
Abstract
Inflammation in the white adipose tissue (WAT) is considered a major player in the development of insulin resistance. The role of macrophages accumulating in the WAT during obesity, promoting WAT inflammation and insulin resistance is well established. In contrast, less is known about the role of lymphocytes. Recent studies have implicated different lymphocyte subsets in WAT inflammation. For instance, cytotoxic CD8+ T cells infiltrating the WAT may contribute to the recruitment, differentiation and activation of macrophages. On the other hand, a differential role for CD4+ Th1 and CD4+ Th2 cells has been suggested. Levels of WAT regulatory T cells decrease during the course of obesity and may represent a crucial factor for the maintenance of insulin sensitivity. Moreover, activation of natural killer T cells, an innate-like T cell population, which recognises lipid antigens, promotes insulin resistance and WAT inflammation. Finally, B cells may infiltrate WAT very early in response to high-fat feeding and worsen glucose metabolism through modulation of T cells and the production of pathogenic antibodies. These interesting new findings however bear controversies and introduce novel, yet unanswered, questions. Here, we review and discuss the impact of the different lymphocyte subsets in obesity-related WAT inflammation and attempt to identify the open questions to be answered by future studies.
Similar content being viewed by others


Introduction
Obesity is a major epidemic of our time and is linked to diseases such as the metabolic syndrome, type 2 diabetes and atherosclerotic cardiovascular disease. Excessive nutrient intake is the cause of the hyperplasia and hypertrophy of the adipose tissue (WAT) and the associated vicious cycle of continuous lipolysis and lipogenesis. It is recognised that the obese WAT displays an inflammatory phenotype, which is at least partly responsible for the metabolic dysfunction and subsequent insulin resistance that leads to the development of type 2 diabetes. The inflammatory phenotype of the obese WAT is mediated by an interplay between cells and components of the innate and adaptive immune systems that may accumulate in the WAT, as well as factors produced by the WAT itself [1–4].
The WAT, including the adipocyte fraction and the stromal vascular fraction (SVF), produces mediators such as leptin and adiponectin in response to perturbations of energy homeostasis. These adipokines orchestrate the expression and function of a complex network of chemokines and cytokines that regulate the accumulation of inflammatory cells in the WAT and, thereby, the development of WAT inflammation. Leptin levels increase with body mass and can, for example, stimulate the production of proinflammatory factors, such as TNF-α (also known as TNF), IL-6 and C-C motif chemokine ligands (CCLs), as well as reactive oxygen species in macrophages and mediate the activation of monocytes/macrophages and T cells [5–7]. In contrast, adiponectin, levels of which are decreased in obese individuals, exerts rather anti-inflammatory activities and ameliorates insulin resistance through an increase in NEFA oxidation and glucose uptake [8].
Several chemokines and chemokine receptors, namely CCL2, CCL5, CCL7, CCL8, C-C motif chemokine receptor (CCR)2, C-X-C chemokine ligand (CXCL)12, CXCL14, C-X-C chemokine receptor (CXCR)2, have been implicated in the process of WAT inflammation in obesity [1, 8, 9]. The causal link between obesity and WAT inflammation was recognised more than a decade ago. Initial findings demonstrated the presence of TNF-α in adipose tissue and its role in mediating insulin resistance [10, 11]. The accumulation of macrophages within the obese WAT [12] was later linked to insulin resistance [2, 4]. Macrophage accumulation in WAT can be triggered by several stimuli present in the local obese WAT microenvironment, including fatty acids, products derived from adipocyte cell death or increased hypoxia owing to the expansion of the WAT [13]. Other cell types that mediate innate immune responses such as mast cells or neutrophils, which may also accumulate in the WAT during obesity, are associated with the weight gain and insulin resistance caused by high-fat feeding [14, 15].
Proinflammatory cytokines such as TNF-α and IL-6, which are derived from inflammatory cells, can interfere with insulin signalling in adipocytes, thereby promoting insulin resistance locally in the WAT as well as systemically [13, 16]. For example, the inhibitor of κB kinase (IKK) and c-Jun N-terminal kinase (JNK) pathways that are activated by such macrophage-derived cytokines can directly impair insulin signalling in adipocytes and in other tissues, such as liver or muscle [17–19]. Distinct roles of different macrophage subpopulations in lean and obese WAT have been defined. Macrophages can be divided into ‘classically activated’ M1 macrophages and ‘alternatively activated’ M2 macrophages, with the latter further subdivided into M2a, M2b and M2c cells [20–24]. M1 macrophages can be generated as a result of stimulation with lipopolysaccharide or T helper cell (Th) type 1 cytokines, such as IFN-γ (known also as IFNG). They secrete proinflammatory factors such as TNF-α, IL-6 and IL-12, and express inducible nitric oxide synthase (iNOS). In contrast, the alternatively activated M2 macrophages can be induced by Th2 cytokines such as IL-4 and IL-13, and produce arginase rather than iNOS. They produce IL-10 and act primarily in an anti-inflammatory fashion, promoting tissue remodelling. Whereas M2a cells are activated by IL-4/IL-13, M2b cells are stimulated by immune complexes and Toll-like receptor ligands or IL-1 receptor agonists, and M2c cells are induced in the presence of IL-10. M2a and M2b cells exhibit immunoregulatory functions, whereas M2c macrophages exert immuno-suppressive and tissue remodelling actions [22, 24–26]. It is important to note that the M1/M2 definitions may represent an oversimplified view of the diversity of macrophages present in tissues. In fact, it is likely that the M1-like and M2-like cell phenotype differs from tissue to tissue and very much depends on the local tissue microenvironment. Whereas lean WAT has more anti-inflammatory M2-like macrophages, during diet-induced obesity (DIO) there is a change in inflammatory programming in favour of proinflammatory M1-like cells, which clearly contribute to the development of insulin resistance. The proinflammatory M1-like cells in the WAT can be characterised as F4/80+CD11b+CD11c+ or F4/80+CD11c+MGL1– (where MGL1 is an abbreviation of macrophage galactose-type C-type lectin 1) [23, 27, 28]. Interestingly, depletion of CD11c+ cells by using transgenic mice expressing the diphtheria toxin receptor under the regulation of the CD11c gene promoter resulted in decreased WAT inflammation and improved insulin sensitivity [29].
The changes in the inflammatory programming of macrophages to a rather proinflammatory phenotype in obese adipose tissue implies that further immune cells, such as lymphocytes, may also participate in WAT inflammation. The aim of the remainder of this review is to discuss the impact of the lymphocyte subsets (see Textbox) in obesity-related WAT inflammation and to identify the open questions to be answered by future studies.
The crosstalk between T cells and the adipose tissue
T cells have been implicated in obesity-mediated insulin resistance and the systemic inflammatory response, with the majority of the reports referring to natural killer T (NKT) cells. However, experimental evidence that lymphocytes are important in obese WAT inflammation is emerging. In 2009 three independent reports [30–32] demonstrated the critical role of T cells infiltrating the WAT for the development of insulin resistance and the associated inflammatory phenotype in genetic obese models or DIO. It has previously been shown that, in obese mice and men, visceral WAT contains more T cells, and this is likely to be due to increased T cell expansion or recruitment [3, 33, 34]. For example, the number of CD3+ T cells in the WAT of human volunteers is correlated with BMI [33]. Early B cell and T cell accumulation in the WAT, preceding macrophage infiltration and insulin resistance, has been described in DIO. The lack of lymphocytes in the Rag2 –/– mice did not seem to affect the onset of obesity or to alter insulin sensitivity [35], whereas both RAG1- and RAG2-deficient mice displayed higher numbers of accumulated M1 macrophages, even when fed a normal diet [32, 36]. RAG2 deficiency was also associated with a higher number of NKT cells in obese WAT [35]. Furthermore, hyperlipidaemic mice expressing dominant-negative transforming growth factor TGF-β receptor II in their CD4+ cells (resulting in the abolishment of the inhibitory actions of TGF-β on T cell activation and in sustained T cell-dependent inflammation) did not show signs of exacerbated insulin resistance, except under conditions of exogenous IL-6 administration [37].
The interplay between T cells and adipocytes may be multi-faceted. Strikingly, a skewed T cell receptor (TCR) repertoire within the obese visceral WAT of mice has been observed, indicating that T cell expansion in the WAT may be driven by adipocyte-specific antigens [32]. Moreover, multiple cytokines and chemokines may regulate the recruitment of lymphocytes to the WAT. The protein known as regulated upon activation, normal T cell expressed and secreted (RANTES), which is secreted by murine and human adipocytes and by cells of the SVF [3], or CCL20, which is produced by human adipocytes [33], and CXCL12, which is mainly secreted by human and murine SVF cells [38], may promote the accumulation of lymphocytes into WAT through binding to their receptors CCR5, CCR6 and CXCR4, respectively. Furthermore, the pathway regulated by the chemokine CXCL10/chemokine IFN-γ inducible protein-10 (IP10), which is expressed by human and murine adipocytes [39], and its receptor CXCR3, which is present on T cells, may alter the balance between effector T cells and regulatory T cells (Tregs) [40] and contribute to the recruitment of T cells into adipose tissue [41]. In turn, lymphocytes may modulate adipokine secretion by adipocytes via activation of the CD40–CD40L pathway [42].
Multiple receptors on T cells mediate activation (e.g. the co-stimulatory pathways), adhesion (integrins, adhesion molecules of the immunoglobulin superfamily) or chemotaxis (several chemokine receptors). Clearly, many investigations remain to be performed and experimental evidence to be generated in order to substantiate the role of T cells in DIO and to understand the mechanisms underlying the direct or indirect interactions between T cells and adipocytes. Notwithstanding this gap in our knowledge, recent studies on the role of distinct T cell subtypes in the WAT have revealed different, in some cases conflicting, functions of these cells in the course of obesity-related WAT inflammation. These issues will be addressed in more detail below.
CD8+ T cells
In 2008, two publications reported for the first time the increased accumulation of cytotoxic CD8+ T cells in the WAT in DIO or in genetically obese mice, suggesting that this cell type may potentially cooperate with macrophages and contribute to the inflammation of the obese WAT [34, 43]. An elegant study by Nishimura et al [31] showed that infiltration of CD8+ T cells precedes that of macrophages during high-fat feeding, and that this T cell subset is essential for macrophage accumulation and activation during WAT inflammation [31]. More specifically, they demonstrated that CD8+ T cells are increased in the epididymal WAT of mice fed a high-fat diet (HFD), whereas CD4+ cells and Tregs were decreased [31]. Genetic or immunological (via anti-CD8 antibody), ablation of CD8+ T cells reduced macrophage infiltration, WAT inflammation and insulin resistance in genetically obese mice and in the model of DIO, whereas adoptive transfer of CD8+ T cells produced the opposite phenotype [31]. Furthermore, co-culture experiments demonstrated that WAT from obese mice can activate CD8+ T cells, which in turn secrete chemokines able to recruit macrophages. Conversely, CD8+ T cells isolated from obese WAT promoted the differentiation of peripheral blood monocytes into F4/80+CD11b+CD68+ macrophages and enhanced the number of TNF-producing macrophages isolated from epididymal WAT [31].
As regards human studies, the presence of mainly CD4+ T cells together with modest levels of CD8+ T cells has been demonstrated in the subcutaneous WAT of type 2 diabetic patients [44]. Other studies have reported an increase in both CD8+ and CD4+ IFN-γ-producing T cells in the WAT of obese individuals, as analysed by flow cytometry [33]. Parallel studies by Koenen et al reported that, in overweight individuals, visceral adipose tissue contains a higher proportion of CD8+ cells than subcutaneous WAT, and the percentage of CD8+ cells in both visceral and subcutaneous adipose tissue was positively correlated with the activity levels of caspase-1, a component of the NLR pyrin domain-containing 3 (NLRP3) inflammasome [45].
Taken together, these results support the hypothesis that, in obese states, WAT-derived factors can activate CD8+ T cells, which then promote macrophage infiltration, thus perpetuating the inflammatory response within the WAT. However, future studies are required to address the molecular mechanisms governing CD8+ T cell recruitment into the WAT and to identify a way of modulating this process for therapeutic purposes.
CD4+ T cells
CD4+ T cells can be divided into Tregs and Th cells. There are three main subsets of Th cells: the Th1 lineage, which results from stimulation by IL-12 and produces proinflammatory cytokines such as IFN-γ; the Th2 lineage, which is generated in response to stimulation by IL-4 and produces IL-4, IL-5, IL-10 and IL-13; and the recently identified IL-17-producing Th17 subpopulation, which differentiates in the presence of IL-6 and TGF-β and is activated by IL-23 [46].
The Th1 and Th2 subsets are both present in the WAT, although the number of Th1 cells is increased, whereas the number of Th2 cells remains stable in visceral WAT of DIO mice [32]. Furthermore, the increased body weight and increased insulin resistance of RAG1-deficient mice relative to wild-type mice were reversed after adoptive transfer of CD4+, but not CD8+, T cells. Th2 cells were the predominant mediators of this metabolic improvement, most likely through activation of IL-10-producing macrophages, although other potential mechanisms cannot be excluded [32]. These results indicate that metabolic dysregulation caused by obesity is modulated by CD4+ cells and that the disrupted balance between Th1 and Th2 cells in the WAT may represent a pathophysiological component of obesity and insulin resistance. Previous studies pertinent to Th1-related cytokines and chemokines such as IFN-γ and RANTES are in keeping with these findings [47]. Briefly, WAT T cells from obese mice express higher levels of Ifn-γ than controls [34], while IFN-γ-deficient mice display reduced levels of TNF-α and CCL2 in WAT, lower numbers of infiltrated immune cells and better insulin sensitivity than wild-type animals [34].
Consistent with the mouse data, WAT T cells from obese human individuals have higher levels of IFN-γ [33, 34, 44, 48], and RANTES expression is elevated in subcutaneous WAT from obese humans [3]. Moreover, in obese humans, levels of RANTES, IFN-γ, TNF-α and IL-2 are higher in WAT lymphocytes than in peripheral blood lymphocytes [33].
The role of the inflammatory Th17 cells in obesity remains intriguing, since the results of different papers regarding the impact of IL-17 and Th17 in obesity-associated WAT inflammation are controversial. Zuniga et al reported that IL-17, which in the WAT is expressed mainly by γδ T cells, inhibits adipogenesis in mouse 3T3-L1 preadipocytes. Moreover, IL-17 downregulated glucose uptake in adipocytes, while young mice deficient in IL-17 displayed better responses to glucose and insulin tolerance tests. In contrast, IL-17 deficiency resulted in more weight gain both under high-fat as well as low-fat feeding conditions [49]. In keeping with these results, IL-17 inhibits adipogenesis from human mesenchymal stem cells and enhances IL-6 and IL-8 expression by these cells [50]. Another study described how Th17 cell numbers are increased in the course of obesity. This might be linked to the faster progress of autoimmunity in murine models of experimental autoimmune encephalomyelitis and colitis upon feeding an HFD [51]. It should be stressed that the functions of IL-17 and Th17 cells do not necessarily overlap, as IL-17 can be produced by additional types of immune cells, such as neutrophils and γδ T cells besides Th17 cells. Moreover, IL-17 may affect several cell populations bearing IL-17 receptors, including adipocytes, neutrophils and endothelial cells [49, 52]. The aforementioned data indicate that the roles of IL-17 and Th17 cells in obesity-related WAT inflammation are complex and require further investigation.

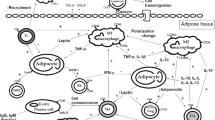
Macrophage accumulation in WAT can be triggered by several stimuli, including fatty acids or products derived from adipocyte cell death. During the course of obesity, macrophages polarise into proinflammatory M1-like cells. Macrophage-derived TNF-α and IL-6 interfere with insulin signalling in adipocytes, promoting insulin resistance (IR) locally in the WAT. Leptin can affect T cell activation by stimulating the development of a Th1-phenotype (characterised by IFN-γ or TNF-α production). Leptin probably inhibits the generation and proliferation of Tregs; Tregs may, in turn, improve insulin sensitivity in an IL-10-dependent manner. CD8+ T cells, which are activated and increased in the WAT of HFD-fed mice secrete chemokines that are able to recruit macrophages. NKT cells, the receptors of which interact with CD1d-related lipid antigens, may co-operate with CD8+ T cells. WAT B cells, which interact with T cells, may produce pathogenic IgG autoantibodies, promoting insulin resistance. Auto-Abs, autoantibodies; CD1d-Ags, CD1d-related antigens; IR, insulin resistance; ΜΦ, macrophage
Regulatory T cells
Tregs, a small T cell subpopulation with anti-inflammatory properties, which are characterised as CD4+/CD25+/forkhead box P3 (FOXP3)+ cells, are considered to be significant players in obesity-related WAT inflammation. A plethora of studies have reported that in WAT the number of Tregs is either decreased or remains stable during the course of obesity, while the number of inflammatory Th1 cells is increased [30, 32, 53].
A study by Feuerer et al [30] has implicated Tregs in the pathogenesis of obese WAT inflammation. Depletion of these cells by engaging mice expressing the diphtheria toxin receptor under the control of promoter for the gene encoding FOXP3 aggravated the metabolic function of even normal mice [30]. Conversely, an increase in the number of Tregs improved insulin sensitivity in an IL-10 dependent manner [30]. Another important finding of this study was that Tregs isolated from WAT showed a gene expression pattern distinct from that in Tregs from a lymph node or the spleen, and this was characterised by the overexpression of genes related to immune cell recruitment, such as Ccr-1, Ccr-2, Ccr-9, Cxcl2 and Il-10 [30]. It is noteworthy that Tregs isolated from fat had a distinct TCR profile compared with Tregs from the spleen or a lymph node or conventional T cells. This phenomenon suggests that the WAT Treg population may undergo antigen selection [30]. In agreement with these studies, WAT Treg numbers are decreased in the WAT of obese mice [53, 54], whereas adoptive transfer of Tregs or their targeted activation and proliferation improved WAT inflammation and ameliorated insulin resistance in ob/ob mice [54, 55].
In contrast with the animal data, the results from human studies are conflicting. Although decreased expression of FOXP3 in the visceral WAT of obese subjects has been reported [53], it has also been demonstrated that the number of Tregs in the same tissue is increased and correlated with general markers of WAT or systemic inflammation, such as C-reactive protein, plasma IL-6 and BMI [56]. One possible explanation for the discrepancies in human studies could be that FOXP3 expression is variable in human Tregs. This suggests that additional Treg markers, such as the low expression of CD127 in these cells, may need to be added to their analysis [57, 58] to give a true reflection of the levels of this important T cell subpopulation in human WAT.
Several studies provide evidence of a connection between leptin and Treg function. Leptin deficiency promotes the generation and proliferation of Tregs in mice, thereby protecting against autoimmune diseases and atherosclerosis [59-61]. This scenario is not only in keeping with the differences between lean and obese mice as regards their levels of leptin and their numbers of Tregs, but may also implicate an additional point of convergence between obesity and autoimmunity.
NKT cells
NKT cells represent a T cell subpopulation with innate-like characteristics capable of producing both Th1 and Th2 cytokines [62]. Specific NKT cell receptors interact with CD1d-related lipid and glycolipid antigens, thus providing a potential link between obesity, autoimmunity and inflammatory diseases [63, 64]. NKT cells could contribute to the development of visceral WAT inflammation during obesity, since HFD-fed NKT-deficient mice (β2-microglobulin knockout mice) show lower macrophage accumulation and better glucose tolerance than control mice, with no differences in body weights [65]. However, it has recently been demonstrated that the aforementioned effects of NKT cell deletion on metabolism can only occur in the absence of CD8+ T cells [66]. Moreover, previous studies have reported that adoptive transfer or agonist activation of NKT cells ameliorate the metabolic dysregulation of the ob/ob mice [55, 67], suggesting that the role of NKT cells in obesity-related inflammation may be complex.
Adipose tissue inflammation and B cells
A few studies have reported that B cells infiltrate the WAT earlier than T cells or macrophages and prior to the appearance of insulin resistance [35]. B cells were recently shown to substantially facilitate the induction of insulin resistance, since mice devoid of B cells (by anti-CD20 administration) displayed lower fasting glucose levels and improved glucose and insulin tolerance tests responses on HFD feeding compared with control B cell-sufficient mice [68]. In the same study, B cell-deficient mice (IgM heavy chain knockout mice) had a reduced number of activated CD8+ T cells and macrophages. However, these effects of B cell deletion were not independent of T cells. In fact, a process of B cell antigen-presentation to T cells mediated by MHC I or MHC II appears likely [68].
Strikingly, high-fat feeding leads to the production of pathogenic IgG autoantibodies, which may promote glucose intolerance. By comparing the IgG autoantibody profiles of insulin-resistant and matched insulin-sensitive human volunteers, Winer et al identified that autoantibodies to golgi SNAP receptor complex member 1 (GOSR1), Bruton agammaglobulinaemia tyrosine kinase (BTK) and glial fibrillary acidic protein (GFAP) displayed a strong association with insulin resistance [68]. These findings should trigger further investigations aiming at clarifying the exact role of adipose-related antigens as contributors to obese WAT inflammatory and metabolic responses (Fig. 1).
The impact of leptin on lymphocyte-related WAT inflammation
Leptin has emerged as a significant factor in both human and mouse obesity, acting as a regulator of food intake and energy expediture. The ob/ob leptin-deficient mice are characterised by hyperphagia, hyperglycaemia, hyperinsulinaemia and insulin resistance [69]. However, leptin levels are increased in humans and in other mouse models of obesity, indicating that leptin resistance is taking place [6]. Besides the unequivocal metabolic role of leptin, which overall prevents obesity, recent evidence points to immuno-modulatory actions of this hormone. In fact, in addition to affecting metabolism, leptin and leptin receptor deficiency (in ob/ob and db/db mice, respectively) also influence aspects of the immune, reproductive and cardiovascular systems [69].
As previously shown, the leptin receptor is present on T cells. Thus, leptin can affect T cell activation by stimulating the development of a Th1 (characterised by IFN-γ or TNF-α production) rather than a Th2 phenotype (characterised by the production of IL-4). Leptin increases IL-2 secretion in conventional T cells and may inhibit T cell apoptosis [6, 19]. Furthermore, the presence of leptin receptor in the thymic environment is essential for T cell development, as shown in db/db mice [70]. In addition, leptin may promote delayed-type hypersensitivity. These effects of leptin on adaptive immunity could provide a common factor linking obesity to autoimmunity and/or cancer. Intriguingly, ob/ob leptin-deficient mice have a reduced susceptibility to arthritis, experimental autoimmune encephalomyelitis and type 1 diabetes (in the NOD mouse model) [6, 7, 19]. As already mentioned, this effect could be attributed to the enhanced proliferation of Tregs in leptin-deficient mice [59-61]. Interestingly, recent studies have described a beneficial role of leptin administration in type 1 diabetes. Although this is an apparent contradiction to the aforementioned reduced susceptibility of leptin-deficient mice to type 1 diabetes, the studies by Wang et al and Denroche et al in insulin-deficient and streptozotocin-treated diabetic mice, respectively [71, 72], have demonstrated that leptin therapy prevents hyperglycaemia, improves lipid metabolism and reduces glucagon secretion. According to a recent paper, the majority of these effects are probably CNS-dependent [73]. Similarly, preliminary data on leptin administration in type 1 diabetic patients are promising [74, 75]; however, additional and prolonged clinical trials are needed to reveal whether leptin could be an adjunct therapy for type 1 diabetes in the future. The impact and therapeutic implications of the described effects of leptin in different cell types, including immune cells, in the context of type 1 diabetes remains to be elucidated.
Interestingly, central leptin signalling can influence the development and maturation of B cells. In particular, the starvation-induced blockade of B cell maturation could be reversed by central administration of leptin, suggesting the orchestrating function of the CNS on immune function in addition to energy homeostasis [76]. These insights underline the necessity of further investigation of the interface between CNS, energy regulation and immunity, and its critical role in the pathogenesis of obesity and the metabolic syndrome.
In contrast to leptin, studies on the role of adiponectin in T cell biology are scarce, despite the fact that lymphocytes express adiponectin receptors and their levels are decreased in obese individuals [77], suggesting a possible role for T cell adiponectin receptors in obesity.
Conclusion and future outlook
Emerging evidence indicates that obesity-related WAT inflammation is not strictly a macrophage-dependent phenomenon. The inflammatory reprogramming from a rather anti-inflammatory M2-like to a proinflammatory M1-like macrophage phenotype in the WAT is undoubtedly a key factor in insulin resistance. However, recent evidence suggests the critical contribution of lymphocytes, especially T cells, in the WAT to the development of insulin resistance. However, many questions remain unanswered. First and foremost, which T cell subset is the most important one in driving obese WAT inflammation and insulin resistance? Although existing data support the hypothesis that CD8+ cells are the predominant T cell subset in the visceral WAT of obese humans and mice [31, 33, 45], CD4+ T cells may also regulate insulin sensitivity and adiposity [32]. In line with this, an increased number of activated CD4+ cells has been identified in morbidly obese humans [78]. Second, the mode of action of activated T cells, independent of their CD4+ or CD8+ identity, seems to be through the induction of proinflammatory cytokines, such as TNF-α, IFN-γ, and chemokines, such as CCL2. Our understanding of this process has evolved from a strictly macrophage-oriented phenomenon to a growing recognition of the effect of T cell activation in obesity, therefore elucidating the specific contribution of each T cell subset to the pathophysiology of this disease and the associated insulin resistance including the cardiovascular sequelae becomes imperative.
A major question that remains unresolved is: what are the molecular interactions that govern the direct or indirect crosstalk between the different immune cell subtypes, such as CD4+ cells, CD8+ cells, Treg cells, macrophages or B cells with each other within the WAT? Moreover, while significant evidence supports the role of macrophages and their products in promoting insulin resistance, it is not clear whether lymphocytes themselves also have such actions or induce insulin resistance in an indirect fashion, e.g. by regulating macrophage accumulation. In addition, how inflammatory cells may promote insulin resistance in insulin target tissues other than the WAT, such as the liver, or even contribute to liver steatosis, is poorly defined and requires attention. The information on the role of activated immune cells infiltrating muscle and liver in the development of tissue-specific insulin resistance therein is limited. Tissue-specific processes may account for the diversity in the degree of liver steatosis and functional impairment among obese individuals, which is strikingly independent of their adiposity and insulin resistance [79].
Furthermore, studies elaborating on the role of specific immune processes within the WAT should be performed. The finding that the TCRs in obese WAT are skewed implicates an antigen-specific response [21]. Some experimental evidence suggests that lymphocytes accumulate in the adipose tissue prior to inflammatory macrophages [35]. Nevertheless, it remains unclear whether T cell accumulation and activation in the WAT is the result of alterations in the inflammatory environment of the adipose tissue, including inflammatory reprogramming of resident macrophages into a proinflammatory phenotype, or the result of antigen recognition by T cells within the WAT. The fact that Tregs from fat have a distinct TCR profile compared with Tregs from other organs [30] supports the second hypothesis. This is consistent with data demonstrating the presence of autoantibodies against WAT antigens [53]. It is also intriguing that NKT cells may react to lipid antigens in the WAT [48]. Clearly, the importance of specific pathways, such as different co-stimulatory systems (e.g. the CD40–CD40L or CD28–B7.1/B7.2 systems) that may mediate T cell activation, as well as the function of adhesion and chemokine receptors critical for recruitment of cells of the adaptive immune response to the WAT need to be elucidated in the future. In addition, the interface between obesity and autoimmunity, which is in parts mediated by effects of adipose-derived factors, such as leptin, on T cell-mediated inflammation is an intriguing phenomenon that requires further investigation.
A further emerging question in the field is whether inflammatory cells, particularly T cells, may prevent the activation of brown adipose tissue (BAT) in the course of obesity and insulin resistance. Whereas BAT activation by muscle-derived circulating factors is intimately linked to insulin sensitivity [80], little is known about the inflammatory cell infiltration of BAT. Reduced expression of proinflammatory cytokines and chemokines in the macrophages of BAT, as compared to macrophages of WAT, has been observed [81], but the role of T cells, including Tregs in the BAT in the course of obesity and insulin resistance remains unclear.
Although the murine models of DIO have proved to be very important for generating several hypotheses and advancing our understanding of obesity and obesity-associated WAT inflammation, many discrepancies between men and mice have been reported with regards to obesity studies. A major bias of the murine studies is that most of them are performed with C57BL/6 mice, a mouse strain that is inherently skewed towards a Th1 cell response. This limitation should be taken into account when attempting to extrapolate the findings from the murine system to humans. In addition, it is not clear to what degree the experimental murine models (experimental diets, WAT isolated and studied, etc.) reflect the human situation. Moreover, the markers of inflammatory cell subtypes that accumulate in obese WAT may differ substantially between the mouse and human system, which limits the conclusions that can be drawn from the murine system.
Despite the biases, discrepancies and limitations of murine and human studies, the major question that arises from the reported experimental data is whether modulation of adaptive immunity could be used therapeutically for preventing insulin resistance and the development of type 2 diabetes. Preclinical studies in animal models are required to demonstrate whether it would be useful to test modulation of adaptive immunity in obesity in future clinical studies. Nevertheless, it is apparent that we are just at the beginning of our exciting journey towards understanding the crosstalk between metabolism and adaptive immunity.

Abbreviations
- BAT:
-
Brown adipose tissue
- CCL:
-
C-C motif chemokine ligand
- CXCL:
-
C-X-C chemokine ligand
- CXCR:
-
C-X-C chemokine receptor
- DIO:
-
Diet-induced obesity
- FOXP3:
-
Forkhead box P3
- HFD:
-
High-fat diet
- iNOS:
-
Inducible nitric oxide synthase
- NKT:
-
Natural killer T
- RANTES:
-
Regulated upon activation normal T cell expressed and secreted
- SVF:
-
Stromal vascular fraction
- TCR:
-
T cell receptor
- Th cell:
-
T-helper cell
- Treg:
-
Regulatory T cell
- WAT:
-
White adipose tissue
References
Anderson EK, Gutierrez DA, Hasty AH (2010) Adipose tissue recruitment of leukocytes. Curr Opin Lipidol 21:172–177
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Wu H, Ghosh S, Perrard XD et al (2007) T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 115:1029–1038
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Fantuzzi G, Faggioni R (2000) Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol 68:437–446
Kaminski DA, Randall TD (2010) Adaptive immunity and adipose tissue biology. Trends Immunol 31:384–390
Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C (2006) Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J Immunol 176:7745–7752
Ouchi N, Parker JL, Lugus JJ, Walsh K (2011) Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11:85–97
Lolmede K, Duffaut C, Zakaroff-Girard A, Bouloumie A (2011) Immune cells in adipose tissue: key players in metabolic disorders. Diabetes Metab 37:283–290
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271:665–668
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS (1997) Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 389:610–614
Bornstein SR, Abu-Asab M, Glasow A et al (2000) Immunohistochemical and ultrastructural localization of leptin and leptin receptor in human white adipose tissue and differentiating human adipose cells in primary culture. Diabetes 49:532–538
Surmi BK, Hasty AH (2008) Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol 3:545–556
Elgazar-Carmon V, Rudich A, Hadad N, Levy R (2008) Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 49:1894–1903
Liu J, Divoux A, Sun J et al (2009) Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 15:940–945
De Taeye BM, Novitskaya T, McGuinness OP et al (2007) Macrophage TNF-α contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am J Physiol Endocrinol Metab 293:E713–E725
Arkan MC, Hevener AL, Greten FR et al (2005) IKK-β links inflammation to obesity-induced insulin resistance. Nat Med 11:191–198
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Matarese G, Procaccini C, De Rosa V, Horvath TL, La Cava A (2010) Regulatory T cells in obesity: the leptin connection. Trends Mol Med 16:247–256
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35
Gordon S, Martinez FO (2010) Alternative activation of macrophages: mechanism and functions. Immunity 32:593–604
Martinez FO, Helming L, Gordon S (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27:451–483
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686
Ricciardi-Castagnoli P, Granucci F (2002) Opinion: interpretation of the complexity of innate immune responses by functional genomics. Nat Rev Immunol 2:881–889
Stein M, Keshav S, Harris N, Gordon S (1992) Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 176:287–292
Schenk S, Saberi M, Olefsky JM (2008) Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 118:2992–3002
Westcott DJ, Delproposto JB, Geletka LM et al (2009) MGL1 promotes adipose tissue inflammation and insulin resistance by regulating 7/4hi monocytes in obesity. J Exp Med 206:3143–3156
Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG (2008) Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8:301–309
Feuerer M, Herrero L, Cipolletta D et al (2009) Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15:930–939
Nishimura S, Manabe I, Nagasaki M et al (2009) CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15:914–920
Winer S, Chan Y, Paltser G et al (2009) Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15:921–929
Duffaut C, Zakaroff-Girard A, Bourlier V et al (2009) Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators. Arterioscler Thromb Vasc Biol 29:1608–1614
Rocha VZ, Folco EJ, Sukhova G et al (2008) Interferon-γ, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res 103:467–476
Duffaut C, Galitzky J, Lafontan M, Bouloumie A (2009) Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun 384:482–485
Lee YS, Li P, Huh JY et al (2011) Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60:2474–2483
Sultan A, Strodthoff D, Robertson AK et al (2009) T cell-mediated inflammation in adipose tissue does not cause insulin resistance in hyperlipidemic mice. Circ Res 104:961–968
Sengenes C, Miranville A, Maumus M, de Barros S, Busse R, Bouloumie A (2007) Chemotaxis and differentiation of human adipose tissue CD34+/CD31– progenitor cells: role of stromal derived factor-1 released by adipose tissue capillary endothelial cells. Stem Cells 25:2269–2276
Herder C, Hauner H, Kempf K, Kolb H, Skurk T (2007) Constitutive and regulated expression and secretion of interferon-gamma-inducible protein 10 (IP-10/CXCL10) in human adipocytes. Int J Obes (Lond) 31:403–410
Heller EA, Liu E, Tager AM et al (2006) Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 113:2301–2312
Krinninger P, Brunner C, Ruiz PA et al (2011) Role of the adipocyte-specific NF-ĸB activity in the regulation of IP-10 and T cell migration. Am J Physiol Endocrinol Metab 300:E304–E311
Poggi M, Jager J, Paulmyer-Lacroix O et al (2009) The inflammatory receptor CD40 is expressed on human adipocytes: contribution to crosstalk between lymphocytes and adipocytes. Diabetologia 52:1152–1163
Rausch ME, Weisberg S, Vardhana P, Tortoriello DV (2008) Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes (Lond) 32:451–463
Kintscher U, Hartge M, Hess K et al (2008) T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 28:1304–1310
Koenen TB, Stienstra R, van Tits LJ et al (2011) The inflammasome and caspase-1 activation: a new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology 152:3769–3778
Stockinger B, Veldhoen M (2007) Differentiation and function of Th17 T cells. Curr Opin Immunol 19:281–286
Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS (2010) T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity (Silver Spring) 18:1918–1925
O’Rourke RW, Metcalf MD, White AE et al (2009) Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int J Obes (Lond) 33:978–990
Zuniga LA, Shen WJ, Joyce-Shaikh B et al (2010) IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol 185:6947–6959
Shin JH, Shin DW, Noh M (2009) Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem Pharmacol 77:1835–1844
Winer S, Paltser G, Chan Y et al (2009) Obesity predisposes to Th17 bias. Eur J Immunol 39:2629–2635
Cua DJ, Tato CM (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 10:479–489
Deiuliis J, Shah Z, Shah N et al (2011) Visceral adipose inflammation in obesity is associated with critical alterations in T regulatory cell numbers. PLoS One 6:e16376
Eller K, Kirsch A, Wolf AM et al (2011) Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 60:2954–2962
Ilan Y, Maron R, Tukpah AM et al (2010) Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci USA 107:9765–9770
Zeyda M, Huber J, Prager G, Stulnig TM (2011) Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity (Silver Spring) 19:743–748
Liu W, Putnam AL, Xu-Yu Z et al (2006) CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 203:1701–1711
Simonetta F, Chiali A, Cordier C et al (2010) Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. Eur J Immunol 40:2528–2538
De Rosa V, Procaccini C, Cali G et al (2007) A key role of leptin in the control of regulatory T cell proliferation. Immunity 26:241–255
Matarese G, Di Giacomo A, Sanna V et al (2001) Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol 166:5909–5916
Taleb S, Herbin O, Ait-Oufella H et al (2007) Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arterioscler Thromb Vasc Biol 27:2691–2698
Getz GS, Vanderlaan PA, Reardon CA (2011) Natural killer T cells in lipoprotein metabolism and atherosclerosis. Thromb Haemost 106:814–819
Borg NA, Wun KS, Kjer-Nielsen L et al (2007) CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 448:44–49
Mallevaey T, Clarke AJ, Scott-Browne JP et al (2011) A molecular basis for NKT cell recognition of CD1d-self-antigen. Immunity 34:315–326
Ohmura K, Ishimori N, Ohmura Y et al (2010) Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol 30:193–199
Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O’Doherty RM (2011) Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS One 6:e19831
Elinav E, Pappo O, Sklair-Levy M et al (2006) Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J Pathol 209:121–128
Winer DA, Winer S, Shen L et al (2011) B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17:610–617
Lindström P (2007) The physiology of obese–hyperglycemic mice [ob/ob mice]. Scientific World Journal 7:666–685
Palmer G, Aurrand-Lions M, Contassot E et al (2006) Indirect effects of leptin receptor deficiency on lymphocyte populations and immune response in db/db mice. J Immunol 177:2899–2907
Denroche HC, Levi J, Wideman RD et al (2011) Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes 60:1414–1423
Wang MY, Chen L, Clark GO et al (2010) Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci USA 107:4813–4819
Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R (2010) Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA 107:17391–17396
Oral EA (2012) Leptin for type 1 diabetes: coming onto stage to be (or not?). Pediatr Diabetes 13:68–73
Park JY, Chong AY, Cochran EK et al (2008) Type 1 diabetes associated with acquired generalized lipodystrophy and insulin resistance: the effect of long-term leptin therapy. J Clin Endocrinol Metab 93:26–31
Tanaka M, Suganami T, Kim-Saijo M et al (2011) Role of central leptin signaling in the starvation-induced alteration of B-cell development. J Neurosci 31:8373–8380
Alberti L, Gilardini L, Girola A, Moro M, Cavagnini F, Invitti C (2007) Adiponectin receptors gene expression in lymphocytes of obese and anorexic patients. Diabetes Obes Metab 9:344–349
van der Weerd K, Dik WA, Schrijver B et al (2012) Morbidly obese human subjects have increased peripheral blood CD4+ T cells with skewing toward a Treg- and Th2-dominated phenotype. Diabetes 61:401–408
Monetti M, Levin MC, Watt MJ et al (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6:69–78
Bostrom P, Wu J, Jedrychowski MP et al (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468
Ortega MT, Xie L, Mora S, Chapes SK (2011) Evaluation of macrophage plasticity in brown and white adipose tissue. Cell Immunol 271:124–133
Appay V, van Lier RA, Sallusto F, Roederer M (2008) Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A 73:975–983
Romagnani S (2006) Regulation of the T cell response. Clin Exp Allergy 36:1357–1366
Romagnani S (2000) T-cell subsets (Th1 versus Th2). Ann Allergy Asthma Immunol 85:9–18
Bluestone JA, Mackay CR, O’Shea JJ, Stockinger B (2009) The functional plasticity of T cell subsets. Nat Rev Immunol 9:811–816
Cipolletta D, Kolodin D, Benoist C, Mathis D (2011) Tissular Tregs: a unique population of adipose-tissue-resident Foxp3+CD4+ T cells that impacts organismal metabolism. Semin Immunol 23:431–437
Godfrey DI, Rossjohn J (2011) New ways to turn on NKT cells. J Exp Med 208:1121–1125
LeBien TW, Tedder TF (2008) B lymphocytes: how they develop and function. Blood 112:1570–1580
Funding
Supported by the Deutsche Forschungsgemeinschaft (CH279/5-1 to TC).
Contribution statement
All authors have contributed to the conception, design and drafting of the manuscript and approved the final version for publication
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Chatzigeorgiou, A., Karalis, K.P., Bornstein, S.R. et al. Lymphocytes in obesity-related adipose tissue inflammation. Diabetologia 55, 2583–2592 (2012). https://doi.org/10.1007/s00125-012-2607-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2607-0