.org/10.1002/jmv.23354The dysregulation of the coagulation cascade is a prominent finding in viral infections.
• Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int J Biol Sci. 2013;9(10):1057–69. https://doi.org/10.7150/ijbs.7502This report described the main endothelial abnormalities that are typically encountered in various human diseases including cancer, diabetes mellitus, atherosclerosis, and viral infections.
Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196(3):430–43. https://doi.org/10.1002/jcp.10333.
•• Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol. 2017;37(9):e108–e14. https://doi.org/10.1161/ATVBAHA.117.309813Εndothelial functions in health and disease are summarized in this well-written informative report.
Gkaliagkousi E, Ritter J, Ferro A. Platelet-derived nitric oxide signaling and regulation. Circ Res. 2007;101(7):654–62. https://doi.org/10.1161/CIRCRESAHA.107.158410.
Fleming I, Bauersachs J, Busse R. Calcium-dependent and calcium-independent activation of the endothelial NO synthase. J Vasc Res. 1997;34(3):165–74. https://doi.org/10.1159/000159220.
Gkaliagkousi E, Douma S, Zamboulis C, Ferro A. Nitric oxide dysfunction in vascular endothelium and platelets: role in essential hypertension. J Hypertens. 2009;27(12):2310–20. https://doi.org/10.1097/HJH.0b013e328330e89a.
List BM, Klosch B, Volker C, Gorren AC, Sessa WC, Werner ER, et al. Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochem J. 1997;323(Pt 1):159–65. https://doi.org/10.1042/bj3230159.
Bevers LM, Braam B, Post JA, van Zonneveld AJ, Rabelink TJ, Koomans HA, et al. Tetrahydrobiopterin, but not L-arginine, decreases NO synthase uncoupling in cells expressing high levels of endothelial NO synthase. Hypertension. 2006;47(1):87–94. https://doi.org/10.1161/01.HYP.0000196735.85398.0e.
Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc Pharmacol. 2018;100:1–19. https://doi.org/10.1016/j.vph.2017.05.005.
Gkaliagkousi E, Gavriilaki E, Triantafyllou A, Douma S. Clinical significance of endothelial dysfunction in essential hypertension. Curr Hypertens Rep. 2015;17(11):85. https://doi.org/10.1007/s11906-015-0596-3.
Gkaliagkousi E, Gavriilaki E, Vasileiadis I, Nikolaidou B, Yiannaki E, Lazaridis A, et al. Endothelial microvesicles circulating in peripheral and coronary circulation are associated with central blood pressure in coronary artery disease. Am J Hypertens. 2019;32(12):1199–205. https://doi.org/10.1093/ajh/hpz116.
• Gkaliagkousi E, Gavriilaki E, Triantafyllou A, Nikolaidou B, Anyfanti P, Koletsos N, et al. Asymmetric dimethylarginine levels are associated with augmentation index across naive untreated patients with different hypertension phenotypes. J Clin Hypertens (Greenwich). 2018;20(4):680–5. https://doi.org/10.1111/jch.13237In this review the mechanisms, clinical significance and methods for the assessment of endothelial dysfunction in patients with essential hypertension are discussed.
Guo Y, Korteweg C, McNutt MA, Gu J. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008;133(1):4–12. https://doi.org/10.1016/j.virusres.2007.01.022.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7. https://doi.org/10.1002/path.1570.
Yang YH, Huang YH, Chuang YH, Peng CM, Wang LC, Lin YT, et al. Autoantibodies against human epithelial cells and endothelial cells after severe acute respiratory syndrome (SARS)-associated coronavirus infection. J Med Virol. 2005;77(1):1–7. https://doi.org/10.1002/jmv.20407.
Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200(3):282–9. https://doi.org/10.1002/path.1440.
Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Modern Pathol: an official journal of the United States and Canadian Academy of Pathology, Inc. 2005;18(1):1–10. https://doi.org/10.1038/modpathol.3800247.
Xiang-Hua Y, Le-Min W, Ai-Bin L, Zhu G, Riquan L, Xu-You Z, et al. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am J Respir Crit Care Med. 2010;182(3):436–7. https://doi.org/10.1164/ajrccm.182.3.436.
Giannis D, Ziogas IA, Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J Clin Virol : the official publication of the Pan American Society for Clinical Virology. 2020;127:104362. https://doi.org/10.1016/j.jcv.2020.104362.
Gralinski LE, Baric RS. Molecular pathology of emerging coronavirus infections. J Pathol. 2015;235(2):185–95. https://doi.org/10.1002/path.4454.
Gralinski LE, Bankhead A 3rd, Jeng S, Menachery VD, Proll S, Belisle SE, et al. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio. 2013;4(4):e00271–13. https://doi.org/10.1128/mBio.00271-13.
Tang BS, Chan KH, Cheng VC, Woo PC, Lau SK, Lam CC, et al. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. J Virol. 2005;79(10):6180–93. https://doi.org/10.1128/JVI.79.10.6180-6193.2005.
Han M, Yan W, Huang Y, Yao H, Wang Z, Xi D, et al. The nucleocapsid protein of SARS-CoV induces transcription of hfgl2 prothrombinase gene dependent on C/EBP alpha. J Biochem. 2008;144(1):51–62. https://doi.org/10.1093/jb/mvn042.
Ng LF, Hibberd ML, Ooi EE, Tang KF, Neo SY, Tan J, et al. A human in vitro model system for investigating genome-wide host responses to SARS coronavirus infection. BMC Infect Dis. 2004;4:34. https://doi.org/10.1186/1471-2334-4-34.
Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK, et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ. 2003;326(7403):1358–62. https://doi.org/10.1136/bmj.326.7403.1358.
• Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348(20):1986–94. https://doi.org/10.1056/NEJMoa030685A report of clinical, laboratory, and radiologic features of severe acute respiratory syndrome (SARS) outbreak in Hong Kong back in 2003.
Yang M, Ng MH, Li CK, Chan PK, Liu C, Ye JY, et al. Thrombopoietin levels increased in patients with severe acute respiratory syndrome. Thromb Res. 2008;122(4):473–7. https://doi.org/10.1016/j.thromres.2007.12.021.
Wu YP, Wei R, Liu ZH, Chen B, Lisman T, Ren DL, et al. Analysis of thrombotic factors in severe acute respiratory syndrome (SARS) patients. Thromb Haemost. 2006;96(1):100–1. https://doi.org/10.1160/TH05-12-0827.
Lew TW, Kwek TK, Tai D, Earnest A, Loo S, Singh K, et al. Acute respiratory distress syndrome in critically ill patients with severe acute respiratory syndrome. JAMA : the journal of the American Medical Association. 2003;290(3):374–80. https://doi.org/10.1001/jama.290.3.374.
Umapathi T, Kor AC, Venketasubramanian N, Lim CC, Pang BC, Yeo TT, et al. Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J Neurol. 2004;251(10):1227–31. https://doi.org/10.1007/s00415-004-0519-8.
Sun W, Wang BL, Liu BL, Zhao FC, Shi ZC, Guo WS, et al. Osteonecrosis in patients after severe acute respiratory syndrome (SARS): possible role of anticardiolipin antibodies. J Clin Rheumatol : practical reports on rheumatic & musculoskeletal diseases. 2010;16(2):61–3. https://doi.org/10.1097/RHU.0b013e3181cf3464.
Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361(9371):1773–8. https://doi.org/10.1016/s0140-6736(03)13413-7.
Chong PY, Chui P, Ling AE, Franks TJ, Tai DY, Leo YS, et al. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: challenges in determining a SARS diagnosis. Arch Pathol Lab Med. 2004;128(2):195–204. https://doi.org/10.1043/1543-2165(2004)128<195:AODDTS>2.0.CO;2.
Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, et al. Complement as a target in COVID-19? Nat Rev Immunol. 2020;20(6):343–4. https://doi.org/10.1038/s41577-020-0320-7.
Gavriilaki E, Touloumenidou T, Sakellari I, Batsis I, Mallouri D, Psomopoulos F, et al. Pretransplant genetic susceptibility: clinical relevance in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2020;120(4):638–46. https://doi.org/10.1055/s-0040-1702225.
Gavriilaki E, Chrysanthopoulou A, Sakellari I, Batsis I, Mallouri D, Touloumenidou T, et al. Linking complement activation, coagulation, and neutrophils in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2019;119(9):1433–40. https://doi.org/10.1055/s-0039-1692721.
Chaturvedi S, Braunstein EM, Yuan X, Yu J, Alexander A, Chen H, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. 2020;135(4):239–51. https://doi.org/10.1182/blood.2019003863.
Gavriilaki E, Brodsky RA. Complementopathies and precision medicine. J Clin Invest. 2020;130(5):2152–63. https://doi.org/10.1172/JCI136094.
Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24(7):4040–7. https://doi.org/10.26355/eurrev_202004_20875.
Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450. https://doi.org/10.1016/j.clim.2020.108450.
Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116:1097–100. https://doi.org/10.1093/cvr/cvaa078.
Wan Y, Shang J, Graham R, Baric RS, Li F. receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127–0. https://doi.org/10.1128/JVI.00127-20.
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. 2008;82(15):7264–75. https://doi.org/10.1128/JVI.00737-08.
Baig AM, Khaleeq A, Ali U, Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11(7):995–8. https://doi.org/10.1021/acschemneuro.0c00122.
Natoli S, Oliveira V, Calabresi P, Maia LF, Pisani A. Does SARS-Cov-2 invade the brain? Translational lessons from animal models. Eur J Neurol. 2020. https://doi.org/10.1111/ene.14277.
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol. 2020;92:699–702. https://doi.org/10.1002/jmv.25915.
Pappas G, Christou L, Akritidis N, Tsianos EV. Quinolones for brucellosis: treating old diseases with new drugs. Clin Microbiol Infect: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2006;12(9):823–5. https://doi.org/10.1111/j.1469-0691.2006.01442.x.
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology. 2020:201187. https://doi.org/10.1148/radiol.2020201187.
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 2020;94:55–8. https://doi.org/10.1016/j.ijid.2020.03.062.
Novi G, Rossi T, Pedemonte E, Saitta L, Rolla C, Roccatagliata L, et al. Acute disseminated encephalomyelitis after SARS-CoV-2 infection. Neurology - Neuroimmunology Neuroinflammation. 2020;7(5):e797. https://doi.org/10.1212/nxi.0000000000000797.
• Gavriilaki E, Brodsky RA. Severe COVID-19 infection and thrombotic microangiopathy: success does not come easily. Br J Haematol. 2020;189(6):e227–e30. https://doi.org/10.1111/bjh.16783This perspective article describes similarities of severe COVID with complement-mediated thrombotic microangiopathy.
• Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res: the journal of laboratory and clinical medicine. 2020. https://doi.org/10.1016/j.trsl.2020.04.007A case-series study describing a possible pathophysiologic mechanism of severe COVID-19 infection unravelled the catastrophic microvascular injury syndrome mediated by complement cascade.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–8. https://doi.org/10.1056/NEJMoa2015432.
Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, Ruggeri A et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc 2020
•• Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology. 2020. https://doi.org/10.1111/his.14134This autopsy study revealed generalized thrombotic microangiopathy and endothelial dysfunction in COVID-19 patients.
Wichmann D, Sperhake JP, Lutgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. 2020. https://doi.org/10.7326/M20-2003.
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–7. https://doi.org/10.1016/j.thromres.2020.04.013.
Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. 2020;18:1421–4. https://doi.org/10.1111/jth.14830.
Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost : JTH. 2020;18:1094–9. https://doi.org/10.1111/jth.14817.
Poissy J, Goutay J, Caplan M, Parmentier E, Duburcq T, Lassalle F, et al. Pulmonary embolism in COVID-19 patients: awareness of an increased prevalence. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.120.047430.
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Muller MCA, et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14888.
Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–98. https://doi.org/10.1007/s00134-020-06062-x.
Llitjos JF, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost : JTH. 2020;18:1743–6. https://doi.org/10.1111/jth.14869.
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan. Italy Thrombosis Research. 2020;191:9–14. https://doi.org/10.1016/j.thromres.2020.04.024.
Artifoni M, Danic G, Gautier G, Gicquel P, Boutoille D, Raffi F, et al. Systematic assessment of venous thromboembolism in COVID-19 patients receiving thromboprophylaxis: incidence and role of D-dimer as predictive factors. J Thromb Thrombolysis. 2020;50:211–6. https://doi.org/10.1007/s11239-020-02146-z.
Barnes GD, Burnett A, Allen A, Blumenstein M, Clark NP, Cuker A, et al. Thromboembolism and anticoagulant therapy during the COVID-19 pandemic: interim clinical guidance from the anticoagulation forum. J Thromb Thrombolysis. 2020;50:72–81. https://doi.org/10.1007/s11239-020-02138-z.
• Spyropoulos AC, Levy JH, Ageno W, Connors JM, Hunt BJ, Iba T et al. Scientific and standardization committee communication: clinical guidance on the diagnosis, prevention and treatment of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. n/a(n/a). https://doi.org/10.1111/jth.14929. This consensus provides practical guidance for the diagnosis, thromboprophylaxis strategies, and treatment of venous thromboembolism in hospitalized patients with suspected or confirmed COVID-19 infection.
Andrea V, Gianluca F, Rodolfo P, Paolo T, Alessandro P, Mauro G. Unheralded lower limb threatening ischemia in a COVID-19 patient. Int J Infect Diseases : IJID : official publication of the International Society for Infectious Diseases. 2020;96:590–2. https://doi.org/10.1016/j.ijid.2020.05.060.
Kaur P, Qaqa F, Ramahi A, Shamoon Y, Singhal M, Shamoon F, et al. Acute upper limb ischemia in a patient with COVID-19. Hematol/Oncol Stem Cell Ther. 2020. https://doi.org/10.1016/j.hemonc.2020.05.001.
Perini P, Nabulsi B, Massoni CB, Azzarone M, Freyrie A. Acute limb ischaemia in two young, non-atherosclerotic patients with COVID-19. Lancet. 2020;395(10236):1546. https://doi.org/10.1016/S0140-6736(20)31051-5.
Fan BE, Chia YW, Sum CLL, Kuperan P, Chan SSW, Ling LM, et al. Global haemostatic tests in rapid diagnosis and management of COVID-19 associated coagulopathy in acute limb ischaemia. J Thromb Thrombolysis. 2020. https://doi.org/10.1007/s11239-020-02165-w.
Bellosta R, Luzzani L, Natalini G, Pegorer MA, Attisani L, Cossu LG, et al. Acute limb ischemia in patients with COVID-19 pneumonia. J Vasc Surg. 2020. https://doi.org/10.1016/j.jvs.2020.04.483.
Bansal M. Cardiovascular disease and COVID-19. Diabetes Metab Syndr Clin Res Rev. 2020;14(3):247–50. https://doi.org/10.1016/j.dsx.2020.03.013.
• Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.0950This cohort study associated cardiac injury with higher risk of inhospital mortality in COVID-19 patients.
Lippi G, Lavie CJ, Sanchis-Gomar F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): evidence from a meta-analysis. Prog Cardiovasc Dis. 2020. https://doi.org/10.1016/j.pcad.2020.03.001.
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. COVID-19 and cardiovascular disease. Circulation. 2020;141(20):1648–55. https://doi.org/10.1161/CIRCULATIONAHA.120.046941.
Bhatraju PK, Ghassemieh BJ, Nichols M, Kim R, Jerome KR, Nalla AK, et al. Covid-19 in critically ill patients in the Seattle region-case series. N Engl J Med. 2020;382(21):2012–22. https://doi.org/10.1056/NEJMoa2004500.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1017.
•• Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5The first cohort study from Wuhan describing COVID-19 patient baseline characteristics, clinical manifestations, and mortality rate.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan China. Jama. 2020;323:1061. https://doi.org/10.1001/jama.2020.1585.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. Bmj. 2020;368:m1091. https://doi.org/10.1136/bmj.m1091.
Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17(5):259–60. https://doi.org/10.1038/s41569-020-0360-5.
Avula A, Nalleballe K, Narula N, Sapozhnikov S, Dandu V, Toom S, et al. COVID-19 presenting as stroke. Brain Behav Immun. 2020. https://doi.org/10.1016/j.bbi.2020.04.077.
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382:e60. https://doi.org/10.1056/NEJMc2009787.
• Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurology. 2020. https://doi.org/10.1001/jamaneurol.2020.1127The first cohort study focused on neurologic clinical manifestations of COVID-19.
UK-wide surveillance of neurological and neuropsychiatric complications of COVID-19: the first 153 patients [database on the Internet]2020. Available from: https://ssrn.com/abstract=3601761 Accessed: 29/5/2020.
Romero-Sánchez CM, Díaz-Maroto I, Fernández-Díaz E, Sánchez-Larsen Á, Layos-Romero A, García-García J, et al. Neurologic manifestations in hospitalized patients with COVID-19: the ALBACOVID registry. Neurology. 2020. https://doi.org/10.1212/WNL.0000000000009937.
Beyrouti R, Adams ME, Benjamin L, Cohen H, Farmer SF, Goh YY, et al. Characteristics of ischaemic stroke associated with COVID-19. J Neurol Neurosurg Psychiatry. 2020. https://doi.org/10.1136/jnnp-2020-323586.
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med. 2020;382(17):e38. https://doi.org/10.1056/NEJMc2007575.
Abdel-Wahab N, Talathi S, Lopez-Olivo MA, Suarez-Almazor ME. Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus. 2018;27(4):572–83. https://doi.org/10.1177/0961203317731532.
Benussi A, Pilotto A, Premi E, Libri I, Giunta M, Agosti C, et al. Clinical characteristics and outcomes of inpatients with neurologic disease and COVID-19 in Brescia, Lombardy, Italy. Neurology. 2020. https://doi.org/10.1212/WNL.0000000000009848.
Yeboah K, Edgell R, Conway J, Alshekhlee A. Interventional stroke management in a COVID-19 patient. Neurology: Clinical Practice. 2020. https://doi.org/10.1212/CPJ.0000000000000884.
Sharifi-Razavi A, Karimi N, Rouhani N. COVID-19 and intracerebral haemorrhage: causative or coincidental? New Microbes New Infect. 2020;35:100669. https://doi.org/10.1016/j.nmni.2020.100669.
Kandemirli SG, Dogan L, Sarikaya ZT, Kara S, Akinci C, Kaya D, Kaya Y, Yildirim D, Tuzuner F, Yildirim MS, Ozluk E, Gucyetmez B, Karaarslan E, Koyluoglu I, Demirel Kaya HS, Mammadov O, Kisa Ozdemir I, Afsar N, Citci Yalcinkaya B, Rasimoglu S, Guduk DE, Kedir Jima A, Ilksoz A, Ersoz V, Yonca Eren M, Celtik N, Arslan S, Korkmazer B, Dincer SS, Gulek E, Dikmen I, Yazici M, Unsal S, Ljama T, Demirel I, Ayyıldız A, Kesimci I, Bolsoy Deveci S, Tutuncu M, Kizilkilic O, Telci L, Zengin R, Dincer A, Akinci IO, Kocer N Brain MRI findings in patients in the intensive care unit with COVID-19 infection. Radiology. 2020;201697. https://doi.org/10.1148/radiol.2020201697.
Brun G, Hak J-F, Coze S, Kaphan E, Carvelli J, Girard N, et al. COVID-19—white matter and globus pallidum lesions. Demyelination or small-vessel vasculitis? Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e777. https://doi.org/10.1212/nxi.0000000000000777.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–13. https://doi.org/10.1016/S0140-6736(20)30211-7.
Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–20. https://doi.org/10.1056/NEJMoa2002032.
Barzegar M, Mirmosayyeb O, Nehzat N, Sarrafi R, Khorvash F, Maghzi AH, et al. COVID-19 infection in a patient with multiple sclerosis treated with fingolimod. Neurol(R) Neuroimmunol Neuroinflammation. 2020;7(4):e753. https://doi.org/10.1212/NXI.0000000000000753.
Dinkin M, Gao V, Kahan J, Bobker S, Simonetto M, Wechsler P, et al. COVID-19 presenting with ophthalmoparesis from cranial nerve palsy. Neurology. 2020. https://doi.org/10.1212/WNL.0000000000009700.
Min J, Qiaoxia T. Rhabdomyolysis as potential late complication associated with COVID-19. Emerg Infect Disease J. 2020;26(7):1618–20. https://doi.org/10.3201/eid2607.200445.
Yin R, Feng W, Wang T, Chen G, Wu T, Chen D, et al. Concomitant neurological symptoms observed in a patient diagnosed with coronavirus disease 2019. J Med Virol. 2020. https://doi.org/10.1002/jmv.25888.
Goh Y, Beh DLL, Makmur A, Somani J, Chan ACY. Pearls and Oy-sters: facial nerve palsy as a neurological manifestation of Covid-19 infection. Neurology. 2020. https://doi.org/10.1212/WNL.0000000000009863.
Filatov A, Sharma P, Hindi F, Espinosa PS. Neurological complications of coronavirus disease (COVID-19): encephalopathy. Cureus. 2020;12(3):e7352. https://doi.org/10.7759/cureus.7352.
Herman C, Mayer K, Sarwal A. Scoping review of prevalence of neurologic comorbidities in patients hospitalized for COVID-19. Neurology. 2020:10.1212/WNL.0000000000009673. https://doi.org/10.1212/WNL.0000000000009673.
• Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 Infection. N Engl J Med. 2020. https://doi.org/10.1056/NEJMc2008597The first European observational series on neurologic features of COVID-19 patients admitted to intensive care units.
Troyer EA, Kohn JN, Hong S. Are we facing a crashing wave of neuropsychiatric sequelae of COVID-19? Neuropsychiatric symptoms and potential immunologic mechanisms. Brain Behav Immun. 2020;87:34–9. https://doi.org/10.1016/j.bbi.2020.04.027.
Kotfis K, Williams Roberson S, Wilson JE, Dabrowski W, Pun BT, Ely EW. COVID-19: ICU delirium management during SARS-CoV-2 pandemic. Crit Care. 2020;24(1):176. https://doi.org/10.1186/s13054-020-02882-x.
Bernard-Valnet R, Pizzarotti B, Anichini A, Demars Y, Russo E, Schmidhauser M, et al. Two patients with acute meningo-encephalitis concomitant to SARS-CoV-2 infection. Eur J Neurol. 2020. https://doi.org/10.1111/ene.14298.
Rábano-Suárez P, Bermejo-Guerrero L, Méndez-Guerrero A, Parra-Serrano J, Toledo-Alfocea D, Sánchez-Tejerina D, et al. Generalized myoclonus in COVID-19. Neurology. 2020. https://doi.org/10.1212/WNL.0000000000009829.
Somani S, Pati S, Gaston T, Chitlangia A, Agnihotri S. De novo status epilepticus in patients with COVID-19. Ann Clin Transl Neurol. 2020. https://doi.org/10.1002/acn3.51071.
Lu L, Xiong W, Liu D, Liu J, Yang D, Li N, et al. New onset acute symptomatic seizure and risk factors in coronavirus disease 2019: a retrospective multicenter study. Epilepsia. 2020;61:e49–53. https://doi.org/10.1111/epi.16524.
Karimi N, Razavi A, Rouhani N. Frequent convulsive seizures in an adult patient with COVID-19: a case report. Iran Red Crescent Med J. 2020;In Press. https://doi.org/10.5812/ircmj.102828.
Gutierrez-Ortiz C, Mendez A, Rodrigo-Rey S, San Pedro-Murillo E, Bermejo-Guerrero L, Gordo-Manas R, et al. Miller fisher syndrome and polyneuritis cranialis in COVID-19. Neurology. 2020:10.1212/WNL.0000000000009619. https://doi.org/10.1212/WNL.0000000000009619.
Alberti P, Beretta S, Piatti M, Karantzoulis A, Piatti ML, Santoro P et al. Guillain-Barre syndrome related to COVID-19 infection. Neurol(R) Neuroimmunol Neuroinflammation. 2020;7(4). https://doi.org/10.1212/NXI.0000000000000741.
Marta-Enguita J, Rubio-Baines I, Gaston-Zubimendi I. Fatal Guillain-Barre syndrome after infection with SARS-CoV-2. Neurologia. 2020;35:265–7. https://doi.org/10.1016/j.nrl.2020.04.004.
Padroni M, Mastrangelo V, Asioli GM, Pavolucci L, Abu-Rumeileh S, Piscaglia MG, et al. Guillain-Barre syndrome following COVID-19: new infection, old complication? J Neurol. 2020;267:1877–9. https://doi.org/10.1007/s00415-020-09849-6.
Ebrahimzadeh SA, Ghoreishi A, Rahimian N. Guillain-Barré syndrome associated with the coronavirus disease 2019 (COVID-19). Neurology: Clinical Practice. 2020. https://doi.org/10.1212/CPJ.0000000000000879.
Chan M, Han SC, Kelly S, Tamimi M, Giglio B, Lewis A. A case series of Guillain-Barré syndrome following Covid-19 infection in New York. Neurology: Clinical Practice. 2020. https://doi.org/10.1212/CPJ.0000000000000880.
Bigaut K, Mallaret M, Baloglu S, Nemoz B, Morand P, Baicry F, et al. Guillain-Barré syndrome related to SARS-CoV-2 infection. Neurology - Neuroimmunology Neuroinflammation. 2020;7(5):e785. https://doi.org/10.1212/nxi.0000000000000785.
Zhao H, Shen D, Zhou H, Liu J, Chen S. Guillain-Barre syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol. 2020;19(5):383–4. https://doi.org/10.1016/S1474-4422(20)30109-5.
Funding
E.G., P.A., and EU.G. are co-financed by Greece and the European Union (European Social Fund-ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning 2014-2020”, in the context of the project “Evaluation of novel markers of endothelial dysfunction and thrombotic microenvironment in patients with rheumatoid arthritis: association with markers of subclinical inflammation and cardiovascular damage (MIS 5047870)”.
Author information
Authors and Affiliations
Contributions
E.G., P.A., M.G., and A.L. drafted the manuscript, tables, and figures. EU.G. edited the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Hypertension and the Heart
Rights and permissions
About this article

Cite this article
Gavriilaki, E., Anyfanti, P., Gavriilaki, M. et al. Endothelial Dysfunction in COVID-19: Lessons Learned from Coronaviruses. Curr Hypertens Rep 22, 63 (2020). https://doi.org/10.1007/s11906-020-01078-6
Published:
DOI: https://doi.org/10.1007/s11906-020-01078-6