
Abstract
COVID-19 pandemia is a major health emergency causing hundreds of deaths worldwide. The high reported morbidity has been related to hypoxia and inflammation leading to endothelial dysfunction and aberrant coagulation in small and large vessels. This review addresses some of the pathways leading to endothelial derangement, such as complement, HIF-1α, and ABL tyrosine kinases. This review also highlights potential targets for prevention and therapy of COVID-19-related organ damage and discusses the role of marketed drugs, such as eculizumab and imatinib, as suitable candidates for clinical trials.
Similar content being viewed by others

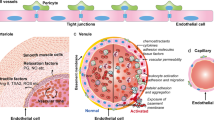

Introduction
The new coronavirus (SARS-COV-2) pandemic has rapidly spread to many countries [1] and caused several thousand deaths. Endothelial derangement and increased permeability are reported to be early hallmarks of organ damage in patients with COVID-10, such as lungs, kidney, and myocardium. Moreover, thrombotic complications are a relevant cause of death in patients with COVID-19. Finally, the interaction of SARS-CoV-2 with ACE2 possibly implies alterations of angiotensin II plasma levels. Therefore, the vascular system is increasingly being addressed as a major therapeutic target for defeating COVID-19 [2, 3].
The present paper in particular reviews some of the pathways leading to endothelial disruption in the course of COVID-19 infection, namely complement activation, hypoxia, platelets, and thyroxin kinases. Furthermore, the paper explores potential therapeutic strategies, particularly commercially available drugs, such as imatinib.
Complement-mediated pathogenesis
Complement system and inflammation
Complement system is a 30 protein network belonging to both the innate and the adoptive immune system: it has opsonization properties, but it also enhances the activity of antibodies and macrophages in eliminating pathogens and damaged cells. C5a anaphylatoxin, a central complement protein, is involved in sepsis and acute lung injury mediated by CC-chemokine receptor 5 [4, 5]. C5a leads to exaggerated early proinflammatory responses and activation of neutrophils and macrophages (by the activation of PI3K/Akt and MAPK signaling pathways), with subsequent release of histones and reactive oxygen species that ultimately lead to endothelial damage, inflammation, and thrombosis; a functional decline of the innate immune system and multiorgan dysfunction follows [6, 7]. Moreover, C5a is involved in pyroptosis, which is a form of programmed cell death by a lytic modality catalyzed by caspase and aimed at eliminating immune cells infected by pathogens. Due to cell membrane break, pyroptosis induces an inflammatory damage and release of caspase, which activates interleukin-1. C5a itself also causes substantial cardiomyocyte dysfunction, which is reversed by complement blockade [8].
Complement and lung injury
Also, C3a complement fraction plays a relevant role in the pathogenesis of infection-related lung injury: high serum C3a predicts evolution to ARDS [9, 10], while both C3a and C5a increase endothelial permeability and activate endothelial cells, thereby increasing the expression of adhesion molecules and cytokines [11, 12], and the distal complement activation product C5 b-9 triggers intracellular fluxes of calcium in epithelial and endothelial cells. The vicious cycle might be sustained by hypoxia, which reduces the expression of CD55, a complement regulator, by HIF-1alpha, IL-2, and TNFα. This downregulation enhances C3a release and caspase 3 deposition onto endothelial cells [13]. The complement damage may also be triggered by some specific infective agents, such as staphylococcus, that provoke shedding of anti-complement surface proteins CD55 and CD59 from endothelial cells thus favoring cell lysis [14].
Complement and vascular damage
Complement also proved to be relevant for survival in patients with heart failure and sepsis-related miocardiopathy [15, 16]. Moreover, tocilizumab proved to reduce C5A receptor in myocardial infarction patients; therefore, its anti-cytokine efficacy in COVID-19 may be partly mediated by attenuation of complement hyperactivation [17]. In fact, SARS-CoV can directly activate complement system by lectin way and particularly acts onto T-lymphocytes in hypertensive patients, since an increased expression of C5a receptor has been reported in Treg Foxp3+ lymphocytes from these individuals [18]. Tissue damage by complement activation seems to be a common way to many pathogen viruses, including H1N1 influenza, SARS, CoV-2, and MERS-CoV [19,20,21,22,23,24]. Preliminary data show that C5a mediates MERS-CoV and SARS-CoV-2-induced pyroptosis in infected macrophage, dendritic cells, and CD4+ lymphocytes [24, 25]. C3 knockout mice proved to be resistant to organ damage induced by SARS-CoV, while the high serum and pulmonary concentrations of C5a and C5b-9 reported in mice infected with MERS-CoV and SARS-CoV could not cause lung damage if a C5a receptor inhibitor was administered [23, 25]. Inhibition of C5a or C3a receptor reduced lung damage and prolonged survival also in mice infected with H1N1 influenza virus [26]. Moreover, a reduced MERS-CoV viral replication was observed after inhibition of C5a receptor [23].
Anti-complement drugs
Specific inhibition of C5a biological activity could gain therapeutic benefit without affecting the protective immune response. In the last few years, several peptide and non-peptide antagonists of C5a have been discovered and tested in relevant pharmacological models.
Treatment with eculizumab, the first-in-class terminal complement inhibitor, reported a rate of meningococcal infection of 0.25/100 patient years and no deaths in patients temporarily treated with the drugs, i.e., patients with atypical hemolytic uremic syndrome, while the risk of meningococcus-related death was 3/10,000 patient years in chronically treated individuals [27]. Eculizumab was successfully used to treat virus-related hemolytic uremic syndrome (cytomegalovirus, human immunodeficiency virus, herpes zoster virus, influenza B virus) with no reported immunosuppressive effects [28,29,30]. The SOLID-C19 (NCT04288713) expanded access study is currently testing 900 mg administration of eculizumab weekly in patients with COVID-19 ARDS, and the PANAMO phase II study in The Netherlands (NCT04333420) is currently testing IFX-1, an anti-complement (C5a) monoclonal antibody.
Endothelial damage
Endothelial cells represent one-third of overall lung cells. Baseline endothelial damage may be chronically caused by increased adiponectins in diabetic and obese patients: this effect is related to the activation of inflammosome NLRP3 and autocrine production of IL-1β [31]. Additional damage to pulmonary endothelial dysfunctional cells is acutely provoked by infections and in turn causes excess thrombin generation and reduced fibrinolysis [32,33,34]. In fact, most dangerous infective agents, such as Dengue 2, adopt an adiponectin-like mechanism of endothelial damage [35].
Thrombin causes further endothelial damage, which can be prevented in vitro by CXCR4 agonists, such as ubiquitin [36]. Moreover, hypoxia may lead to increased hypoxia inducible factor-1α (HIF-1α) expression and hypercoagulability [37]. Therefore, a higher rate of thrombotic episodes is reported in patients with COVID-19 pneumonia, while increased vascular permeability seems to be strongly related to increased thrombosis. In particular, in patients with lymphopenic pneumonia and organ failure increased vascular permeability was strongly correlated with severe lymphopenia [30].
Lung computed tomography of patients infected with SARS-CoV-2 shows an earlier interstitial edema followed by a subsequent alveolar edema, which questioned the similarity of COVID-19 lung damage and ARDS. In pathology samples from COVID-19 patients, diffuse microcirculatory and macrovascular thrombosis have been detected in lung tissue, which is not typical for ARDS, and when a few endothelial cells were directly infected by SARS, several macrophages were instead [38]. However, no evidence of vasculitis has been reported by pathologists and no laboratory signs of diffuse intravascular coagulopathy were found in most of the patients, since antithrombin-III, fibrinogen, and platelet count rarely decline in the first phases of the disease, while D-dimer usually shows a progressive increase during hospital stay and predicts a higher mortality [39]. Angiopoietin 2, thrombomodulin, ICAM 1, endothelin, and E-selectin are usually increased in cases of endothelial damage; however, quantitative data from COVID-19 patients are still awaited. Rather, severe early hypoalbuminemia and subsequent increased CPK, myoglobin, creatinine, and uric acid are observed in the majority of patients with COVID-19, resembling systemic capillary leak syndrome and explaining some cases of renal failure. Hypoalbuminemia is also a relevant prognostic factor of the lung injury prediction score in sepsis [40].
Apoptosis of human pulmonary microvascular endothelial cell may be chronically triggered by inflammation, such as in COPD, or acutely induced by ARDS; the latter is mediated by Bruton kinase (BTK), IL-17, and macrophage stimulating-1, while IL-35 seems protective [41,42,43,44]. LDH is typically released into the serum by apoptotic endothelial cells [42]. A different kind of damage to lung microvascular endothelial cells, namely autophagy, may be induced by NADPH oxidase 2 during viral infections, but prevented by NADPH oxidase 4 as a protecting way against oxidative stress [45, 46]. Furthermore, endothelial cells of lymph vessels in the lungs are particularly sensible to oxidative stress and might be the most damaged cell population in SARS-CoV-2 infection [47].
Drugs and endothelium
Several drugs can damage the endothelium. Endothelial damage secondary to ponatinib is mediated by NOTCH1 hyperactivation, but also propranolol and sirolimus inhibit endothelial proliferation and carteolol induces apoptosis in corneal endothelial cells by caspase- and mitochondria-dependent pathways [48,49,50]. Also, ACE2, a SARS-Cov-2 target, inhibits proliferation of endothelial cells; however, it also reduces endothelial inflammation [51]. Finally, steroids induce apoptosis in bone endothelial cells causing osteonecrosis, but such an effect has not been proven in pulmonary capillary cells.
A shorter list of drugs prevent endothelial apoptosis, namely recombinant adenosine deaminase, registered for SCID ADA deficient patients, casin, and plerixafor, which markedly expand endothelial proliferation in vitro, inducing also tubing and sprouting [51, 52]. However, cyclin-dependent kinases are upregulated by plerixafor, and endothelial permeability is therefore reduced [36].
Adrecizumab is a first-in-class monoclonal antibody targeting ad stabilizing plasma bioactive adrenomedullin (bio-ADM ®), a key hormone secreted by endothelial cells showing vasoprotective activity, preserving endothelial barrier function (i.e., keeping tight junctions) and preventing vascular leakage (ref shock 2018)[53]. Conversely, in the interstitium ADM has dangerous vasodilatory properties at high concentrations. The ongoing AdrenOSS-2 trial (NTC03085758) has enrolled 301 patients with early septic shock and high serum levels of bio-ADM: patients were randomized to receive adrecizumab or placebo in addition to standard care. Preliminary data confirmed the treatment safety (BJCP phase I) and showed a trend to a reduced 28-day mortality as compared with 28% in the placebo group (www.adrenomed.com).
Platelets
The role of platelets in inducing or amplifying the endothelial damage in COVID-19 patients is unknown. A low platelet count, possibly due to consumption, bone marrow infection, or autoimmune phenomena, was reported to increase COVID-19 mortality rate by five-fold, even though the rates were very heterogeneous among the analyzed studies [54,55,56]. Moreover, the opposite is more common in COVID-19 patients, being its platelet counts quite high or higher than in patients with sepsis or ARDS; increased serum levels of thrombopoietin caused by pulmonary inflammation have been supposed to explain this phenomenon [57].
Anti-thrombotic drugs
Organ damage to skin, CNS, heart, and kidney appears to be related to transient thrombotic or ischemic phases (hypoperfusion, hypoxemia). Heparin proved to protect tight junctions in alveolar endothelial cells from IL-6 induced hyperpermeability and thus may possibly improve the outcome of COVID-19 patients [39, 58]. It also antagonizes histones released from damaged cells by histone methylation and interaction with MAPK and NF-kB pathways [59, 60]. Furthermore, heparin has well-documented anti-inflammatory properties that include binding to inflammatory cytokines, inhibiting neutrophil chemotaxis and migration, neutralizing the positively charged C5a, and sequestering acute phase proteins [61]. Heparin polyanionic nature also seems to interact with SARS-CoV-2 spike S1 protein receptor binding domain (https://www.biorxiv.org/content/10.1101/2020.02.29.971093v1.full). Several COVID-19 cases reported atypical thromboses (renal veins, uterine veins, mesenteric vessels) and myocardial micro-thrombotic vessels; however, a large portion of thromboembolic events occurred in patients receiving prophylactic heparin doses. Furthermore, some centers reported the need for high heparin doses to achieve a target aPTT ratio in these patients and no standard level of anticoagulation has yet been established. Early, i.e., domiciliary, antithrombotic prophylaxis, and subsequent dose titration according to ARDS severity still need to be validated, especially in selected patients, such as older patients, those receiving antiplatelet therapies, and those with renal failure.
Defibrotide and rTPA have also been proposed to rescue patients with severe thrombotic events. Full (UFH 15,000 U/day or enoxaparin 60 mg/day) or intermediate (UFH 10,000/day or enoxaparin 40 mg/day) heparin dose was reported to benefit COVID-19 patients with higher platelet counts and higher D-dimer levels [57]. An English trial is currently assigning patients to either aspirin 75 mg, clopidogrel 75 mg, rivaroxaban 2.5 mg, or omeprazole versus standard of care, in order to prevent cardiac complications of COVID-19 8 (NCT04333407). Other antiplatelet agents, such as prasugrel, a P2Y12 inhibitor, proved to reduce prost-myocardial infarction inflammation and might be a potential drug to be tested in the context of COVID-19-induced endothelial inflammation [62].
Hypoxia-mediated endothelial damage
Also, hypoxia is expected to play a central role in leading inflammation and endothelial activation: cells adapt to hypoxia by activating hypoxia-inducible factors HIF-1 and HIF-2 which in turn induce the expression of a number of genes promoting energy metabolism and cell survival and specifically endothelial cell adaptation (migration, growth, differentiation) (Whyte–hypoxia). A transition from HIF-1 to HIF-2 governs adaptation from acute to prolonged hypoxia, despite most genes may be regulated by both [63]. HIF-2α upon activation increases the expression of vascular-endothelial protein thyrosine phosphatase, which in turn decreases VE-caderin phosphorylation, supporting the integrity of adherens junctions and preventing loss of endothelial barrier function [64]. Contrarily, expression of HIF-1α in alveolar epithelial cells enhances lung inflammation in a NF-kB mediated way [65] and favors a cell-mediated inflammation (CD4+ CD8+) and proinflammatory cytokines (IL-2 and TNFα), which proportionately downregulate CD55 and augment complement-mediated endothelial damage [8]. Moreover, myeloid cell HIF-1α is a key driver of myeloid cell response in hypoxic and inflammatory microenvironments by modulating cellular energetics, upregulating glycolytic enzymes and glucose transporters to permit ATP generation under conditions of hypoxia, and preventing apoptosis of innate immunity cells. In chronic infections, however, HIF-1α was reported to prevent excessive lymphocyte recruitment into lung interstitium and avert immunopathological consequences to the host [66]. Despite there are no currently marketed drugs achieving a selective control of HIF-1α, imatinib proved to reduce HIF-1α levels and pulmonary hypertension caused by chronic hypoxia [67, 68].
The activation and damage of pulmonary endothelium is a hallmark of ARDS, and ACE+ circulating endothelial microparticles are markers of pulmonary endothelial injury predicting development of ARDS in septic patients [69]. Endothelial damage has been shown to induce the recruitment of circulating endothelial cells: circulating mature endothelial cells increased in patients with moderate or severe ARDS as compared with mild ARDS or non-ARDS septic patients [70]. Increased circulating endothelial progenitor cells (EPC) proved to positively correlate with survival [71]. However, also overall CD34+ CD45+ circulating cell count is associated with improved survival in sepsis-related ARDS. Inhaled nitric oxide was reported to induce mobilization of endothelial cell progenitors from bone marrow into circulation, contributing to repair lung vascular injury in vivo, while autotransplanted endothelial progenitor cells protected mice from LPS-induced lung injury [72, 73]. No data are yet available regarding phase-related mobilization of mature or progenitor endothelial cells during COVID-19 infection. Beta-blockers favor migration of endothelial progenitor cells mediated by SDF-1 thanks to the inhibition of beta2adrenergic receptors onto circulating mononuclear cells and bone marrow stem cells [74].
Finally, an increased synthesis of hyaluronic acid seems to explain the huge shift of liquids into the interstitium of patients with SARS-CoV-2-related endothelial damage and respiratory failure. This phenomenon, however, might be counteracted by marketed hymecromone or intranasal hyaluronidase [75,76,76,77].
Imatinib
The permeabilizing action of VEGF on the endothelium of the pulmonary capillaries, typical of ARDS, can be counteracted by inhibiting ABL2 (Arg). Imatinib is an ABL1 and ABL2 tyrosine kinase inhibitor with EMA designation of orphan drug for the treatment of ARDS (January 12, 2015) by virtue of its demonstrated in vitro and in vivo ability to attenuate the expression of VCAM-1 in endothelial cells, restoring VE junctions-caderine and thus reducing the ARDS secondary to sepsis, mechanical ventilation, and ischemia and mediated by VEGF, thrombin, and histamine [78,79,80,81]. Also, the anti-cytokine action of imatinib, mediated by the inhibition of the NF-kB pathway in myeloid-derived cells, has been widely demonstrated with efficacy in asthma, rheumatoid arthritis, systemic sclerosis, and pulmonary hypertension [82, 83]. Several cases of acute lung inflammation induced by toxic agents (bleomycin, gemcitabine) or sepsis got rapid clinical improvement after treatment with low doses of imatinb [84, 85]. Furthermore, the immunomodulatory action of imatinib is proven: the inhibition exerted on c-Kit in bone marrow stem cells favors the mobilization of myeloid cells already at low doses [86]. In primates with concomitant HIV infection and Mycobacterium tuberculosis, imatinib has demonstrated an effective action directly to the host [87]. A phase II clinical trial (IMPACT-TB, NCT03891901) is enrolling patients with mycobacteriosis TB (± HIV) to concomitant imatinib therapy.
Finally, a direct anti-viral action of imatinib has also been documented: it appears to inhibit the fusion of SARS-CoV2 with the late endosome membrane at micromolar doses (EC 50 9.8 μM/L), a phase necessary for subsequent viral replication [88,89,90]. Based on the above rational, the COUNTER-COVID-randomized, placebo-controlled, clinical trial is currently enrolling in Amsterdam (EudraCT number, 2020-001236-10).
Discussion
Several pathways have been identified as possible drivers in the pathogenesis of SARS-CoV-2: some pathways, such as the complement, may be targeted by available drugs, while some others are orphan of specific therapies, such as HIF-1α and HIF-2α. Several drugs currently marketed for blood diseases, such as imatinib, ruxolitinib, heparins, tocilizumab, and eculizumab, are currently being tested for patients with COVID-19 symptoms. Hematologists, who are expert on such drugs, are therefore called into multidisciplinary teams for managing patient selection and therapy monitoring. While awaiting vaccines and effective antiviral therapies, their expertise needs to be combined with the updated evidence on the pathogenesis of COVID-19, as they have the chance to contribute to amelioration of patient outcomes. Appropriate trial designs still need to be developed in order to address some fundamental issues, such as COVID-19 patient stratification and testing of multiple drugs. Furthermore, trials are challenged by heterogeneous support therapies for COVID-19 and partial non-comparability of patients belonging to different clinical centers. Nevertheless, most of the above cited candidate drugs have a favorable safety profile, especially in the short term, and are promising tools for defeating COVID-19.
References
Siordia JA (2020) Epidemiology and clinical features of COVID-19: a review of current literature. J Clin Virol 127:104357
Giannis D, Ziogas IA, Gianni P (2020) Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERSD-CoV and lessons from the past. J Virol 127:104362
Esher R, Breakey N, Lammle B (2020) Severe COVID-19 infection associated with endothelial activation. Thromb Res 190:62
Fattahi F, Zetoune FS, Ward PA (2020) Complement as a major induced of harmful events in infectious sepsis. Shock. https://doi.org/10.1097/SHK.0000000000001531
Russkamp NF, Ruemmler R, Roewe J, Moore BB, Ward PA, Bosmann M (2015) Experimental design of complement component C5a-induced acute lung injury (C5a-ALI): a role of CC-chemokine receptor type 5 during immune activation by anaphylatoxin. FASEB J 29:3762–3772
Barrett CD, Hsu AT, Ellson CD et al (2018) Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clin Exp Immunol 194:103–117
Ward PA, Fattahi F, Bosmann M (2016) New insights into molecular mechanisms of immune complex-induced injury in lung. Front Immunol 7:86
Fattahi F, Kalbitz M, Malan EA, Abe E, Jajou L, Huber-Lang MS, Bosmann M, Russell MW, Zetoune FS, Ward PA (2017) Complement-induced activation of MAPKs and Akt during sepsis: role in cardiac dysfunction. FASEB J 31:4129–4139
Donnelly TJ, Meade P, Jagels M et al (1994) Cytokine, complement, and endotoxin profiles associated with the development of the adult respiratory distress syndrome after severe injury. Crit Care Med 22:768–776
Ip WKE, Chan KH, Law HKW, Tso GHW, Kong EKP, Wong WHS, To YF, Yung RWH, Chow EY, Au KL, Chan EYT, Lim W, Jensenius JC, Turner MW, Peiris JSM, Lau YL (2005) Mannose-binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis 191:1697–1704
Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, Re F (2018) Caspase-1-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog 14:e1007105
Wang YC, Liu QX, Liu T, Xu XE, Gao W, Bai XJ, Li ZF (2018) Caspase-1-dependent pyroptosis of peripheral blood mononuclear cells predicts the development of sepsis in severe trauma patients: a prospective observational study. Medicine (Baltimore) 97:e9859
Khan MA, Shamma T, Kazmi S, Altuhami A, Ahmed HA, Assiri AM, Broering DC (2020) Hypoxia-induced complement dysregulation is associated with microvascular impairments in mouse tracheal transplants. J Transl Med 18:147
Lin YC, Liao YJ, Lee YH (2019) Staphylococcal phosphatidylinositol-specific phospholipase C potentiates lung injury via complement sensitization. Cell Microbiol 21:e13085
Fattahi F, Frydrych LM, Bian G (2018) Role of complement C5a and histones in septic cardiomyopathy. Mol Immunol 102:32–41
Orrrem HL, Nilsson PH, Piscke SE (2018) IL-6 receptor inhibition by tocilizumab attenuated expression of C5a receptor 1 and 2 in non-ST-elevation myocardial infarction. Front Immunol 9:2035
Wang W, Cai D (2020) Complement components sC5b-9 and CH50 predict prognosis in heart failure patients combined with hypertension. Am J Hypertens 33:53–60
Chen X-H, Ruan C-C, Ge Q, Ma Y, Xu JZ, Zhang ZB, Lin JR, Chen DR, Zhu DL, Gao PJ (2018) Deficiency of complement C3 and C5 receptors prevents angiotensin II-induced hypertension via regulatory T cells. Circ Res 122:970–983
Ng WF, To KF, Lam WWL et al (2006) The comparative pathology of severe acute respiratory syndrome and avian influenza a subtype H5N1-a review. Hum Pathol 37:381–390
Ohta R, Torii Y, Imai M, Kimura H, Okada N, Ito Y (2011) Serum concentrations of complement anaphylatoxins and proinflammatory mediators in patients with 2009 H1N1 influenza. Microbiol Immunol 55:191–198
Wang R, Xiao H, Guo R, Li Y, Shen B (2015) The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infect 4:e28
Gralinski LE, Sheahan TP, Morrison TE, et al. (2018) Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 9. doi:https://doi.org/10.1128/mBio.01753-18
Jiang Y, Li J, Teng Y, et al. (2019) Complement receptor C5aR1 inhibition reduces pyroptosis in hDPP4-trangenic mice infected with MERS-CoV. Viruses, 11. doi: https://doi.org/10.3390/v11010039
Yang M (2020) Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. SSRN. https://doi.org/10.2139/ssrn.3527420
Pang RTK, Poon TCW, Chan KCA, Lee NLS, Chiu RWK, Tong YK, Wong RMY, Chim SSC, Ngai SM, Sung JJY, Lo YMD (2006) Serum proteomic fingerprints of adult patients with severe acute respiratory syndrome. Clin Chem 52:421–429
Sun S, Zhao G, Liu C, Wu X, Guo Y, Yu H, Song H, du L, Jiang S, Guo R, Tomlinson S, Zhou Y (2013) Inhibition of complement activation alleviates acute lung injury induced by highly pathogenic avian influenza H5N1 virus infection. Am J Respir Cell Mol Biol 49:221–230
Socié G, Caby-Tosi MP, Marantz JL, Cole A, Bedrosian CL, Gasteyger C, Mujeebuddin A, Hillmen P, Vande Walle J, Haller H (2019) Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haemtol 185:297–310
Condom P, Mansury JM, Decramer S et al (2017) Atypical hemolytic uremic syndrome triggered by varicella infection. IDCases 9:89–90
Fraga-Rodriguez GM, Brio’-Sanguistin S, Turon-Vinas E, et al. (2017) Eculizumab in a child with atypical haemolytic uraemic syndrome and haemophagocytic lymphohistiocytosis triggered by cytomegalovirus infection. BMJ Case Rep
Méndez R, Menéndez R, Amara-Elori I et al (2019) Lymphopenic community-acquired pneumonia is associated with a dysregulated immune response and increased severity and mortality. J Inf Secur 78:423–431
Romacho T, Valencia I, Ramos-Gonzales M et al (2020) Visfatin/eNampt induces endothelial dysfunction in vivo: a role for toll-like receptor 4 and NLRP3 inflammasome. Sci Rep 10:5386
Dolmatova EV, Wang K, Mandavilli R, et al. (2020) The effects of sepsis on endothelium and clinical implications. Cardiovasc Res (in press)
Levi M, van der Poll T (2017) Coagulation and sepsis. Thromb Res 149:38–44
Schmitt FCF, Manolov V, Morgenstern J, Fleming T, Heitmeier S, Uhle F, al-Saeedi M, Hackert T, Bruckner T, Schöchl H, Weigand MA, Hofer S, Brenner T (2019) Acute fibrinolysis shutdown occurs early in septic shock and is associated with increased morbidity and mortality: results of an observational pilot study. Ann Intensive Care 9:19
Shrivastava G, Visoso-Carvajal G, Carcia-Cordero J et al (2020) Dengue virus serotype 2 and its non-structural proteins 2A and 2B activate TLRP3 inflammasome. Front Immunol 11:352
Babu FS, LaPorte HM, Nassoiy SP, Majetschak M (2019) Chemokine (C-X-C motif) receptor 4 regulates lung endothelial barrier permeability during resuscitation from hemorrhagic shock. Physiol Res 68:675–679
Gupta N, Zhao YY, Evans CE (2019) The stimulation of thrombosis by hypoxia. Thromb Res 181:77–83
Ye J, Zhang B, Xu J, Chang Q, McNutt MA, Korteweg C, Gong E, Gu J (2007) Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am J Pathol 170:538–545
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18(4):844–847
Chan JCK, Tsui ELH, Wong VCW et al (2007) Prognostication in severe acute respiratory syndrome: a retrospective time-course analysis of 1312 laboratory-confirmed patients in Hong Kong. Respirology 12:531–542
Zhou P, Ma B, Xu S, Zhang S, Tang H, Zhu S, Xiao S, Ben D, Xia Z (2014) Knockdown of Bruton’s kinase tyrosine kinase confers potent protection against sepsis-induced acute lung injury. Cell Biochem Biophys 70:1265–1275
Xu L, Geng T, Zang G, Bo L, Liang Y, Zhou H, Yan J (2020) Exosome derived from CD137-modified endothelial cells regulates the Th17 responses in atherosclerosis. J Cell Mol Med 24:4659–4667
Lu K, Zhao J, Liu W et al (2020) Macrophage stimulating 1-induced inflammation response promotes aortic aneurysm formation through triggering endothelial cells death and activating NF-kB signaling pathway. J Recept Signal Transduction 40:374–382
Li X, Fang P, Sun Y et al (2020) Anti-inflammatory cytokines IL-35 and IL-10 block atherogenic lysophosphatidylcholine-induced, mitochondrial ROS-mediated innate immune activation, but spare innate immune memory signature in endothelial cells. Redox Biol 28:101273
Agarwal S, Sharma H, Chen L, Dhillon NK (2020) NADPH oxidase mediated endothelial injury in HIV and opioid induced pulmonary arterial hypertension. Am J Phys Lung Cell Mol Phys 318:L1097–L1108
He H, Xiao S, Xu G et al (2020) the NADPH oxidase 4 protects vascular endothelial cells from copper oxide nanoparticles-induced oxidative stress and cell death. Life Sci 252:117571
Cui Y, Chen G, Yang Z (2020) Mitochondrial superoxide mediates PM2.5-induced cytotoxicity in human pulmonary lymphatic endothelial cells. Environ Pollut 263:114423
Connor DE, Gerbelli J, Chew AN (2020) Sirolimus and propranolol inhibit endothelial proliferation while detergent sclerosants induce endothelial activation, microparticle release and apoptosis in vitro. Phlebology 268355520913384
Su W, Zhao J, Fan TJ (2020) Dose- and time-dependent cytotoxycity of cartelol in corneal endothelial cells and the underlying mechanisms. Front Pharmacol 11:202. https://doi.org/10.3389/fphar.2020.00202
Song J, Hu B, Qu H et al (2020) Upregulation of angiotensin converting enzyme 2 by shear stress reduced inflammation and proliferation in vascular endothelial cells. Biochem Biophys Res Commun 525:812–818
Zhang Y, Zhu H, Layritz F (2020) Recombinant adenosine deaminase ameliorates inflammation, vascular disease and fibrosis in preclinical models of systemic sclerosis. Arthritis Rheum. https://doi.org/10.1002/art.41259
Kalkan BM, Akol S, Ak D et al (2020) Casin and ADM3100 enhance endothelial cell proliferation, tube formation and sprouting. Microvasc Res 130:104001
Geven C, Bergmann A, Kox M, Pickkers P (2018) Vascular effects of adrenomedullin and the anti-adrenomedullin antibody adrecizumab in sepsis. Shock 50:132–140
Lippi G, Plebani M, Henry BM (2020) Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID −19) infections: a meta-analysis. Clin Chem Acta 506:145–148
Xu P, Zhou Q, Xu J (2020) Mechanism of thrombocytopenia in COVID-19 patients. Version 2. Ann Hematol 99(6):1205–1208. https://doi.org/10.1007/s00277-020-04019-0
Liu X, Zhang R, He G (2020) Hematological findings in coronavirus disease 2019: indications of progression of disease. Ann Hematol. https://doi.org/10.1007/s00277-020-04103-5 Epub ahead of print
Yin S, Huang M, Li D (2020) Difference of coagulation features between severe pneumonia induced by SARS-SoV2 and non-SARS-CoV2. J Thromb Thrombolysis
Bellingan G, Jacono F, Bannard SJ et al (2019) Primary analysis of a phase 1/2 study to assess MultiStem® cell therapy, a regenerative advanced medicinal product (ATMP) in acute respiratory distress syndrome (MUST-ARDS). Am J Respir Crit Care Med 199:A7353
Iba T, Hashiguci N, Nagaoka I et al (2015) Heparins attenuated histone-mediated cytotoxicity in vitro and improved the survival in a rat model of histone-induced organ dysfunction. Intensive Care Med Exp 31:36
Liu Y, Mu S, Li X, Liang Y, Wang L, Ma X (2019) Unfractionated heparin alleviates sepsis-induced acute lung injury by protecting tight junctions. J Surg Res 238:175–185
Potterucha TJ, Libby P, Goldhaber SZ (2017) More than an anticoagulant: do heparins have direct anti-inflammatory effects? Thromb Haemost 28(117):437–444
SeuNg H, Wadle C, Hopp T, et al. (2018) P2Y12 is involved in emergency hematopoiesis after myocardial infarction. Eur Heart J 39 suppl 1: 694
Bartoszewski R, Moszyńska A, Serocki M, Cabaj A, Polten A, Ochocka R, Dell'Italia L, Bartoszewska S, Króliczewski J, Dąbrowski M, Collawn JF (2019) Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J 33:7929–7941
Gong H, Rehman J, Tang H, Wary K, Mittal M, Chatturvedi P, Zhao Y, Komorova YA, Vogel SM, Malik AB (2015) HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 125:652–664
Suresh MV, Malijepalli S, Zhang B (2019) Hypoxia-inducible factor (HIF)-1alpha promotes inflammation and injury following aspiration-induced lung injury in mice. Shock 52:612–621
Kimura Y, Inoue K, Abe M, Nearman J, Baranowska-Kortylewicz J (2007) PDGFRβ and HIF-1α inhibition with imatinib and radioimmunotherapy of experimental prostate cancer. Cancer Biol Ther 6:1763–1772
Veith C, Zakrzewicz D, Dahal BK (2014) Hypoxia- or PDGF-BB-dependent paxillin tyrosine phosphorilation in pulmonary hypertension is reversed by HIF-1alpha depletion or imatinib treatment. Thromb Haemost 112:1288–1303
Takei Y, Yamada M, Saito K et al (2019) Increase in circulating ACE-positive endothelial microparticles during acute lung injury. Eur Respir J 54:4
Moussa MD, Santonocito C, Fagnoul D et al (2014) Evaluation of endothelial damage in sepsis-related ARDS using circulating endothelial cells. Intensive Care Med 2:231–238
Rafat N, Nanusch C, Brikkoetter PT et al (2007) Increased circulating endothelial progenitor cells in septic patients: correlation with survival. Crit Care Med 7:1677–1684
Qi Y, Qian L, Sun B et al (2017) Inhaled NO contributes to lung repair in piglets with acute respiratory distress syndrome via increasing circulating endothelial progenitor cells. PLoS One 3:e33859
Gao X, Chen W, Liang Z, Chen L (2011) Autotransplantation of circulating endothelial progenitor cells protects against lipopolysaccharide-induced acute lung injury in rabbit. Int Immunopharmacol 11:1584–1590
Liu X, Tan W, Liu Y, Lin G, Xie C (2013) The role of the beta2 adrenergic receptor on endothelial progenitor cells dysfunction of proliferation and migration in chronic obstructive pulmonary disease patients. Expert Opin Ther Targets 17:485–500
Hallgren R, Samuelsson T, Laurent TC et al (1989) Accumulation of hyaluronan (hyaluronic acid) in the lung in adult respiratory distress syndrome. Am Rev Respir Dis 139:682–687
Bell TJ, Borgan DJ, Salek-Ardakani S et al (2018) Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol 80:14–28
Collum SD, Chen NY, Hernandez AM et al (2017) Inhibition of hyaluronan synthesis attenuates pulmonary hypertension associated with lung fibrosis. Br J Pharmacol 174:284–301
Letsiou E, Rizzo AN, Sammani S, Naureckas P, Jacobson JR, Garcia JGN, Dudek SM (2015) Differential and opposing effects of imatinib on LPS- and ventilator-induced lung injury. Am J Phys Lung Cell Mol Phys 308:L259–L269
Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, Groeneveld ABJ, Vonk Noordegraaf A, van Hinsbergh VWM, van Nieuw Amerongen GP (2012) Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 126:2728–2738
An R, Ammani S, Esquinca AE et al (2015) Imatinib attenuates inflammation and vascular leak in a clinically relevant two-hit model of acute lung injury. Am J Phys Lung Cell Mol Phys 309:L1294–L1304
Koning NJ, de Lange F, van Meurs M, Jongman RM, Ahmed Y, Schwarte LA, van Nieuw Amerongen GP, Vonk ABA, Niessen HW, Baufreton C, Boer C (2018) Reduction of vascular leakage by imatinib is associated with preserved microcirculatory perfusion and reduced renal injury markers in a rat model of cardiopulmonary bypass. Br J Anaesth 120:1165–1175
Sato H, Sugimura K, Miura M, Konno R, Kozu K, Yaoita N, Shimizu T, Yamamoto S, Aoki T, Tatebe S, Satoh K, Shimokawa H (2019) Beneficial effects of imatinib in a patient with suspected pulmonary veno-occlusive disease. Tohoku J Exp Med 247:69–73
Overbeek MJ, van Nieuw Amerongen GP, Boonstra A, Smit EF, Vonk-Noordegraaf A (2008) Possible role of imatinib in clinical pulmonary veno-occlusive disease. Eur Respir J 32:232–235
Feniocchio E, Depetris I, Campanella D et al (2016) Successful treatment of gemcitabine-induced acute interstitial pneumonia with imatinib mesylate: a case report. BMC Cancer 16:793
Carnevale-Schianca F (2010) S. Gallo, D. Rota-Scalabrini, D. Complete resolution of life-threatening bleomycin-induced pneumonitis after treatment with imatinib mesylate in a patient with Hodgkin’s lymphoma: hope for severe chemotherapy-induced toxicity? J Clin Oncol 29:e691–e693
Napier RJ, Norris RA, Swimm A (2015) Low dose of imatinib induce myelopoiesis and enhance host anti-microbial immunity. PLoS Pathog 11:e1004770
Giver CR, Shaw PA, Fletcher H et al (2019) IMPACT-TB: a phase II trial assessing the capacity of low dose imatinib to induce myelopoiesis and enhance host anti-microbial immunity against tuberculosis. Imatinib Mesylate per Oral As a Clinical Therapeutic for TB. Blood 134:1050
Dyall J, Coleman CM, Hart BJ, Venkataraman T, Holbrook MR, Kindrachuk J, Johnson RF, Olinger GG Jr, Jahrling PB, Laidlaw M, Johansen LM, Lear-Rooney CM, Glass PJ, Hensley LE, Frieman MB (2014) Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother 58:4885–4893
Coleman CM, Sisk JM, Mingo RM, Nelson EA, White JM, Frieman MB (2016) Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus fusion. J Virol 90:8924–8933
Sisk JM, Frieman MB, Machmer CE (2018) Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J Gen Virol 99:619–630
Chan JF, Lau SK, To KK et al (2015) Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev 28:465–522
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MM has received speaker honorarium from Novartis and consultant fees from Gilead.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Marchetti, M. COVID-19-driven endothelial damage: complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann Hematol 99, 1701–1707 (2020). https://doi.org/10.1007/s00277-020-04138-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-020-04138-8