
Abstract
Plasma glycine level is low in patients with obesity or diabetes and the improvement of insulin resistance increases plasma glycine concentration. In prospective studies, hypoglycinemia at baseline predicts the risk of developing type 2 diabetes and higher serum glycine level is associated with decreased risk of incident type 2 diabetes. Consistently, plasma glycine concentration is lower in the lean offspring of parents with type 2 diabetes compared to healthy subjects. Among patients with type 2 diabetes, hypoglycinemia occurs before clinical manifestations of the disease, but the pathophysiological mechanisms underlying glycine deficit and its potential clinical repercussions are unclear. Glycine participates in several metabolic pathways, being required for relevant human physiological processes. Humans synthesize glycine from glyoxylate, glucose (via serine), betaine and likely from threonine and during the endogenous synthesis of L-carnitine. Glycine conjugates bile acids and other acyl moieties producing acyl-glycine derivatives. The glycine cleavage system catalyzes glycine degradation to carbon dioxide and ammonium while tetrahydrofolate is converted into 5,10-methylene-tetrahydrofolate. Glycine is utilized to synthesize serine, sarcosine, purines, creatine, heme group, glutathione, and collagen. Glycine is a major quantitative component of collagen. In addition, the role of glycine maintaining collagen structure is critical, as glycine residues are required to stabilize the triple helix of the collagen molecule. This quality of glycine likely contributes to explain the occurrence of medial arterial calcification and the elevated cardiovascular risk associated with diabetes and chronic kidney disease, as emerging evidence links normal collagen content with the initiation and progression of vascular calcification in humans.
Similar content being viewed by others

Glycine and insulin resistance
Plasma glycine level is consistently lower in patients with obesity and diabetes mellitus compared to healthy individuals. In addition, the improvement of insulin resistance with weight loss, exercise or drugs increases plasma glycine concentration in subjects with obesity or diabetes. Further, prospective studies indicate that low plasma glycine level precedes the clinical onset of diabetes mellitus. Consistently, plasma glycine concentration is lower in the offspring of parents with type 2 diabetes compared to healthy individuals. Human glycine metabolism is defective among people with obesity and diabetes mellitus and this impairment may be crucial to explain the elevated cardiovascular risk associated with these conditions.
It has long been observed that plasma glycine level is reduced in patients with obesity (Felig and Cahill 1969; Glynn et al. 2015; Takashina et al. 2016) and diabetes mellitus (Felig et al. 1970; Wahren et al. 1972; Drabkova et al. 2015) compared to control subjects. In a study recruiting obese Saudi participants, the urinary metabolic phenotype associated with obesity is also characterized by lower glycine concentration (Ahmad et al. 2017). Further, plasma glycine level is lower in obese diabetic African-American women compared to obese non-diabetic African-American women (Fiehn et al. 2010).
Plasma glycine concentration correlates positively with insulin sensitivity and negatively with insulin resistance determined by the homeostasis model assessment of insulin resistance (Takashina et al. 2016). Similarly, serum glycine concentration is positively correlated with insulin sensitivity estimated during a hyperinsulinemic–euglycemic clamp by glucose disposal rate among non-diabetic subjects. In patients with type 2 diabetes, plasma glycine level is lower and the positive association with insulin sensitivity is attenuated (Thalacker-Mercer et al. 2014).
Plasma glycine levels increase after weight loss in obese patients (Felig and Cahill 1969) and after a 6-month exercise program in overweight insulin-resistant subjects, (Glynn et al. 2015) compared to baseline values.
In a randomized trial including obese or diabetic patients with insulin resistance, insulin sensitizer therapy with pioglitazone + metformin increases plasma glycine concentration compared to placebo (Irving et al. 2015).
Prospective studies indicate that reduced plasma glycine level is a predictor for impaired glucose tolerance and type 2 diabetes. (Wang-Sattler et al. 2012; Floegel et al. 2013; Palmer et al. 2015) A prospective analysis on the population-based Cooperative Health Research in the Region of Augsburg cohort shows that low levels of glycine (and lysophosphatidylcholine) at baseline predict the development of impaired glucose tolerance and type 2 diabetes even 7 years before the clinical onset. (Wang-Sattler et al. 2012) Higher serum glycine is associated with decreased risk of incident type 2 diabetes in the European Prospective Investigation into Cancer and Nutrition. Accordingly, reduced plasma glycine is an independent predictor of type 2 diabetes. Choline-containing phospholipids are also associated with type 2 diabetes risk in this study (Floegel et al. 2013). In a cohort from the Insulin Resistance Atherosclerosis Study, logistic regression analysis reveals decreased serum glycine level in insulin-resistant subjects. In addition, patients who develop type 2 diabetes have reduced plasma glycine level compared to individuals who do not develop type 2 diabetes (Palmer et al. 2015).
Plasma glycine concentration is lower in the lean offspring of parents with type 2 diabetes compared to healthy control subjects (Perseghin et al. 1997).
Glucagon secretion modifies plasma glycine concentration in healthy humans. Glucagon deficiency is associated with an elevation of circulating glycine in plasma whereas excess glucagon diminishes plasma glycine level (Boden et al. 1984).
Glycine receptors have been found in human pancreatic β-cells, but the effect of glycine on insulin secretion is uncertain (Yan-Do et al. 2016).
Some glycine derivatives have also been found to be associated with insulin resistance and risk of type 2 diabetes mellitus. Reduced plasma level of betaine (trimethylglycine) is a predictor of diabetes mellitus risk. The Diabetes Prevention Program study found that both an intensive lifestyle intervention and metformin, relative to placebo, reduce the incidence of diabetes in obese individuals with high risk of developing type 2 diabetes. In samples from this study, lower plasma betaine level at baseline is associated with incident type 2 diabetes. In addition, the lifestyle intervention increases plasma betaine level and this elevation is associated with lower incidence of diabetes at 2 years. The relationship between baseline levels of serum betaine and incident diabetes mellitus does not differ among participants randomized to placebo, metformin or lifestyle arms (Walford et al. 2016).
Cross-sectional studies have shown that plasma glycine level is inversely associated with risk of acute myocardial infarction. Higher plasma glycine at baseline is associated with lower prevalence of obesity and diabetes mellitus and more favorable lipid profile. After multivariate adjustment for traditional coronary heart disease risk factors, plasma glycine is inversely associated with risk of acute myocardial infarction (Ding et al. 2015).
In healthy Mexican women from the Mexican–American cohort study, plasma acetyl-glycine and acetylcholine levels at baseline predict significant weight gain during 5-year follow-up (Zhao et al. 2016).
Genetic variants at the DMGDH locus encoding dimethylglycine dehydrogenase modulate the concentration of dimethylglycine in plasma. The major allele of rs2431332 at the DMGDH locus is associated with lower dimethylglycine levels in plasma. The same genetic variant is also slightly associated with higher plasma insulin concentration, increased homeostasis model assessment of insulin resistance and an increased risk of incident diabetes during follow-up (Magnusson et al. 2015). However, common genetic variants associated with metabolites in glycine metabolism are not conclusively associated with insulin resistance or diabetes in a genome-wide association study including nondiabetic participants (Xie et al. 2013).
The underlying mechanisms causing hypoglycinemia among patients with obesity and diabetes are unclear. The potential clinical repercussions and the potential usefulness of hypoglycinemia as diagnostic tool have not been investigated.
Glycine metabolism in humans
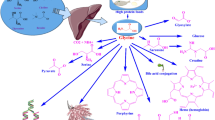
Glycine (2-aminoacetate) is a versatile amino acid connected with several metabolic pathways and crucial for a number of human physiological processes (Fig. 1). Humans obtain glycine from dietary sources and from endogenous synthesis. Glycine may be generated from glyoxylate, glucose (via serine), betaine (trimethylglycine) and likely from threonine and during the endogenous synthesis of L-carnitine. In turn, glycine may be utilized in a number of metabolic pathways. The glycine cleavage system catalyzes glycine degradation to carbon dioxide and ammonium. Glycine may be used to produce serine via the reversible reaction catalyzed by serine hydroxymethyl transferase. Glycine may be methylated to generate N-methylglycine (sarcosine). Glycine is used to conjugate acyl groups derived from acyl-coA esters to generate acyl-glycine derivatives. Glycine is required to synthesize creatine, heme group, glutathione, and collagen. Glycine is fundamental to maintain the helical structure of the collagen molecule; therefore, glycine-deficient collagen is defective and likely predisposed to degradation and secondary calcification, which may be instrumental to explain the development of arterial medial calcification and the elevated cardiovascular risk associated to diabetes mellitus.

Summary of glycine metabolism
Pathways of glycine formation
Humans may synthesize glycine via several enzymatic reactions. Serine hydroxymethyl transferase catalyzes the reversible synthesis of glycine + 5,10-methylene-tetrahydrofolate from serine + tetrahydrofolate. Alanine:glyoxylate aminotransferase catalyzes the transamination of alanine and glyoxylate to produce pyruvate and glycine. Glycine may be generated from betaine via dimethylglycine and N-methylglycine (sarcosine). Threonine may likely produce glycine through the sequential action of two enzymes. The biochemical pathway resulting in the synthesis of L-carnitine produces glycine in a reaction catalyzed by an unidentified enzyme.
Glycine formation from l-threonine
l-threonine may be converted into glycine by a two-step enzymatic pathway involving the enzymes l-threonine dehydrogenase and glycine C-acetyltransferase. First, l-threonine dehydrogenase catalyzes the conversion of threonine into 2-amino-3-ketobutyrate while nicotinamide adenine dinucleotide (NAD+) is reduced to NADH. The human gene encoding l-threonine dehydrogenase is located at 8p23.1. However, this gene is not capable of encoding a functional l-threonine dehydrogenase protein and therefore humans lack a functioning l-threonine dehydrogenase enzyme (Edgar 2002). Second, the enzyme glycine C-acetyltransferase catalyzes the synthesis of glycine and acetyl-coA from 2-amino-3-ketobutyrate and coenzyme A (coA). On a Northern blot of human tissues, glycine C-acetyltransferase mRNA appears as a single transcript being strongly expressed in heart, brain, liver, and pancreas. Glycine C-acetyltransferase is a vitamin B6 (pyridoxal 5′-phosphate)-dependent enzyme. The human GCAT gene is located on 22q13.1 (Edgar and Polak 2000). The relevance of this pathway to synthesize glycine in humans has not been investigated. Congenital deficiency of human threonine dehydrogenase and glycine C-acetyltransferase enzymes has not been documented.
Glycine formation from glyoxylate
Human alanine:glyoxylate aminotransferase is a pyridoxal 5′-phosphate (PLP)-dependent enzyme localized to peroxisomes that catalyzes the transamination of l-alanine and glyoxylate to form pyruvate and glycine. Glyoxylate is derived from degradation of 4-hydroxyproline, a component of collagen. The X-ray structure of human alanine:glyoxylate aminotransferase in complex with the competitive inhibitor amino-oxyacetic acid has been solved, revealing a homodimeric structure (Cellini et al. 2007; Oppici et al. 2015). The gene AGXT located to 2q37.3 encodes alanine:glyoxylate aminotransferase. Congenital deficiency of this enzyme causes primary hyperoxaluria type 1. In patients with this disease, the transamination of glyoxylate into glycine is impaired and glyoxylate is oxidized to oxalate leading to deposition of calcium oxalate in tissues, particularly the kidney. Nephrocalcinosis and renal failure occur (Cellini et al. 2007; Oppici et al. 2015).
Glycine formation from glucose
Glycine may be synthesized from l-serine by the enzyme serine hydroxymethyl transferase, which catalyzes the reversible interconversion between serine and glycine. Humans may synthesize l-serine from d-glucose through the action of three enzymes from 3-phosphoglycerate, an intermediate in glycolysis. In the first step, 3-phosphoglycerate is oxidized by the enzyme phosphoglycerate dehydrogenase to form 3-phospho-hydroxypyruvate. In the second step, the enzyme phosphoserine phosphatase converts 3-phospho-hydroxypyruvate into phosphoserine. Phosphoserine aminotransferase-1 catalyzes the third and final step, converting phosphoserine into serine. Then, serine hydroxymethyl transferase may synthesize glycine from serine (Fig. 2).

Glycine synthesis from glucose
3-Phosphoglycerate dehydrogenase
The enzyme 3-phosphoglycerate dehydrogenase catalyzes the oxidation of 3-phosphoglycerate to 3-phospho-hydroxypyruvate while NAD+ is reduced to NADH. Two different transcripts of 3-phosphoglycerate dehydrogenase are detected in human tissues. The larger one (2.1 kb) is expressed at high levels in brain, liver, kidney, pancreas, prostate, testis, and ovary, and weakly expressed in thymus, colon, and heart. A smaller transcript (710 bp) appears as a weaker band where the 2.1 kb mRNA is expressed. In addition, the smaller transcript is more significant that the 2.1 kb mRNA in heart and skeletal muscle (Cho et al. 2000).
The gene PHGDH located to 1p12 encodes 3-phosphoglycerate dehydrogenase. Congenital deficiency of this enzyme is a rare disease first described in 1996 that usually presents in the neonatal period, although clinical onset may occur later in life. Clinical manifestations of 3-phosphoglycerate dehydrogenase deficiency include congenital microcephaly, mental retardation, and therapy-resistant seizures. However, patients with normocephaly at birth and milder clinical presentation have been reported (Jaeken et al. 1996; Hausler et al. 2001). Brain magnetic resonance imaging demonstrates reduced myelination, white matter attenuation, and cerebral atrophy (Jaeken et al. 1996).
Patients with 3-phosphoglycerate dehydrogenase deficiency show decreased plasma and cerebrospinal fluid concentration of serine and glycine (Jaeken et al. 1996). Oral therapy with serine alone or in combination with glycine reduces the frequency of seizures and improves psychomotor development (Jaeken et al. 1996). However, high-dose serine has been associated with an arrest of head growth that improves by reducing the serine supply. In addition, serine therapy might decrease the concentration of methionine, isoleucine, and ornithine in the cerebrospinal fluid (Hausler et al. 2001). An association between a single nucleotide polymorphism near the PHGDH gene and plasma serine levels has been documented in non-diabetic individuals (Xie et al. 2013).
3-Phosphoserine aminotransferase
3-Phosphoserine aminotransferase is a pyridoxal 5′-phosphate-dependent enzyme that catalyzes the reversible transamination of 3-phospho-hydroxypyruvate to 3-phosphoserine using glutamate as the nitrogen donor, so that glutamate is converted into 2-ketoglutarate (Baek et al. 2003).
Two isoforms of human 3-phosphoserine aminotransferase can be produced by alternative splicing, α and β, but the β-isoenzyme is the physiologically functional enzyme required for the serine synthesis pathway. Northern-blot analysis shows that human 3-phosphoserine aminotransferase mRNA is expressed at high levels in the brain, liver, kidney and pancreas. Very weak expression is observed in the thymus, prostate, testis and colon. Both 3-phosphoserine aminotransferase and 3-phosphoglycerate dehydrogenase transcripts are expressed in human brain, liver, kidney and pancreas (Baek et al. 2003). The gene PSAT1 that encodes human 3-phosphoserine aminotransferase is located on 9p21.2. Congenital deficiency of 3-phosphoserine aminotransferase has been reported in two siblings. The index patient presented with acquired microcephaly, intractable seizures, hypertonia, and psychomotor retardation and died at age 7 months despite supplementation with serine and glycine. Both patients showed low concentration of serine and glycine in plasma and cerebrospinal fluid (Hart et al. 2007).
Phosphoserine phosphatase
Phosphoserine phosphatase catalyzes the irreversible dephosphorylation of 3-phosphoserine to yield l-serine and phosphate (Collet et al. 1997). The crystal structure of human phosphoserine phosphatase complexed with inhibitors has been described showing a dimeric protein (Kim et al. 2002). The enzymatic reaction of human phosphoserine phosphatase is Mg2+-dependent and the enzyme is inactive when Mg2+ is replaced by Ca2+ in the active site (Moro-Furlani and Hopkinson 1980; Kim et al. 2002). The enzyme activity is inhibited by the product of the reaction, l-serine (Collet et al. 1997). Phosphoserine phosphatase displays a widespread distribution in human tissues, having been identified in all the human tissues tested, although it is most active in adult kidney and liver (Moro-Furlani and Hopkinson 1980). Cytosolic phosphoserine phosphatase has been purified from human brain. The PSPH gene encodes human phosphoserine phosphatase, being located to 7p11.2. Congenital deficiency of phosphoserine phosphatase has been rarely reported. Clinical presentation usually occurs in infancy or childhood. Clinical phenotype is characterized by growth and motor retardation, intellectual disability, microcephaly, epilepsy, hypertonia, and axonal neuropathy complicated by non-healing wounds. Patients with congenital deficiency of phosphoserine phosphatase show reduced level of serine and glycine in plasma and cerebrospinal fluid (Jaeken et al. 1997).
Serine hydroxymethyl transferase
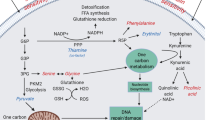
Serine hydroxymethyl transferase is a pyridoxal 5′-phosphate-dependent enzyme that catalyzes the reversible conversion serine + tetrahydrofolate ↔ glycine + 5,10-methylene-tetrahydrofolate. Tetrahydrofolate is a derivative of folate that may carry one of several chemical groups containing one carbon atom, such as methyl, methenyl, methylene, and formyl groups. These so-called “one-carbon units” attached at either position 5 or 10 of tetrahydrofolate may be converted into each other. The enzyme methylene-tetrahydrofolate dehydrogenase catalyzes the oxidation of 5,10-methylene-tetrahydrofolate to 10-formyl-tetrahydrofolate while the enzyme methylene-tetrahydrofolate reductase catalyzes the reduction of 5,10-methylene-tetrahydrofolate to 5-methyl-tetrahydrofolate (Fig. 3) (Stover et al. 1997; Renwick and Baumann 1998; Lamers et al. 2009).

Methylene-tetrahydrofolate reductase (MTHFR) and methylene-tetrahydrofolate dehydrogenase (MTHFD) reactions
The crystal structure of human serine hydroxymethyl transferase has been solved, showing a homotetramer with each monomer binding a single pyridoxal-5′-phosphate molecule. Tetramer formation results from the association of two dimers. Each dimer contains two active sites (Stover et al. 1997; Renwick and Baumann 1998). There are two isoforms of human serine hydroxymethyl transferase encoded by the genes SHMT1 and SHMT2, located on 17p11.2 and 12q13.3, respectively, but their differential function is unknown (Stover et al. 1997; Garrow et al. 1993). Patients with congenital deficiency of serine hydroxymethyl transferase have not been reported. Polymorphisms in the genes SHMT1 and MTHFR (that encodes methylene-tetrahydrofolate reductase) might interact to modulate cardiovascular risk in humans. In a nested case–control study within the all-male Normative Aging Study cohort, a gene–gene interaction between the SHMT1 gene and the MTHFR gene has been observed, such that the association of MTHFR 677C → T polymorphism and incident cardiovascular disease varied by the SHMT1 genotype (Lim et al. 2005). Two meta-analyses indicate that there is no association between the C1420T polymorphism in the SHMT1 gene and risk of cancer in humans (Zhong et al. 2014; Wang et al. 2014).
In the direction of glycine formation, serine hydroxymethyl transferase transfers the hydroxymethyl group of serine to tetrahydrofolate to generate glycine + 5,10-methylene-tetrahydrofolate. 5,10-methylene-tetrahydrofolate may be involved in several metabolic pathways. First, it participates in the synthesis of thymidylate (deoxy-thymidine monophosphate). Second, methylene-tetrahydrofolate dehydrogenase oxidizes 5,10-methylene-tetrahydrofolate to 10-formyl-tetrahydrofolate, which is needed for purine synthesis. Third, methylene-tetrahydrofolate reductase reduces 5,10-methylene-tetrahydrofolate to 5-methyl-tetrahydrofolate, which participates in methionine synthesis.
The enzyme thymidylate synthase catalyzes the methylation of 2′-deoxyuridine monophosphate (dUMP) to 2′-deoxythymidine monophosphate (dTMP) or thymidylate, a pyrimidine derivative required for DNA formation. In this reaction, 5,10-methylene-tetrahydrofolate serves as methyl donor and dihydrofolate is generated. Inhibition of thymidylate synthase impairs DNA synthesis by curtailing thymidylate formation. The TYMS gene located to 18p11.32 encodes thymidylate synthase. Fluorouracil is an inhibitor of thymidylate synthase (Lamers et al. 2009). The enzyme dihydrofolate reductase catalyzes the NADPH-dependent reduction of dihydrofolate to generate tetrahydrofolate. Methotrexate inhibits dihydrofolate reductase. The gene DHFR located on 5q14.1 encodes dihydrofolate reductase (Stover et al. 1997; Renwick and Baumann 1998; Anderson and Stover 2009).
The enzyme methylene-tetrahydrofolate dehydrogenase (MTHFD) catalyzes the oxidation of 5,10-methylene-tetrahydrofolate to 10-formyl-tetrahydrofolate while NADP+ is reduced to NADPH (Fan et al. 2014). Two isoenzymes of human methylene-tetrahydrofolate dehydrogenase are known to exist, MTHFD1 and MTHFD2, although the physiological relevance of the latter is unclear (Pelletier and MacKenzie 1994). The product of the methylene-tetrahydrofolate dehydrogenase reaction, 10-formyl-tetrahydrofolate, is required for the de novo synthesis of purines (Stover et al. 1997; Renwick and Baumann 1998; Lamers et al. 2009) In addition, in vitro studies suggest that NADPH production via the MTHFD reaction is a significant source of intracellular NADPH. Knockdown of either isoenzyme of methylene-tetrahydrofolate-dehydrogenase, MTHFD1 or MTHFD2, decreases NADPH production in HEK293T cells (Fan et al. 2014). NADPH is required for a number of metabolic processes, including fatty acid synthesis and the reaction catalyzed by dihydrofolate reductase which consumes one molecule of NADPH (Anderson and Stover 2009). Two separate loci encode the isoenzymes of methylene-tetrahydrofolate dehydrogenase. The gene MTHFD1 located on 14q23.3 encodes MTHFD1 whereas the gene MTHFD2 located on 2p13.1 encodes MTHFD2. Mutations in the MTHFD1 gene have been very rarely reported. In one family, MTHFD1 mutations caused neural tube defects (Hol et al. 1998). In one patient, mutations in the MTHFD1 gene resulted in megaloblastic anemia and severe combined immunodeficiency (Field et al. 2015). It has been suggested that some polymorphisms in the MTHFD1 locus might modulate cardiovascular risk (Ding et al. 2016).
The enzyme methylene-tetrahydrofolate reductase (MTHFR) catalyzes the reduction of 5,10-methylene-tetrahydrofolate to 5-methyl-tetrahydrofolate. Methylene-tetrahydrofolate reductase is a flavoprotein that requires flavin adenine dinucleotide (FAD), a derivative of riboflavin, as cofactor (Caudill et al. 2009). 5-methyl-tetrahydrofolate is the methyl donor for the methylation of homocysteine to methionine catalyzed by the enzyme methionine synthase, producing tetrahydrofolate (Fig. 4). Vitamin B12 (cobalamin) is required for this reaction to take place (Stover et al. 1997; Knowles and Walter 2016) The gene MTHFR located to 1p36.22 encodes methylene-tetrahydrofolate reductase. Congenital deficiency of this enzyme presents usually in infancy or childhood. Clinical phenotype is predominantly neurological and includes microcephaly, seizures, progressive ataxia, dysarthria, tremor, developmental delay, and psychiatric symptoms. Thrombotic events may occur. Common findings on brain magnetic resonance imaging are cerebral atrophy with white-matter changes secondary to demyelination or vascular pathology (Bishop et al. 2008; Knowles and Walter 2016) The biochemical phenotype is characterized by elevated plasma and urinary level of homocysteine due to impaired methylation. Plasma concentration of methionine and S-adenosylmethionine are low or normal. Plasma cysteine–homocysteine disulfide may be present (Bishop et al. 2008). Treatment of methylene-tetrahydrofolate reductase deficiency consists of administration of betaine, vitamin B12 and vitamin B6 (pyridoxine), but its effectiveness is unclear. Betaine provides methyl groups for methylation of homocysteine to synthesize methionine. Vitamin B12 is a cofactor for methionine synthase. Vitamin B6 is used to maximize the trans-sulfuration pathway as a cofactor for cystathionine β-synthase. Aspirin might be considered to improve thrombotic events (Bishop et al. 2008; Knowles and Walter 2016).

Methylation of homocysteine by methionine synthase (MS) or betaine homocysteine methyltransferase (BHMT) and trans-sulfuration pathway
Glycine formation from betaine (trimethylglycine)
In humans, a second enzyme catalyzes the methylation of homocysteine to generate methionine: betaine homocysteine methyltransferase. This enzyme uses betaine (trimethylglycine) as methyl donor, yielding methionine and dimethylglycine (Figs. 4, 5) (Holme and Ronge 1989; Vanek et al. 2009). Betaine is a nutrient obtained from dietary food. Endogenous synthesis of betaine through the oxidation of choline via the enzymes choline dehydrogenase and betaine aldehyde dehydrogenase has not been demonstrated in humans and no deficiency of these enzymes has been reported. Betaine homocysteine methyltransferase is highly expressed in human liver where it has been estimated that catalyzes up to 50% of homocysteine remethylation (Feng et al. 2011). Two loci, BHMT and BHMT2, located to 5q14.1, encode two isoenzymes of betaine homocysteine methyltransferase, but their differential physiological role is unclear. Molecular defects in these genes have not been reported. A single nucleotide polymorphism near the BHMT gene has been associated with betaine levels in a genome-wide association study with non-diabetic participants (Xie et al. 2013).

Glycine formation from betaine (trimethylglycine) BHMT betaine homocysteine methyltransferase, DMGD dimethylglycine dehydrogenase, SD sarcosine dehydrogenase
Dimethylglycine generated from betaine is demethylated to sarcosine (N-methylglycine) by the enzyme dimethylglycine dehydrogenase and then into glycine by the enzyme sarcosine dehydrogenase (Fig. 5).
Dimethylglycine dehydrogenase is a flavoprotein that catalyzes the oxidative demethylation of dimethylglycine to sarcosine while FAD serves as electron acceptor, being reduced to FADH2. The methyl group derived from dimethylglycine is incorporated to tetrahydrofolate which is converted into 5,10-methylene-tetrahydrofolate (Augustin et al. 2016). The crystal structure of the human wild-type dimethylglycine dehydrogenase has been elucidated (Augustin et al. 2016). The gene DMGDH, located to 5q14.1, encodes dimethylglycine dehydrogenase. Genetic variants at this locus may modulate the level of dimethylglycine in plasma (Magnusson et al. 2015). Congenital deficiency of dimethylglycine dehydrogenase due to a homozygous mutation in the DMGDH gene has been reported in one patient, a 38-year-old man with fishlike body odor since childhood, muscle fatigue and increased serum creatine kinase. Plasma level of dimethylglycine was increased approximately 100-fold in this patient (Moolenaar et al. 1999).
Sarcosine dehydrogenase is a flavoprotein that catalyzes the oxidative demethylation of sarcosine (N-methyl-glycine) to produce glycine. A methyl group is incorporated into tetrahydrofolate to generate 5,10-methylene-tetrahydrofolate. The SARDH gene located on 9q34.2 encodes sarcosine dehydrogenase. Mutations in this gene cause sarcosinemia. In one patient, maternal isodisomy in the region of SARDH locus has been detected (Bar-joseph et al. 2012). Sarcosinemia is a rare disorder clinically characterized by cardiomyopathy, developmental delay, mental retardation, dystonia, deafness, and visual disturbance. The disease presents usually in the neonatal period or infancy, but later presentation may occur. Patients affected with sarcosinemia show elevated concentration of sarcosine in plasma and urine (Benarrosh et al. 2013).
Glycine formation during the synthesis of L-carnitine
Humans synthesize L-carnitine via a four-step enzymatic pathway. Glycine is generated in the second step of this pathway, the aldolytic cleavage of 3-hydroxy-trimethyl-lysine. The human gene coding the aldolase that catalyzes this reaction has not been identified (Rebouche and Engel 1980).
Pathways of glycine utilization
Glycine may be used in a number of human metabolic pathways. The glycine cleavage system catalyzes glycine degradation to carbon dioxide and ammonium; in this reaction, tetrahydrofolate is converted into 5,10-methylene-tetrahydrofolate. Glycine may be utilized to synthesize serine by the reversible reaction catalyzed by serine hydroxymethyl transferase (serine + tetrahydrofolate ↔ glycine + 5,10-methylene-tetrahydrofolate). Glycine may be methylated to generate sarcosine (N-methylglycine). Glycine is used to conjugate bile acids and other acyl moieties derived from acyl-coA esters to generate acyl-glycine derivatives. Glycine is required to synthesize purines, creatine, heme, glutathione, and collagen. The contribution of glycine to maintain collagen structure is crucial.
Glycine cleavage system
The glycine cleavage system is a multienzyme complex that catalyzes the degradation of glycine to generate ammonium and carbon dioxide. At the same time, tetrahydrofolate is converted to 5,10-methylene-tetrahydrofolate, and NAD+ is reduced to NADH (Fig. 6).

The glycine cleavage system reaction
The reaction catalyzed by the glycine cleavage system is reversible in vitro, but deficient activity of the human complex leads to hyperglycinemia, suggesting that the reaction in vivo proceeds predominantly in the direction of glycine breakdown. The glycine cleavage system consists of four components: P-protein, T-protein, H-protein, and L-protein and its reaction proceeds in several steps. First, the P-protein (glycine decarboxylase) catalyzes the decarboxylation of glycine, releasing carbon dioxide. Human P-protein is a homodimer containing pyridoxal phosphate. Second, the decarboxylated moiety of glycine (aminomethyl intermediate) is attached to lipoate in the H-protein. Third, the T-protein (aminomethyl transferase) transfers a methyl group from the aminomethyl intermediate to tetrahydrofolate, resulting in 5,10-methylene-tetrahydrofolate and ammonium. This reaction leaves the H protein bearing dihydrolipoate (reduced lipoate) instead of lipoate. The crystal structure of the human T-protein has been determined, revealing that the T-protein forms a complex with the H-protein in a 1:1 molar ratio (Okamura-Ikeda et al. 2005). Finally, L-protein (dihydrolipoamide dehydrogenase) catalyzes the oxidation of dihydrolipoate attached to the H-protein whereas NAD+ is reduced to NADH. The L protein is a component of the glycine cleavage system shared with other enzymatic complexes, including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain ketoacid dehydrogenase (Okamura-Ikeda et al. 2005). Components of the glycine cleavage system are encoded by separate human genes. The genes GLDC (glycine decarboxylase), GCSH (glycine cleavage system protein H), AMT (aminomethyl transferase), and DLD (dihydrolipoamide dehydrogenase) encode P-protein, H-protein, T-protein, and L-protein, respectively (Azize et al. 2014). They are located on 9p24.1, 16q23.2, 3p21.31, and 7q31.1, respectively. Molecular changes in some of these genes cause glycine encephalopathy (nonketotic hyperglycinemia). Mutations in the GCSH gene that encodes the H-protein have not been detected in patients with this disease so far (Azize et al. 2014). More than 85% of patients with glycine encephalopathy have deficiency of the P-protein (glycine decarboxylase), whereas the remaining are deficient in the T-protein (aminomethyl transferase) (Okamura-Ikeda et al. 2005). Glycine encephalopathy is a rare disorder in most populations, but it is more frequent in Northern Finland and British Columbia populations. Clinical presentation usually occurs in the neonatal period or infancy, but atypical late onset in childhood or adulthood may occur. Clinical phenotype is predominantly neurological, with lethargy, hypotonia, psychomotor retardation, developmental delay, learning disabilities, behavioral problems, treatment-resistant epilepsy, choreiform movements, spastic paraparesia, optic atrophy and vacuolating leukoencephalopathy. Dilated cardiomyopathy and hypertrophic cardiomyopathy have been documented (Azize et al. 2014; Swanson et al. 2015). Patients with glycine encephalopathy show elevated glycine level in plasma and urine. Plasma serine concentration is usually in the normal range (Swanson et al. 2015). Treatment involves sodium benzoate to lower plasma glycine levels and dextromethorphan to reduce the stimulating activity of glycine on N-methyl-d-aspartate receptors, with unclear effects on outcome (Swanson et al. 2015). In patients with deficiency of β-ketothiolase, isovaleryl-coA dehydrogenase, propionyl-coA carboxylase, and methylmalonyl-coA mutase, secondary hyperglycinemia is commonly observed (Adibi 1980).
Glycine utilization to form serine
The enzyme serine hydroxymethyl transferase catalyzes the reversible conversion serine + tetrahydrofolate ↔ glycine + 5,10-methylene-tetrahydrofolate. In the direction of serine synthesis, the enzyme transfers one-carbon group from 5,10-methylene-tetrahydrofolate to glycine forming tetrahydrofolate and serine (Stover et al. 1997; Renwick and Baumann 1998; Lamers et al. 2009) In human healthy volunteers, serine synthesis by serine hydroxymethyl transferase accounts for approximately 41% of the whole body glycine flux (Lamers et al. 2007).
Glycine utilization to form sarcosine (N-methyl-glycine)
Methionine is converted into S-adenosylmethionine by the enzyme l-methionine S-adenosyltransferase. The reaction involves the transfer of the adenosyl group of ATP to the sulfur atom of l-methionine, forming S-adenosylmethionine (Caudill et al. 2009; Furujo et al. 2012). S-adenosylmethionine serves as methyl donor for several methylation reactions (Caudill et al. 2009). After donating the methyl group, S-adenosylmethionine is sequentially converted into S-adenosylhomocysteine and homocysteine, which can be either remethylated to methionine or enter the so-called trans-sulfuration pathway, being converted first to cystathionine by cystathionine β-synthase and then to cysteine by cystathionine γ-lyase, two vitamin B6 (pyridoxal 5′-phosphate)-dependent enzymes (Fig. 4) (Luka and Wagner 2009). Besides cysteine, cystathionine γ-lyase generates 2-ketobutyrate, which is thought to be the precursor of 2-hydroxybutyrate in humans. Plasma concentration of 2-hydroxybutyrate is higher in obese African American women with type 2 diabetes compared to obese non-diabetic African American women (Fiehn et al. 2010). In addition, plasma level of 2-hydroxybutyrate is negatively correlated with insulin sensitivity. In plasma samples from the Relationship of Insulin Sensitivity to Cardiovascular Risk (RISC) study, a cohort of nondiabetic subjects representing a broad spectrum of insulin sensitivity and glucose tolerance, 2-hydroxybutyrate is able to separate insulin resistant from insulin sensitive subjects independently of body mass index (Gall et al. 2010).
The enzyme glycine N-methyltransferase catalyzes the transfer of a methyl group from S-adenosylmethionine to glycine to form sarcosine (N-methylglycine) and S-adenosyl-homocysteine (Fig. 7) (Chen et al. 2000). The physiological mission of this reaction has not been elucidated. Glycine N-methyltransferase is a homotetramer that has been identified in liver, pancreas, and prostate in Northern blot analysis of human tissues (Chen et al. 2000). The crystal structure of human glycine N-methyltransferase has been reported and this enzyme has been identified in the nucleus and cytosol of human liver cells (Luka et al. 2007). The gene GNMT encodes glycine N-methyltransferase, being localized to 6p21.1 (Chen et al. 2000). Congenital deficiency of glycine N-methyl-transferase has been rarely documented. The clinical phenotype is mild, the only abnormally being hepatomegaly. Affected patients show chronic elevation of serum transaminases and elevated plasma level of methionine and S-adenosylmethionine. Plasma concentration of S-adenosylhomocysteine, total homocysteine, and sarcosine is normal (Luka and Wagner 2009). Genetic variants in the gene GNMT may influence plasma homocysteine level in healthy women (Beagle et al. 2005).

Sarcosine (N-methylglycine) formation from glycine by the enzyme glycine N-methyltransferase (GNMT)
Glycine conjugation: glycine N-acyltransferase isoenzymes
Glycine conjugates with a number of substrates derived from acyl-coA esters, including bile acids. Glycine N-acyltransferase catalyzes the transfer of the acyl group from acyl-coA esters to the amino group of glycine, generating acyl-glycine derivatives. Several isoforms of human glycine acyltransferase have been identified, namely glycine acyltransferase, glycine acyltransferase-like-1, and glycine acyltransferase-like-2. In humans there is a gene cluster on chromosome 11q12.1 that contains the genes GLYAT, GLYATL1 and GLYATL2, encoding the corresponding glycine N-acyltransferases. Another gene named GLYATL3 is located on chromosome 6p12.3 (Waluk, Schultz, and Hunt 2010). The enzyme bile acid-coA:amino acid N-acyltransferase catalyzes the conjugation (amidation) of bile acids with glycine or taurine before their excretion into the bile.
Glycine acyltransferase
Human glycine acyltransferase catalyzes the transfer of a number of substrates from acyl-coA esters to glycine. This enzyme is a mitochondrial monomeric protein predominantly expressed in liver and kidney. Preferred substrates for human glycine acyltransferase are benzoate and short- and medium-chain fatty acids (Matsuo et al. 2012).
Glycine acyltransferase catalyzes the transfer of benzoate from benzoyl-coA to glycine, generating hippurate that is excreted in the urine and thus reducing plasma glycine level. Benzoate may also be combined with carnitine forming benzoyl-carnitine that is excreted in urine (Matsuo et al. 2012). Human diet contains benzoate in variable amounts, predominantly in soft drinks. It has been estimated that about 83–90% of ingested benzoate is excreted as its glycine conjugate (hippurate). Plasma level of hippurate increases in response to ingestion of sodium benzoate (Lennerz et al. 2015). The increase in plasma concentration of hippurate observed after an oral glucose load likely reflects the presence of the benzoate as preservative (Shaham et al. 2008). Sodium benzoate administration to overweight subjects in amounts that can be encountered in a normal diet (480 mg) has no effect on glucose homeostasis in a randomized controlled crossover study. Sodium benzoate exposure led to a fall in the plasma level of acetyl-glycine and a trend for glycine to fall (Lennerz et al. 2015).
In addition to benzoate, human glycine acyltransferase catalyzes the transfer of acyl groups from acyl-coA esters to glycine, generating acyl-glycine derivatives. Butyryl-coA, hexanoyl-coA, octanoyl-coA, decanoyl-coA, isovaleryl-coA, 3-methylcrotonyl-coA, 2-methylbutyryl-coA, and isobutyryl-coA are substrates for glycine acyltransferase, forming the corresponding acyl-glycine derivatives (Gregersen, Kolvraa, and Mortensen 1986). The formation of acyl-glycine derivatives is crucial to reduce the concentration of the active acyl-coA esters in patients affected with congenital deficiencies of enzymes involved in the oxidation of fatty acids and branched-chain amino acids. Plasma and urine accumulation of acyl-glycine moieties is used as diagnostic marker for these disorders. Methods to perform the quantitative analysis of urine acyl-glycines have been described (Hobert et al. 2016). In the leucine catabolic pathway, congenital deficiency of isovaleryl-coA dehydrogenase and 3-methylcrotonyl-coA carboxylase produces accumulation of isovaleryl-glycine and 3-methylcrotonyl-glycine, respectively. In the isoleucine catabolic pathway, congenital deficiency of short-branched chain acyl-coA dehydrogenase produces accumulation of 2-methylbutyryl-glycine. The urinary excretion of tiglyl-glycine is increased both in 17-β-hydroxy-steroid dehydrogenase type 10 deficiency and in 3-ketothiolase deficiency. The human enzyme that catalyzes the formation of tiglyl-glycine has not been investigated. In the valine catabolic pathway, the urinary excretion of isobutyryl-glycine is elevated in patients with isobutyryl-coA dehydrogenase deficiency (Gregersen and Mortensen 1986). In patients with medium-chain acyl-coA dehydrogenase deficiency, glycine conjugation is the major pathway for the disposal of C6 to C8 acyl moieties. L-carnitine supplementation increases the excretion of C6 to C10 acyl-carnitine esters and causes a 60% reduction in acyl-glycine excretion. However, even with L-carnitine supplementation, acyl-glycine excretion is 10 times greater than that of acyl-carnitine (Rinaldo et al. 1993).
Salicylate conjugates with glycine to generate salicylurate (2-hydroxyhippurate). It has been estimated that about 75–84% of ingested salicylate is excreted as glycine conjugate. Patients with salicylate overdose show lower plasma glycine concentration compared to healthy volunteers, suggesting depletion of available glycine after conjugation with salicylate. The urinary excretion of salicylurate is higher in patients who receive glycine compared to control subjects, suggesting that exogenous glycine increases the rate of formation of salicylurate in salicylate overdose (Patel et al. 1990). The enzyme that catalyzes the conjugation of salicylate and glycine has not been investigated.
Glycine acyltransferase-like-1
Glycine acyltransferase-like-1 utilizes glutamine as acceptor, being the first human glutamine N-acyltransferase reported. This enzyme catalyzes most effectively the transfer of phenylacetate from phenylacetyl-coA to glutamine (Matsuo et al. 2012). The physiological relevance of this enzyme is uncertain.
Glycine acyltransferase-like-2
Human glycine N-acyltransferase-like-2 catalyzes the transfer of acyl groups from oleoyl-coA, arachidonoyl-coA, linoleoyl-coA, decanoyl-coA, and lauroyl-coA to glycine, generating the corresponding acyl-glycines. Human glycine N-acyltransferase-like-2 is localized to the endoplasmic reticulum and its mRNA shows highest expression in salivary gland, trachea, spinal cord, and skin fibroblasts (Waluk and Hunt 2010). Lysine acetylation of human glycine N-acyltransferase-like-2 inhibits the enzyme activity (Waluk et al. 2012).
Bile acid-coA:amino acid N-acyltransferase
The bile acids cholic acid and chenodeoxycholic acid are primary bile acids with 24 carbon atoms (C24) synthesized in hepatocytes from cholesterol. Before their excretion into bile, they are first converted into their respective acyl-coA esters by acyl-coA synthetases and then conjugated with glycine or taurine. The enzyme bile acid-coA:amino acid N-acyltransferase catalyzes the conjugation (amidation) of C24 bile acids to glycine or taurine by transferring bile acid moieties from acyl-coA esters to glycine or taurine forming the corresponding glycine- or taurine-derivatives. This enzyme has been purified from human liver and its cDNA has been isolated and characterized. Bile acid-coA:amino acid N-acyltransferase is a monomeric enzyme sited in the cytosol that may utilize glycine and taurine as substrates (Johnson et al. 1991). Tissue expression studies show strong expression of bile acid-coA:amino acid N-acyltransferase in liver. In addition to catalyzing the conjugation of C24 bile acids, recombinant human bile acid-coA:amino acid N-acyltransferase conjugates other acyl-coA esters to glycine in vitro. Furthermore, this enzyme may possess activity as acyl-coA thioesterase. Acyl-coA thioesterases hydrolyze acyl-coA esters to fatty acids and free coenzyme A (coA-SH). Human bile acid-coA:amino acid N-acyltransferase shows sequence homology to type 1 acyl-coA thioesterases and thioesterase activity with saturated long- and very long-chain acyl-coA esters. Introduction of double bonds resulted in a strikingly lower activity (O’Byrne et al. 2003). The human gene BAAT located on 9q31.1 encodes bile acid-coA:amino acid N-acyltransferase.
Glycine contribution to the synthesis of creatine, heme group, glutathione, and collagen
Glycine is required for the synthesis of several physiologically relevant compounds, including creatine, heme group, glutathione, purines, and collagen.
Creatine synthesis
Phosphocreatine represents a reserve of intracellular high-energy phosphate crucial for brain function and skeletal muscle response to exercise that is generated through the phosphorylation of creatine by creatine kinase. Creatine is synthesized by a two-step biochemical pathway and glycine is required for the first step to take place.
The enzyme l-arginine:glycine amidinotransferase transfers an amidino group from l-arginine to glycine resulting in l-ornithine and guanidinoacetate. The crystal structure of human l-arginine:glycine amidinotransferase has been determined. The gene that encodes l-arginine:glycine amidinotransferase has been mapped to 15q15.3 (Humm et al. 1997). l-arginine:glycine amidinotransferase deficiency is a rare disease first described in 2001, clinically characterized by developmental delay, mental retardation, and myopathy. The concentration of guanidinoacetate in plasma and urine is low. Treatment with creatine supplement may prevent intellectual disability and may improve muscular function but has limited effect ameliorating established cognitive deterioration (Item et al. 2001). The enzyme guanidinoacetate methyltransferase catalyzes the second and last step in the synthesis de creatine, the transfer of a methyl group from S-adenosylmethionine to guanidinoacetate, forming creatine (Stockler-Ipsiroglu et al. 2015) Guanidinoacetate methyltransferase deficiency is a rare disease with an estimated incidence of 1:250,000 newborns, first described in 1996. In data collected from 48 patients, usual presentation occurs in infancy or childhood and the most frequent clinical manifestations are developmental delay, intellectual disability, epilepsy, movement disorders, speech delay and behavioral problems. Biochemical phenotype is characterized by elevated guanidinoacetate levels in blood, urine, and cerebrospinal fluid. Proton magnetic resonance spectroscopy shows reduced creatine in the basal ganglia. Treatment recommendations consist of supplements of creatine and ornithine, sodium benzoate to reduce plasma glycine level, and arginine-restricted diet. Treatment is usually associated with stabilization of symptoms, but does not normalize intellectual disability or guanidinoacetate accumulation in the central nervous system (Stockler-Ipsiroglu et al. 2014).
Heme synthesis
The first step in heme synthesis requires glycine and succinyl-co-A. The pyridoxal 5′-phosphate-dependent enzyme 5-aminolevulinate synthase catalyzes the decarboxylative condensation of glycine and succinyl-coA to yield 5-aminolevulinic acid, the precursor of the heme group. There are two isoenzymes of human 5-aminolevulinate synthase, one is ubiquitously expressed and the other is erythroid specific (Astner et al. 2005). A three-dimensional model of 5-aminolevulinate synthase from human erythrocytes (ALAS2) has been constructed (Shoolingin-Jordan et al. 2003). The ALAS2 gene located to Xp11.21 encodes the erythroid isoform. Mutations in this gene cause X-linked sideroblastic anemia (Astner et al. 2005).
Glutathione synthesis
Glutathione is a tripeptide (γ-glutamyl-cysteinyl-glycine) formed from glutamate, cysteine, and glycine. Two consecutive ATP-dependent enzymatic reactions lead to glutathione synthesis in humans. First, γ-glutamylcysteine synthetase (glutamate cysteine ligase) catalyzes the condensation of glutamate and cysteine to form γ-glutamylcysteine. Second, glutathione synthetase catalyzes the combination of glycine and γ-glutamylcysteine to generate glutathione (Njalsson et al. 2004; Lee et al. 2005). Human glutathione synthetase is a homodimer (Njalsson et al. 2004). The GSS gene located on chromosome 20q11.2 encodes glutathione synthetase (Lee et al. 2005). An alternative splicing variant of the GSS gene has been identified and detected in human colon, kidney, lung, liver, peripheral blood, uterus, and placenta. This variant is not found in heart, skeletal muscle, and spleen (Uchida et al. 2010). Congenital deficiency of glutathione synthetase is a rare disorder. Clinical presentation usually occurs in the neonatal period or infancy. The clinical phenotype varies widely including central nervous system dysfunction with motor disturbances and developmental delay and increased susceptibility to infections. Patients with this disease suffer from hemolytic anemia and metabolic acidosis in the neonatal period. Deficiency of glutathione synthetase leads to accumulation of γ-glutamylcysteine, which is converted to 5-oxoproline and cysteine by γ-glutamyl cyclotransferase. 5-oxoproline is excreted in urine (5-oxoprolinuria) (Dinescu et al. 2010). Selective tandem mass spectrometry-based newborn screening reveals an elevation of 5-oxoproline in dried blood samples (Simon et al. 2009).
Diabetic patients have lower erythrocyte concentration of glycine and glutathione compared with control subjects. In addition, patients with uncontrolled type 2 diabetes show deficient synthesis of glutathione that may be attributed to limited precursor availability, as dietary supplementation of the glycine and cysteine restores glutathione synthesis (Sekhar et al. 2011).
Purine synthesis
Glycine is required for de novo synthesis of purines providing two carbons and a nitrogen of the purine ring (Renwick and Baumann 1998; Lamers et al. 2009; Fan et al. 2014).
Collagen
Collagens are abundant extracellular proteins in humans. Each collagen polypeptide is designated α-chain. A mature collagen molecule contains three α chains arranged in a triple helix (superhelix). The collagen superhelix is stable because glycine residues are sited at every third position on the three α chains. Glycine moieties are critical to stabilize the three-chain superhelix. Any moiety other than glycine would impede the adjacent chains from coming together into the superhelix structure. Glycine has to be in every third residue to allow different chains to be in close proximity in the structure of collagen. Therefore, glycine is not only a major quantitative constituent of collagen, but also a fundamental residue required to form any type of healthy collagen structure in the human body. This attribute of glycine may be crucial to explain the development of medial calcification in the arteries of diabetic patients that results in elevated cardiovascular risk among these patients. Collagen is an important component of the arterial wall in humans. The media layer of human arteries contains collagen type I and type III. Type IV collagen is a constituent of basement membranes in the arterial wall (Shekhonin et al. 1985). In vitro studies using three-dimensional collagen hydrogels that mimic structural features of the atherosclerotic fibrous cap show that calcification morphology and normal collagen content of the plaque are interlinked, such that increasing the concentration of normal collagen within the hydrogels slows the calcification process compared to the samples without normal collagen. In human carotid atherosclerotic plaques, there is a similar inverse relationship between microcalcification area and normal collagen content within the fibrous cap. Normal collagen inhibits calcification formation and progression in human arteries. However, collagen degradation promotes initiation of calcification and allows microcalcification to progress to larger calcification spots (Hutcheson et al. 2016). The deficiency of plasma glycine observed in diabetic patients may lead to the formation of defective collagen in the vessel wall. The abnormal glycine-deficient collagen may promote subsequent calcification. Medial calcification and elevated cardiovascular risk are also typical features of chronic kidney failure. Metabolism of serine and glycine is profoundly altered in patients with kidney dysfunction, as healthy kidney synthesizes serine while extracting glycine from the blood stream. Plasma serine level is characteristically low in patients with chronic kidney disease whereas plasma glycine level is variable (Owen and Robinson 1963).
Summary
Plasma glycine level is lower in subjects with diabetes mellitus compared with healthy individuals and correlates positively with insulin sensitivity. Accordingly, serum glycine concentration increases after ameliorating insulin sensitivity with weight loss, exercise or drugs such as metformin. The pathophysiological mechanisms underlying glycine depletion and the clinical consequences of hypoglycinemia associated with insulin resistance remain to be elucidated.
Glycine is generated predominantly from glucose (via serine) and dietary betaine, but the activity of human serine hydroxymethyl transferase or other enzymes involved in glycine synthesis has not been investigated in conditions associated with inadequate insulin action and there is no evidence for a reduction in glycine synthesis among diabetic patients.
Glycine utilization is likely enhanced in patients with diabetes, as these subjects undergo excessive fatty acyl group formation. Glycine is an acceptor of acyl groups that buffers excess acyl groups generating acyl-glycine derivatives. Glycine consumption to this purpose may deplete free glycine pool and generate hypoglycinemia. It has been suggested that increased gluconeogenesis associated with diabetes leads to glycine consumption and might justify glycine depletion, but no biochemical pathway from glycine to glucose has been identified in humans.
The clinical consequences of glycine deficiency associated with insulin resistance are currently speculative. Epidemiological evidence suggests that diabetes mellitus is associated with increased frequency of some types of human cancer and in vitro studies indicate that the proliferation of cancer cells is supported by serine consumption. Disturbed glycine metabolism might contribute to explain cancer propensity in subjects with diabetes, although pathogenic mechanisms remain to be understood. Glycine is considered to be an inhibitory neurotransmitter to the nervous system, but the role of this amino acid in diabetic neuropathy has not been investigated. Glycine participates in the synthesis of sarcosine, creatine, heme group, and glutathione. Glycine depletion in patients with diabetes may jeopardize its contribution to these metabolic pathways, although the clinical repercussions are uncertain.
Glycine is a major quantitative component of collagen molecules. Furthermore, glycine is vital to stabilize the collagen molecule to form a triple helix. Therefore, glycine-deficient collagen is unstable and unable to maintain together the three chains that compose the collagen helix. This feature of glycine may be fundamental to explain the development of medial calcification that typically occurs in the arteries of diabetic patients. Glycine-lacking collagen in the arterial wall is likely predisposed to degradation and collagen debris promotes medial calcification, contributing to explain the elevated cardiovascular risk associated with diabetes. Consistently, serine and glycine metabolism is markedly altered in chronic kidney disease, a condition typically associated with medial calcification and high cardiovascular mortality as well.
References
Adibi SA (1980) Roles of branched-chain amino acids in metabolic regulation. J Lab Clin Med 95(4):475–484
Ahmad MS, Alsaleh M, Kimhofer T, Ahmad S, Jamal W, Wali SO, Nicholson JK, Damanhouri ZA, Holmes E (2017) Metabolic phenotype of obesity in a saudi population. J Proteome Res 16(2):635–644. https://doi.org/10.1021/acs.jproteome.6b00710
Anderson DD, Stover PJ (2009) SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLoS ONE 4(6):e5839. https://doi.org/10.1371/journal.pone.0005839
Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW (2005) Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J 24(18):3166–3177. https://doi.org/10.1038/sj.emboj.7600792
Augustin P, Hromic A, Pavkov-Keller T, Gruber K, Macheroux P (2016) Structure and biochemical properties of recombinant human dimethylglycine dehydrogenase and comparison to the disease-related H109R variant. FEBS J 283(19):3587–3603. https://doi.org/10.1111/febs.13828
Azize NA, Ngah WZ, Othman Z, Md Desa N, Chin CB, Md Yunus Z, Mohan A, Hean TS, Syed Zakaria SZ, Lock-Hock N (2014) Mutation analysis of glycine decarboxylase, aminomethyltransferase and glycine cleavage system protein-H genes in 13 unrelated families with glycine encephalopathy. J Hum Genet 59(11):593–597. https://doi.org/10.1038/jhg.2014.69
Baek JY, Jun DY, Taub D, Kim YH (2003) Characterization of human phosphoserine aminotransferase involved in the phosphorylated pathway of l-serine biosynthesis. Biochem J 373(Pt 1):191–200. https://doi.org/10.1042/bj20030144
Bar-joseph I, Pras E, Reznik-Wolf H, Marek-Yagel D, Abu-Horvitz A, Dushnitzky M, Goldstein N, Rienstein S, Dekel M, Pode-Shakked B, Zlotnik J, Benarrosh A, Gillery P, Hofliger N, Auray-Blais C, Garnotel R, Anikster Y (2012) Mutations in the sarcosine dehydrogenase gene in patients with sarcosinemia. Hum Genet 131(11):1805–1810. https://doi.org/10.1007/s00439-012-1207-x
Beagle B, Yang TL, Hung J, Cogger EA, Moriarty DJ, Caudill MA (2005) The glycine N-methyltransferase (GNMT) 1289 C → T variant influences plasma total homocysteine concentrations in young women after restricting folate intake. J Nutr 135(12):2780–2785
Benarrosh A, Garnotel R, Henry A, Arndt C, Gillery P, Motte J, Bakchine S (2013) A young adult with sarcosinemia. No benefit from long duration treatment with memantine. JIMD Rep 9:93–96. https://doi.org/10.1007/8904_2012_185
Bishop L, Kanoff R, Charnas L, Krenzel C, Berry SA, Schimmenti LA (2008) Severe methylenetetrahydrofolate reductase (MTHFR) deficiency: a case report of nonclassical homocystinuria. J Child Neurol 23(7):823–828. https://doi.org/10.1177/0883073808315410
Boden G, Rezvani I, Owen OE (1984) Effects of glucagon on plasma amino acids. J Clin Invest 73(3):785–793
Caudill MA, Dellschaft N, Solis C, Hinkis S, Ivanov AA, Nash-Barboza S, Randall KE, Jackson B, Solomita GN, Vermeylen F (2009) Choline intake, plasma riboflavin, and the phosphatidylethanolamine N-methyltransferase G5465A genotype predict plasma homocysteine in folate-deplete Mexican-American men with the methylenetetrahydrofolate reductase 677TT genotype. J Nutr 139(4):727–733. https://doi.org/10.3945/jn.108.100222
Cellini B, Bertoldi M, Montioli R, Paiardini A, Borri Voltattorni C (2007) Human wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: functional properties and physiological implications. Biochem J 408(1):39–50. https://doi.org/10.1042/bj20070637
Chen YM, Chen LY, Wong FH, Lee CM, Chang TJ, Yang-Feng TL (2000) Genomic structure, expression, and chromosomal localization of the human glycine N-methyltransferase gene. Genomics 66(1):43–47. https://doi.org/10.1006/geno.2000.6188
Cho HM, Jun DY, Bae MA, Ahn JD, Kim YH (2000) Nucleotide sequence and differential expression of the human 3-phosphoglycerate dehydrogenase gene. Gene 245(1):193–201
Collet JF, Gerin I, Rider MH, Veiga-da-Cunha M, Van Schaftingen E (1997) Human L-3-phosphoserine phosphatase: sequence, expression and evidence for a phosphoenzyme intermediate. FEBS Lett 408(3):281–284
Dinescu A, Brown TR, Barelier S, Cundari TR, Anderson ME (2010) The role of the glycine triad in human glutathione synthetase. Biochem Biophys Res Commun 400(4):511–516. https://doi.org/10.1016/j.bbrc.2010.08.081
Ding Y, Svingen GF, Pedersen ER, Gregory JF, Ueland PM, Tell GS, Nygard OK (2015) Plasma glycine and risk of acute myocardial infarction in patients with suspected stable angina pectoris. J Am Heart Assoc 5(1):e002621. https://doi.org/10.1161/jaha.115.002621
Ding Y, Pedersen ER, Svingen GF, Helgeland O, Gregory JF, Loland KH, Meyer K, Tell GS, Ueland PM, Nygard OK (2016) Methylenetetrahydrofolate dehydrogenase 1 polymorphisms modify the associations of plasma glycine and serine with risk of acute myocardial infarction in patients with stable angina pectoris in WENBIT (Western Norway B Vitamin intervention trial). Circ Cardiovasc Genet 9(6):541–547. https://doi.org/10.1161/circgenetics.116.001483
Drabkova P, Sanderova J, Kovarik J, Kandar R (2015) An assay of selected serum amino acids in patients with type 2 diabetes mellitus. Adv Clin Exp Med 24(3):447–451. https://doi.org/10.17219/acem/29223
Edgar AJ (2002) The human l-threonine 3-dehydrogenase gene is an expressed pseudogene. BMC Genet 3:18
Edgar AJ, Polak JM (2000) Molecular cloning of the human and murine 2-amino-3-ketobutyrate coenzyme A ligase cDNAs. Eur J Biochem 267(6):1805–1812
Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD (2014) Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510(7504):298–302. https://doi.org/10.1038/nature13236
Felig P, Marliss E, Cahill GF Jr (1969) Plasma amino acid levels and insulin secretion in obesity. N Engl J Med 281(15):811–816. https://doi.org/10.1056/nejm196910092811503
Felig P, Marliss E, Ohman JL, Cahill CF Jr (1970) Plasma amino acid levels in diabetic ketoacidosis. Diabetes 19(10):727–728
Feng Q, Kalari K, Fridley BL, Jenkins G, Ji Y, Abo R, Hebbring S, Zhang J, Nye MD, Leeder JS, Weinshilboum RM (2011) Betaine-homocysteine methyltransferase: human liver genotype-phenotype correlation. Mol Genet Metab 102(2):126–133. https://doi.org/10.1016/j.ymgme.2010.10.010
Fiehn O, Garvey WT, Newman JW, Lok KH, Hoppel CL, Adams SH (2010) Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE 5(12):e15234. https://doi.org/10.1371/journal.pone.0015234
Field MS, Kamynina E, Watkins D, Rosenblatt DS, Stover PJ (2015) Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proc Natl Acad Sci USA 112(2):400–405. https://doi.org/10.1073/pnas.1414555112
Floegel A, Stefan N, Yu Z, Muhlenbruch K, Drogan D, Joost HG, Fritsche A, Haring HU, Hrabe de Angelis M, Peters A, Roden M, Prehn C, Wang-Sattler R, Illig T, Schulze MB, Adamski J, Boeing H, Pischon T (2013) Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 62(2):639–648. https://doi.org/10.2337/db12-0495
Furujo M, Kinoshita M, Nagao M, Kubo T (2012) Methionine adenosyltransferase I/III deficiency: neurological manifestations and relevance of S-adenosylmethionine. Mol Genet Metab 107(3):253–256. https://doi.org/10.1016/j.ymgme.2012.08.002
Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, Ryals JA, Milburn MV, Nannipieri M, Camastra S, Natali A, Ferrannini E, RISC Study Group (2010) alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE 5(5):e10883. https://doi.org/10.1371/journal.pone.0010883
Garrow TA, Brenner AA, Whitehead VM, Chen XN, Duncan RG, Korenberg JR, Shane B (1993) Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J Biol Chem 268(16):11910–11916
Glynn EL, Piner LW, Huffman KM, Slentz CA, Elliot-Penry L, AbouAssi H, White PJ, Bain JR, Muehlbauer MJ, Ilkayeva OR, Stevens RD, Porter Starr KN, Bales CW, Volpi E, Brosnan MJ, Trimmer JK, Rolph TP, Newgard CB, Kraus WE (2015) Impact of combined resistance and aerobic exercise training on branched-chain amino acid turnover, glycine metabolism and insulin sensitivity in overweight humans. Diabetologia 58(10):2324–2335. https://doi.org/10.1007/s00125-015-3705-6
Gregersen N, Kolvraa S, Mortensen PB (1986) Acyl-CoA: glycine N-acyltransferase: in vitro studies on the glycine conjugation of straight- and branched-chained acyl-CoA esters in human liver. Biochem Med Metab Biol 35(2):210–218
Hart CE, Race V, Achouri Y, Wiame E, Sharrard M, Olpin SE, Watkinson J, Bonham JR, Jaeken J, Matthijs G, Van Schaftingen E (2007) Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet 80(5):931–937. https://doi.org/10.1086/517888
Hausler MG, Jaeken J, Monch E, Ramaekers VT (2001) Phenotypic heterogeneity and adverse effects of serine treatment in 3-phosphoglycerate dehydrogenase deficiency: report on two siblings. Neuropediatrics 32(4):191–195. https://doi.org/10.1055/s-2001-17373
Hobert JA, Liu A, Pasquali M (2016) Analysis by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Curr Protoc Hum Genet 91:17.25.1–17.25.12. https://doi.org/10.1002/cphg.19
Hol FA, van der Put NM, Geurds MP, Heil SG, Trijbels FJ, Hamel BC, Mariman EC, Blom HJ (1998) Molecular genetic analysis of the gene encoding the trifunctional enzyme MTHFD (methylenetetrahydrofolate-dehydrogenase, methenyltetrahydrofolate-cyclohydrolase, formyltetrahydrofolate synthetase) in patients with neural tube defects. Clin Genet 53(2):119–125
Holme E, Kjellman B, Ronge E (1989) Betaine for treatment of homocystinuria caused by methylenetetrahydrofolate reductase deficiency. Arch Dis Child 64(7):1061–1064
Humm A, Fritsche E, Steinbacher S, Huber R (1997) Crystal structure and mechanism of human l-arginine:glycine amidinotransferase: a mitochondrial enzyme involved in creatine biosynthesis. EMBO J 16(12):3373–3385. https://doi.org/10.1093/emboj/16.12.3373
Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S, Aikawa E (2016) Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater 15(3):335–343. https://doi.org/10.1038/nmat4519
Irving BA, Carter RE, Soop M, Weymiller A, Syed H, Karakelides H, Bhagra S, Short KR, Tatpati L, Barazzoni R, Nair KS (2015) Effect of insulin sensitizer therapy on amino acids and their metabolites. Metabolism 64(6):720–728. https://doi.org/10.1016/j.metabol.2015.01.008
Item CB, Stockler-Ipsiroglu S, Stromberger C, Muhl A, Alessandri MG, Bianchi MC, Tosetti M, Fornai F, Cioni G (2001) Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet 69(5):1127–1133. https://doi.org/10.1086/323765
Jaeken J, Detheux M, Van Maldergem L, Foulon M, Carchon H, Van Schaftingen E (1996) 3-Phosphoglycerate dehydrogenase deficiency: an inborn error of serine biosynthesis. Arch Dis Child 74(6):542–545
Jaeken J, Detheux M, Fryns JP, Collet JF, Alliet P, Van Schaftingen E (1997) Phosphoserine phosphatase deficiency in a patient with Williams syndrome. J Med Genet 34(7):594–596
Johnson MR, Barnes S, Kwakye JB, Diasio RB (1991) Purification and characterization of bile acid-CoA:amino acid N-acyltransferase from human liver. J Biol Chem 266(16):10227–10233
Kim HY, Heo YS, Kim JH, Park MH, Moon J, Kim E, Kwon D, Yoon J, Shin D, Jeong EJ, Park SY, Lee TG, Jeon YH, Ro S, Cho JM, Hwang KY (2002) Molecular basis for the local conformational rearrangement of human phosphoserine phosphatase. J Biol Chem 277(48):46651–46658. https://doi.org/10.1074/jbc.M204866200
Knowles L, Morris AA, Walter JH (2016) Treatment with mefolinate (5-methyltetrahydrofolate), but not folic acid or folinic acid, leads to measurable 5-methyltetrahydrofolate in cerebrospinal fluid in methylenetetrahydrofolate reductase deficiency. JIMD Rep 29:103–107. https://doi.org/10.1007/8904_2016_529
Lamers Y, Williamson J, Gilbert LR, Stacpoole PW, Gregory JF 3rd (2007) Glycine turnover and decarboxylation rate quantified in healthy men and women using primed, constant infusions of [1,2-(13)C2]glycine and [(2)H3]leucine. J Nutr 137(12):2647–2652
Lamers Y, Williamson J, Ralat M, Quinlivan EP, Gilbert LR, Keeling C, Stevens RD, Newgard CB, Ueland PM, Meyer K, Fredriksen A, Stacpoole PW, Gregory JF 3rd (2009) Moderate dietary vitamin B-6 restriction raises plasma glycine and cystathionine concentrations while minimally affecting the rates of glycine turnover and glycine cleavage in healthy men and women. J Nutr 139(3):452–460. https://doi.org/10.3945/jn.108.099184
Lee TD, Yang H, Whang J, Lu SC (2005) Cloning and characterization of the human glutathione synthetase 5′-flanking region. Biochem J 390(2):521–528. https://doi.org/10.1042/bj20050439
Lennerz BS, Vafai SB, Delaney NF, Clish CB, Deik AA, Pierce KA, Ludwig DS, Mootha VK (2015) Effects of sodium benzoate, a widely used food preservative, on glucose homeostasis and metabolic profiles in humans. Mol Genet Metab 114(1):73–79. https://doi.org/10.1016/j.ymgme.2014.11.010
Lim U, Peng K, Shane B, Stover PJ, Litonjua AA, Weiss ST, Gaziano JM, Strawderman RL, Raiszadeh F, Selhub J, Tucker KL, Cassano PA (2005) Polymorphisms in cytoplasmic serine hydroxymethyltransferase and methylenetetrahydrofolate reductase affect the risk of cardiovascular disease in men. J Nutr 135(8):1989–1994
Luka Z, Pakhomova S, Luka Y, Newcomer ME, Wagner C (2007) Destabilization of human glycine N-methyltransferase by H176 N mutation. Protein Sci 16(9):1957–1964. https://doi.org/10.1110/ps.072921507
Luka Z, Mudd SH, Wagner C (2009) Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J Biol Chem 284(34):22507–22511. https://doi.org/10.1074/jbc.R109.019273
Magnusson M, Wang TJ, Clish C, Engstrom G, Nilsson P, Gerszten RE, Melander O (2015) Dimethylglycine deficiency and the development of diabetes. Diabetes 64(8):3010–3016. https://doi.org/10.2337/db14-1863
Matsuo M, Terai K, Kameda N, Matsumoto A, Kurokawa Y, Funase Y, Nishikawa K, Sugaya N, Hiruta N, Kishimoto T (2012) Designation of enzyme activity of glycine-N-acyltransferase family genes and depression of glycine-N-acyltransferase in human hepatocellular carcinoma. Biochem Biophys Res Commun 420(4):901–906. https://doi.org/10.1016/j.bbrc.2012.03.099
Moolenaar SH, Poggi-Bach J, Engelke UF, Corstiaensen JM, Heerschap A, de Jong JG, Binzak BA, Vockley J, Wevers RA (1999) Defect in dimethylglycine dehydrogenase, a new inborn error of metabolism: NMR spectroscopy study. Clin Chem 45(4):459–464
Moro-Furlani AM, Turner VS, Hopkinson DA (1980) Genetical and biochemical studies on human phosphoserine phosphatase. Ann Hum Genet 43(4):323–333
Njalsson R, Carlsson K, Bhansali V, Luo JL, Nilsson L, Ladenstein R, Anderson M, Larsson A, Norgren S (2004) Human hereditary glutathione synthetase deficiency: kinetic properties of mutant enzymes. Biochem J 381(Pt 2):489–494. https://doi.org/10.1042/bj20040114
O’Byrne J, Hunt MC, Rai DK, Saeki M, Alexson SE (2003) The human bile acid-CoA:amino acid N-acyltransferase functions in the conjugation of fatty acids to glycine. J Biol Chem 278(36):34237–34244. https://doi.org/10.1074/jbc.M300987200
Okamura-Ikeda K, Hosaka H, Yoshimura M, Yamashita E, Toma S, Nakagawa A, Fujiwara K, Motokawa Y, Taniguchi H (2005) Crystal structure of human T-protein of glycine cleavage system at 2.0 a resolution and its implication for understanding non-ketotic hyperglycinemia. J Mol Biol 351(5):1146–1159. https://doi.org/10.1016/j.jmb.2005.06.056
Oppici E, Fargue S, Reid ES, Mills PB, Clayton PT, Danpure CJ, Cellini B (2015) Pyridoxamine and pyridoxal are more effective than pyridoxine in rescuing folding-defective variants of human alanine:glyoxylate aminotransferase causing primary hyperoxaluria type I. Hum Mol Genet 24(19):5500–5511. https://doi.org/10.1093/hmg/ddv276
Owen EE, Robinson RR (1963) Amino acid extraction and ammonia metabolism by the human kidney during the prolonged administration of ammonium chloride. J Clin Invest 42:263–276. https://doi.org/10.1172/JCI104713
Palmer ND, Stevens RD, Antinozzi PA, Anderson A, Bergman RN, Wagenknecht LE, Newgard CB, Bowden DW (2015) Metabolomic profile associated with insulin resistance and conversion to diabetes in the Insulin Resistance Atherosclerosis study. J Clin Endocrinol Metab 100(3):E463–E468. https://doi.org/10.1210/jc.2014-2357
Patel DK, Ogunbona A, Notarianni LJ, Bennett PN (1990) Depletion of plasma glycine and effect of glycine by mouth on salicylate metabolism during aspirin overdose. Hum Exp Toxicol 9(6):389–395. https://doi.org/10.1177/096032719000900606
Pelletier JN, MacKenzie RE (1994) Binding of the 2′,5′-ADP subsite stimulates cyclohydrolase activity of human NADP(+)-dependent methylenetetrahydrofolate dehydrogenase/cyclohydrolase. Biochemistry 33(7):1900–1906
Perseghin G, Ghosh S, Gerow K, Shulman GI (1997) Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes 46(6):1001–1009
Rebouche CJ, Engel AG (1980) Tissue distribution of carnitine biosynthetic enzymes in man. Biochim Biophys Acta 630(1):22–29
Renwick SB, Snell K, Baumann U (1998) The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure 6(9):1105–1116
Rinaldo P, Schmidt-Sommerfeld E, Posca AP, Heales SJ, Woolf DA, Leonard JV (1993) Effect of treatment with glycine and L-carnitine in medium-chain acyl-coenzyme a dehydrogenase deficiency. J Pediatr 122(4):580–584
Sekhar RV, McKay SV, Patel SG, Guthikonda AP, Reddy VT, Balasubramanyam A, Jahoor F (2011) Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 34(1):162–167. https://doi.org/10.2337/dc10-1006
Shaham O, Wei R, Wang TJ, Ricciardi C, Lewis GD, Vasan RS, Carr SA, Thadhani R, Gerszten RE, Mootha VK (2008) Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol Syst Biol 4:214. https://doi.org/10.1038/msb.2008.50
Shekhonin BV, Domogatsky SP, Muzykantov VR, Idelson GL, Rukosuev VS (1985) Distribution of type I, III, IV and V collagen in normal and atherosclerotic human arterial wall: immunomorphological characteristics. Coll Relat Res 5(4):355–368
Shoolingin-Jordan PM, Al-Daihan S, Alexeev D, Baxter RL, Bottomley SS, Kahari ID, Roy I, Sarwar M, Sawyer L, Wang SF (2003) 5-Aminolevulinic acid synthase: mechanism, mutations and medicine. Biochim Biophys Acta 1647(1–2):361–366
Simon E, Vogel M, Fingerhut R, Ristoff E, Mayatepek E, Spiekerkotter U (2009) Diagnosis of glutathione synthetase deficiency in newborn screening. J Inherit Metab Dis 32(Suppl 1):S269–S272. https://doi.org/10.1007/s10545-009-1213-x
Stockler-Ipsiroglu S, van Karnebeek C, Longo N, Korenke GC, Mercimek-Mahmutoglu S, Marquart I, Barshop B, Grolik C, Schlune A, Angle B, Araujo HC, Coskun T, Diogo L, Geraghty M, Haliloglu G, Konstantopoulou V, Leuzzi V, Levtova A, Mackenzie J, Maranda B, Mhanni AA, Mitchell G, Morris A, Newlove T, Renaud D, Scaglia F, Valayannopoulos V, van Spronsen FJ, Verbruggen KT, Yuskiv N, Nyhan W, Schulze A (2014) Guanidinoacetate methyltransferase (GAMT) deficiency: outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab 111(1):16–25. https://doi.org/10.1016/j.ymgme.2013.10.018
Stockler-Ipsiroglu S, Apatean D, Battini R, DeBrosse S, Dessoffy K, Edvardson S, Eichler F, Johnston K, Koeller DM, Nouioua S, Tazir M, Verma A, Dowling MD, Wierenga KJ, Wierenga AM, Zhang V, Wong LJ (2015) Arginine:glycine amidinotransferase (AGAT) deficiency: clinical features and long term outcomes in 16 patients diagnosed worldwide. Mol Genet Metab 116(4):252–259. https://doi.org/10.1016/j.ymgme.2015.10.003
Stover PJ, Chen LH, Suh JR, Stover DM, Keyomarsi K, Shane B (1997) Molecular cloning, characterization, and regulation of the human mitochondrial serine hydroxymethyltransferase gene. J Biol Chem 272(3):1842–1848
Swanson MA, Coughlin CR Jr, Scharer GH, Szerlong HJ, Bjoraker KJ, Spector EB, Creadon-Swindell G, Mahieu V, Matthijs G, Hennermann JB, Applegarth DA, Toone JR, Tong S, Williams K, Van Hove JL (2015) Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol 78(4):606–618. https://doi.org/10.1002/ana.24485
Takashina C, Tsujino I, Watanabe T, Sakaue S, Ikeda D, Yamada A, Sato T, Ohira H, Otsuka Y, Oyama-Manabe N, Ito YM, Nishimura M (2016) Associations among the plasma amino acid profile, obesity, and glucose metabolism in Japanese adults with normal glucose tolerance. Nutr Metab (Lond) 13:5. https://doi.org/10.1186/s12986-015-0059-5
Thalacker-Mercer AE, Ingram KH, Guo F, Ilkayeva O, Newgard CB, Garvey WT (2014) BMI, RQ, diabetes and sex affect the relationships between amino acids and clamp measures of insulin action in humans. Diabetes 63(2):791–800. https://doi.org/10.2337/db13-0396
Uchida M, Sugaya M, Kanamaru T, Hisatomi H (2010) Alternative RNA splicing in expression of the glutathione synthetase gene in human cells. Mol Biol Rep 37(4):2105–2109. https://doi.org/10.1007/s11033-009-9675-3
Vanek V, Budesinsky M, Kabeleova P, Sanda M, Kozisek M, Hanclova I, Mladkova J, Brynda J, Rosenberg I, Koutmos M, Garrow TA, Jiracek J (2009) Structure-activity study of new inhibitors of human betaine-homocysteine S-methyltransferase. J Med Chem 52(12):3652–3665. https://doi.org/10.1021/jm8015798
Wahren J, Felig P, Cerasi E, Luft R (1972) Splanchnic and peripheral glucose and amino acid metabolism in diabetes mellitus. J Clin Invest 51(7):1870–1878. https://doi.org/10.1172/jci106989
Walford GA, Ma Y, Clish C, Florez JC, Wang TJ, Gerszten RE (2016) Metabolite profiles of diabetes incidence and intervention response in the diabetes prevention program. Diabetes 65(5):1424–1433. https://doi.org/10.2337/db15-1063
Waluk DP, Schultz N, Hunt MC (2010) Identification of glycine N-acyltransferase-like 2 (GLYATL2) as a transferase that produces N-acyl glycines in humans. Faseb J 24(8):2795–2803. https://doi.org/10.1096/fj.09-148551
Waluk DP, Sucharski F, Sipos L, Silberring J, Hunt MC (2012) Reversible lysine acetylation regulates activity of human glycine N-acyltransferase-like 2 (hGLYATL2): implications for production of glycine-conjugated signaling molecules. J Biol Chem 287(20):16158–16167. https://doi.org/10.1074/jbc.M112.347260
Wang Q, Lu K, Du H, Zhang Q, Chen T, Shu Y, Hua Y, Zhu L (2014) Association between cytosolic serine hydroxymethyltransferase (SHMT1) gene polymorphism and cancer risk: a meta-analysis. Biomed Pharmacother 68(6):757–762. https://doi.org/10.1016/j.biopha.2014.08.002
Wang-Sattler R, Yu Z, Herder C, Messias AC, Floegel A, He Y, Heim K, Campillos M, Holzapfel C, Thorand B, Grallert H, Xu T, Bader E, Huth C, Mittelstrass K, Doring A, Meisinger C, Gieger C, Prehn C, Roemisch-Margl W, Carstensen M, Xie L, Yamanaka-Okumura H, Xing G, Ceglarek U, Thiery J, Giani G, Lickert H, Lin X, Li Y, Boeing H, Joost HG, de Angelis MH, Rathmann W, Suhre K, Prokisch H, Peters A, Meitinger T, Roden M, Wichmann HE, Pischon T, Adamski J, Illig T (2012) Novel biomarkers for pre-diabetes identified by metabolomics. Mol Syst Biol 8:615. https://doi.org/10.1038/msb.2012.43
Xie W, Wood AR, Lyssenko V, Weedon MN, Knowles JW, Alkayyali S, Assimes TL, Quertermous T, Abbasi F, Paananen J, Häring H, Hansen T, Pedersen O, Smith U, Laakso M, Dekker JM, Nolan JJ, Groop L, Ferrannini E, Adam KP, Gall WE, Frayling TM, Walker M, MAGIC Investigators, DIAGRAM Consortium, GENESIS Consortium, and RISC Consortium (2013) Genetic variants associated with glycine metabolism and their role in insulin sensitivity and type 2 diabetes. Diabetes 62(6):2141–2150. https://doi.org/10.2337/db12-0876
Yan-Do R, Duong E, Manning Fox JE, Dai X, Suzuki K, Khan S, Bautista A, Ferdaoussi M, Lyon J, Wu X, Cheley S, MacDonald PE, Braun M (2016) A glycine-insulin autocrine feedback loop enhances insulin secretion from human β-cells and is impaired in type 2 diabetes.”. Diabetes 65(8):2311–2321
Zhao H, Shen J, Djukovic D, Daniel-MacDougall C, Gu H, Wu X, Chow WH (2016) Metabolomics-identified metabolites associated with body mass index and prospective weight gain among Mexican American women. Obes Sci Pract 2(3):309–317. https://doi.org/10.1002/osp4.63
Zhong SL, Zhang J, Hu Q, Chen WX, Ma TF, Zhao JH (2014) C1420T polymorphism of cytosolic serine hydroxymethyltransferase and risk of cancer: a meta-analysis. Asian Pac J Cancer Prev 15(5):2257–2262
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
There was no financial support for this work.
Additional information
Handling Editor: J. D. Wade.
Rights and permissions
About this article

Cite this article
Adeva-Andany, M., Souto-Adeva, G., Ameneiros-Rodríguez, E. et al. Insulin resistance and glycine metabolism in humans. Amino Acids 50, 11–27 (2018). https://doi.org/10.1007/s00726-017-2508-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-017-2508-0