
ABSTRACT
In Brazil, Leishmania braziliensis is the main causative agent of the neglected tropical disease, cutaneous leishmaniasis (CL). CL caused by L. braziliensis can present on a spectrum of disease severity with a high rate of treatment failure. Yet the parasite factors that contribute to disease presentation and treatment outcome are not well understood, in part because successfully isolating and culturing parasites from patient lesions remains a major technical challenge. Here we describe the development of selective whole genome amplification (SWGA) for Leishmania and show that this method enables culture-independent analysis of parasite genomes obtained directly from primary patient skin samples, allowing us to circumvent artifacts associated with adaptation to culture. We show that SWGA can be applied to multiple Leishmania species residing in different host species, suggesting that this method is broadly useful in both experimental infection models and clinical studies. Finally, we show that parasite genome sequencing data generated by SWGA of skin biopsies collected from patients in Corte de Pedra, Bahia, Brazil, exhibit substantial genetic diversity and can be integrated with published whole genome data from parasite isolates to identify variants associated with high treatment failure rates observed in Northeast Brazil.
AUTHOR SUMMARY Leishmania braziliensis is the main cause of cutaneous leishmaniasis in Brazil. Due to limitations in culturing, it is important to study the parasite in a culture-independent manner. We use selective whole genome amplification (SWGA) to explore parasite genomic diversity directly from patient biopsies. This method is inexpensive and can be broadly used to generate parasite genome sequence data sampled from different Leishmania species infecting different mammalian hosts. We found high diversity among the L. braziliensis genomes from Bahia, Brazil, which correlated with geographic location. By integrating these data with publicly available genome sequences from other studies spanning four countries in South America, we identified variants unique to Northeast Brazil that may be linked to high regional rates of treatment failure.
INTRODUCTION
Leishmania constitutes a genus of intracellular protozoan parasites whose species are all transmitted by the bite of an infected phlebotomine sand fly and can lead to leishmaniasis. This neglected tropical disease has a spectrum of clinical presentations, including visceral and cutaneous, which vary in severity and are influenced by parasite species and strain genetics [1,2]. The most common form of disease caused by these parasites is cutaneous leishmaniasis (CL), which is usually characterized by a single or multiple localized skin ulcers. Moreover, up to 10% of patients can develop more severe forms of the disease, such as mucosal leishmaniasis (ML) and disseminated leishmaniasis (DL)[2]. Worldwide there are 700,000 to 1 million new cases of CL annually [3]. Although mortality is low for patients with CL, the disease is disfiguring, leads to chronic and systemic inflammation [4], and adversely impacts quality of life.
In Brazil, CL cases are largely caused by Leishmania braziliensis. Previous population genetics studies of this species have relied on low-resolution techniques, such as multilocus sequence typing and restriction fragment length polymorphism, both of which only consider a small set of genetic loci. Collectively, these studies have shown that the genetic diversity of L. braziliensis is higher in and around the Amazon rainforest than near the coast [5,6]. Moreover, recent whole genome sequencing studies have determined that L. braziliensis exhibits higher intraspecies genetic variation than other Leishmania species [7,8]. Variation in virulence, drug resistance, and clinical phenotype among strains has been observed in many parasites. A recent study using random amplified polymorphic DNA analysis showed that L. braziliensis genotypes are associated with disease presentation in patients [9]. Collectively, these studies underscore the importance of generating high-resolution genotyping data from L. braziliensis to identify genetic variants linked to disease severity and treatment outcome in CL patients.
We recently showed that L. braziliensis burden in patients is a strong predictor of inflammation, pathology, and poor response to chemotherapy, yet the parasite factors that contribute to differences in parasite load between patients have been difficult to address [10]. Technical and biological factors associated with culture adaptation of L. braziliensis complicate efforts to generate high-resolution genomic data from this important species. Unlike other Leishmania species, L. braziliensis is characterized by relatively slow growth and low parasitemia, which pose a major challenge to isolating parasites from patient lesions [11,12]. Even when parasites are successfully adapted to culture, some studies suggest that drug resistance markers identified from in vitro assays may not be driving drug resistance observed in the clinic, and the process of isolating parasites from primary patient samples may transiently alter chromosomal copy number [13–15]. There is an urgent need for culture-independent methods to circumvent these issues. Since parasitemia at the site of infection in the skin is extremely low during L. braziliensis infection, a direct metagenomic sequencing approach is not a viable culture-independent methodology. Enrichment of Leishmania donovani genomes from primary patient samples was recently published using Agilent SureSelect arrays which utilize custom RNA ‘bait’ sequences used to capture Leishmania genomic DNA for subsequent amplification [15]. However, this method is expensive, requires specialized reagents, and is specific to L. donovani.
In this study, we develop a selective whole genome amplification (SWGA) protocol to selectively amplify L. braziliensis directly from primary patient samples. SWGA is based on the use of organism-specific, short oligonucleotide primers and a high-fidelity, highly processive polymerase to preferentially amplify large segments of the target genome. Effective SWGA protocols have resulted in high-throughput sequencing-ready samples that are enriched for specific target microbial genomes and have been used to address biologically important questions in several microorganisms, including Mycobacterium tuberculosis, Wolbachia spp., Plasmodium spp., Neisseria meningitidis, Coxiella burnetii, Wuchereria bancrofti, and Treponema pallidum [16–30]. The ability to carry out SWGA without specialized equipment or reagents makes it more feasible to implement this method in low- and middle-income countries (LMICs) where laboratory resources may be limited [16,21]. Here we report the development of SWGA for Leishmania and show that this method enables robust amplification of L. braziliensis DNA from complex metagenomic samples obtained from patients and experimental mouse models of infection. We investigate SNPs and somy in the parasite genomes directly sequenced from primary patient samples. Ultimately, we reveal the population genetic structure of L. braziliensis in Corte de Pedra, Bahia, Brazil, and compare these genomes to previously published L. braziliensis genomes.
RESULTS
Validation of SWGA for Leishmania in silico and using synthetic controls
We used the improved SWGA algorithm, swga2.0, which employs machine learning to design primer sets that preferentially bind to a target genome, compared to one or more background genomes ([31]; see Methods). We used L. braziliensis (MHOM/BR/75/M2904 2019) as the target genome and the human genome as background. Genomes from Staphylococcus aureus and Streptococcus pyogenes were also included as background since both are skin commensals that we previously reported to be common members of the dysbiotic skin microbiome on L. braziliensis lesions [32]. We calculated the expected number of perfect match binding sites – across a range of parasite and host genomes – for each of the 23, 8-mer primer sequences designed by the SWGA algorithm (Supplementary Table 1). This in silico analysis showed that our SWGA primers had a median of 15 (8.4 - 27.5) ‘hits’, or exact matches, per million base pairs (Mbp) of the L. braziliensis genome and a median of 0.22 (0.16 - 0.60) hits per Mbp of the human genome (Figure 1A) – a nearly 60-fold (27-to 100-fold) enrichment in predicted binding to the parasite genome compared to host (Figure 1B). We next tested whether our SWGA primers would be predicted to work when applied to other Leishmania species and/or when other host species were involved. Multiple species of Leishmania cause disease in humans, and several infect canines that are sympatric with humans. In addition, many Leishmania species are used to experimentally infect rodent models for research. L. major, L. donovani, L. infantum, and L. amazonensis all exhibited similar results with our SWGA primers as L. braziliensis, with median hits per Mbp of 16.8, 15.5, 16.0, and 16.3, respectively (Figure 1A).
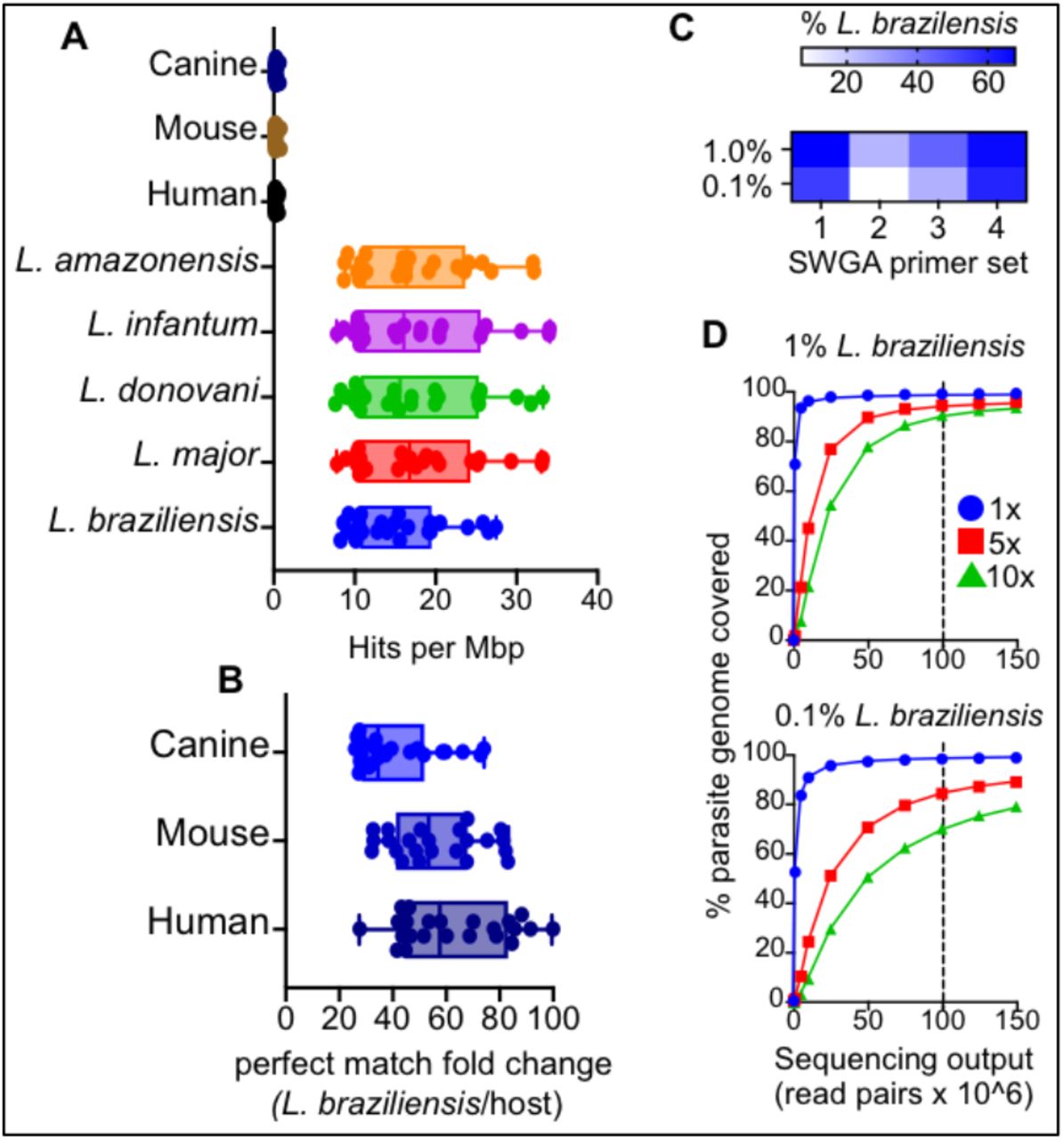
(A) The number of exact match ‘hits’ per megabase (Mbp) for each of the 23 identified SWGA primers against Leishmania and host reference genomes, and (B) the fold difference in exact matches against L. braziliensis compared to human, mouse, or canine genomes. (C) Heatmap showing percent reads aligning to L. braziliensis for each of the four SWGA primer sets used to carry out SWGA on known ratios of L. braziliensis DNA spiked into human genomic DNA (0.1 and 1% final parasite DNA). (D) The number of reads is shown in relation to the percentage of the parasite genome covered at ≥1x (blue line), 5x (red line) and 10x (green line). Vertical dashed line indicates a sequencing effort of 100 × 106 150bp paired-end reads.
Similarly, when our primers were tested against the mouse or canine reference genomes, we observed 53-fold and 34-fold enrichment, respectively, of predicted primer binding to the L. braziliensis genome over these hosts (Figure 1B). Taken together, these in silico data suggest that primers designed using SWGA are valuable in a wide range of contexts, from natural infection of humans and canines to experimental infections of mice. L. braziliensis is known to be present at low levels in skin lesions. Less than 1% of total reads from RNA-seq studies of lesions map to the parasite [10]. To evaluate the efficacy of our SWGA assay in a controlled setting that mimics patient samples, we prepared purified human DNA spiked with either 1% or 0.1% (w/w) purified L. braziliensis genomic DNA. Using high-throughput sequencing, we evaluated the ability of four separate SWGA primer sets, each consisting of 10 SWGA primers, to selectively amplify parasite DNA in these synthetic samples. After a 16-hour isothermal SWGA reaction, we found that multiple primer sets resulted in substantial amplification of the synthetic samples. Primer set 1 (PS1) and PS4 yielded the best results, achieving ≥60% of parasite-mapping reads in samples that started with only 1% or 0.1% L. braziliensis DNA (Figure 1C). We next examined the depth and breadth of coverage following SWGA of these synthetic samples. For each sample, sequencing data from individual SWGA reactions (PS1, PS2, PS3, PS4) were combined and mapped to the parasite genome, and depth and breadth of coverage were evaluated at different sequencing efforts. In the 1% spike-in control, after SWGA, a sequencing effort of ∼100M paired-end reads (Figure 1D, top, vertical dashed line) yielded 10x coverage across nearly 90% of the parasite genome, and 5x coverage across over 94% of the genome. Similarly, when the synthetic sample containing only 0.1% parasite DNA was used, the same sequencing effort resulted in 10x coverage across over 70% of the parasite genome and 5x coverage across 84% of the genome (Figure 1D, bottom). These data show that even when L. braziliensis DNA is present at incredibly low levels, and in the presence of abundant contaminating human DNA, SWGA yields an excellent breadth of coverage across the 32Mbp parasite genome.
Validation of SWGA assay on primary human and mouse samples
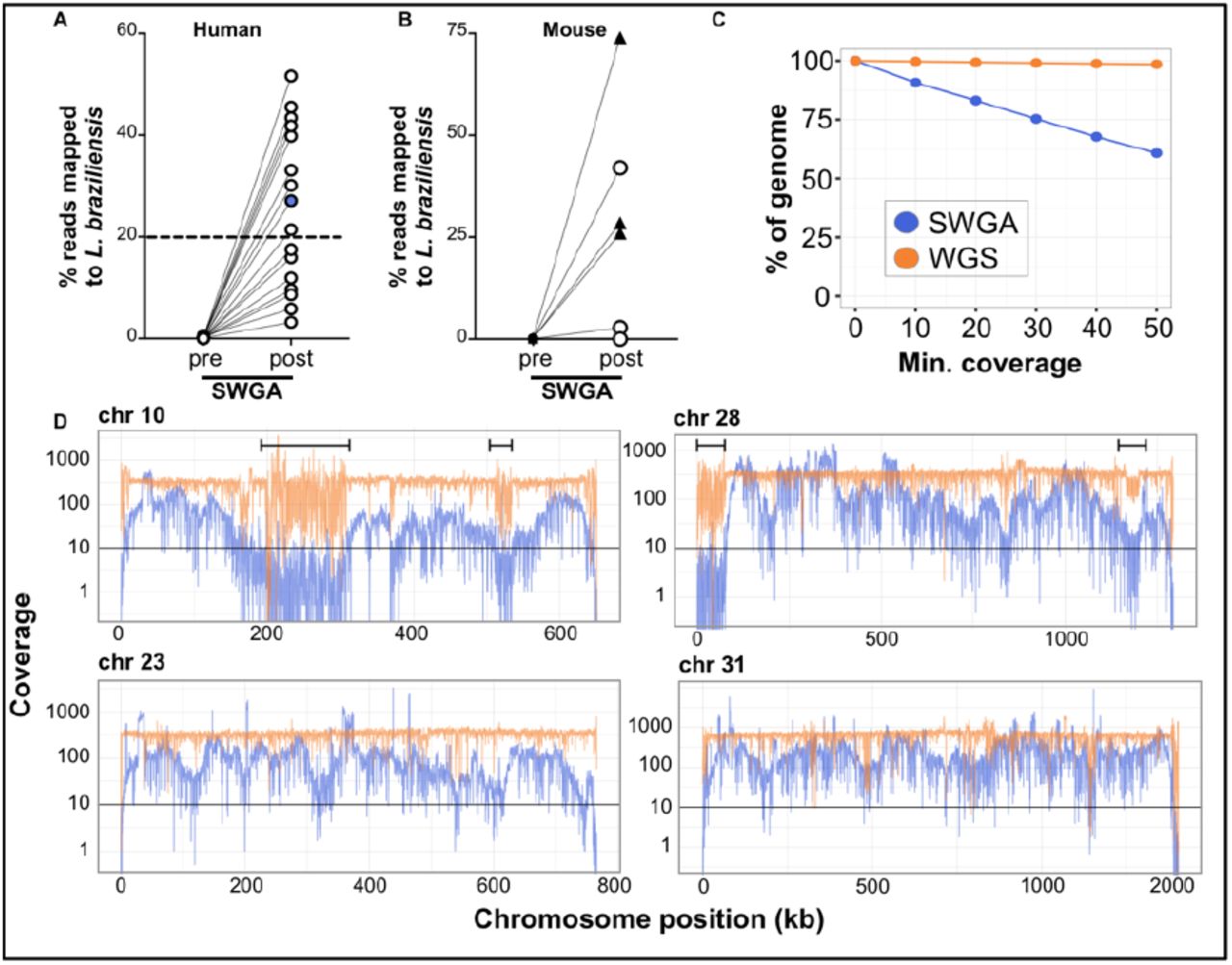
We next tested our SWGA protocol on primary patient samples. DNA extracted from skin punch biopsies from 16 L. braziliensis patients was subjected to high-throughput sequencing before and after SWGA. Reads from these pre- and post-SWGA samples for each patient were mapped to the parasite genome to evaluate depth and breadth of coverage. Direct sequencing of DNA extracted from lesions showed that less than 0.5% of reads mapped to the parasite before SWGA (Figure 2A), consistent with previous reports of extremely low parasite burden in L. braziliensis lesions [10]. However, following SWGA, these same samples showed dramatic increases in the proportion of parasite-mapping reads, ranging from 2% to 55%, with over half of the patient samples (9/16) having ≥20% of reads mapping to the parasite (Figure 2A, dashed line).

Percentage of reads mapping to L. braziliensis genome in DNA from (A) patient lesion biopsies or (B) infected mouse ears (n=3 animals infected with L. major, triangles; n=5 animals infected with L. braziliensis, circles), sequenced before (pre) and after (post) SWGA. Data shown are from the SWGA primer set the yielded the best amplification for each sample. (C) Genome coverage for SWGA data from a single patient sample (patient #7, blue point from panel A). (D) Coverage of four selected L. braziliensis chromosomes in SWGA data from a single patient (#7; blue lines) compared to whole genome sequencing (WGS) of pure, cultured L. braziliensis (orange lines). Data shown in panel C and D are merged from all SWGA primer sets to maximize coverage.
Next, we selected SWGA data from a single patient sample (#7; Figure 2A, blue point) and measured coverage across the parasite genome (Figure 2C), which showed that over 80% of the genome was covered at 10x depth by SWGA, and over 50% of the genome at 50x. Based on these data, we reasoned that SWGA may be a useful tool for monitoring parasite genotypes linked to drug resistance and disease phenotypes. SWGA data from the same patient sample was evaluated for coverage across the full length of each of the 35 parasite chromosomes (Supplemental Figure 1). We focused our initial analysis on chromosomes 10, 23, and 31 since they encode the GP63, MRPA, and AQP1 genes, respectively, which have previously been linked to drug resistance in other Leishmania species [33–36]. In addition, chromosome 31 is known to have extra copies that have been linked to parasite adaptation to stress [37–39]. Lastly, we examined chromosome 28 because it has been linked to atypical manifestations of CL [40,41]. We observed over 10x coverage across most of the length of each of these chromosomes following SWGA (Figure 2D, blue lines). Regions of poor coverage in our SWGA sample often corresponded to ends of chromosomes or to regions (Figure 2D, brackets) that were poorly covered in whole genome sequencing (WGS) of pure cultures of L. braziliensis (Figure 2D, orange lines). This result likely reflects low complexity regions that pose a challenge to genome sequencing for L. braziliensis, rather than issues specific to SWGA. These data indicate that SWGA directly applied to primary patient samples generates high-quality data suitable for high-resolution parasite genotyping.

Based on our in silico analysis (Figure 1A-B), we predicted that our SWGA primer sets would be effective in other species of Leishmania, as well as, in other host backgrounds. To formally test this, we infected mice with either L. braziliensis (same target parasite species, but different host species background) or L. major (different parasite and different background) and carried out SWGA on DNA extracted from whole ears recovered from these mice. Like human primary samples, tissues from experimentally infected mice have nearly undetectable levels of parasite sequences prior to SWGA (Figure 2B). After SWGA, however, the proportion of parasite reads increased to over 20% in one animal infected with L. braziliensis and three animals infected with L. major (Figure 2B, circles and triangles, respectively). These data are consistent with the notion that L. major has a higher parasite burden than L. braziliensis.
Somy analysis with SWGA
Leishmania parasites exhibit mosaic aneuploidy, and it has been suggested that modulating chromosomal copy number provides the parasite with a mechanism for regulating gene dosage in the absence of promoter-driven gene expression [42,43]. Previous attempts to use allele frequency to estimate somy of L. infantum were unsuccessful due to a low number of heterozygous SNPs in this parasite species [44]. Since L. braziliensis has been reported to have a higher number of SNPs than other Leishmania species [7], we tested whether the alternate allele read depth proportion (AARDP), as determined by SWGA, could be used to infer chromosome copy number. We first examined AARDP in DNA isolated from pure L. braziliensis cultures and subjected to either traditional WGS or SWGA (Figure 3A and 3B, respectively). Sharp peaks in the WGS sample centered over an AARDP of 0.5 indicated that chromosomes 10, 23, and 28 were disomic. In contrast, three distinct peaks were observed for chromosome 31 centered on an allele frequency of 0.25, 0.50, and 0.75, consistent with multiple previous reports that this chromosome is supernumerary, and potentially tetrasomic [37]. SWGA of the same pure culture closely resembled the WGS data, albeit with allele frequency peaks that were slightly less sharp (Figure 3B). In contrast to the data from pure cultures, AARDP analysis of SWGA data from three separate patient samples showed broad peak profiles, making it difficult to determine somy (Figure 3C). The high amount of host contamination present in our patient samples could impact the quality of the allele frequency data. To evaluate this, we turned back to our synthetic controls (Figure 1C-D). AARDP analyses of SWGA data from the synthetic controls showed reasonably sharp peaks when 1% parasite DNA was used, but greatly diminished signal with 0.1% parasite DNA, suggesting that SWGA generates data of sufficient quality for somy determination in L. braziliensis but that host DNA contamination impacts the ability to determine allele frequency from SWGA experiments. Our patient samples have <0.5% parasite DNA prior to SWGA (Figure 2A) and are therefore more similar to the 0.1% synthetic control data.

Alternate Allele Read Depth Proportion (AARDP) histograms for L. braziliensis chromosomes 10, 23, 28, and 31, for (A) whole genome sequencing (WGS) of pure cultured parasites, (B) SWGA of pure cultures, (C) SWGA on three patient samples from Figure 2A, or (D) synthetic controls consisting of 1% or 0.1% parasite DNA spiked into human DNA. Peaks centered on 0.5 indicate disomic chromosomes, while peaks at approximately 0.25, 0.5 and 0.75 indicate trisomic chromosomes.
A high-throughput screen of patient samples using SWGA
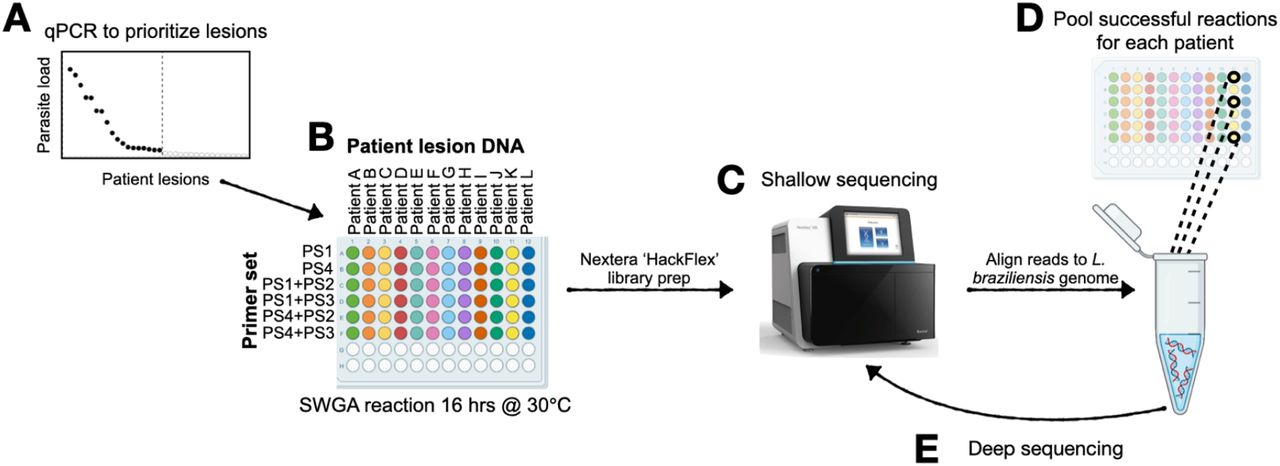
Routine diagnosis of L. braziliensis infection is carried out by collection of a punch biopsy from the site of the skin lesion followed by DNA extraction and parasite-specific PCR. We reasoned that this original DNA extract from a diagnostic biopsy, which is often archived for retesting purposes, could be sufficient for large-scale generation of parasite genomes by SWGA. To test this, we devised a screening approach that allowed us to scale our SWGA assay by an order of magnitude. 165 archived patient samples, of which 51 were intact skin biopsies and 114 were diagnostic DNA samples, were acquired from the health clinic in Corte de Pedra, Brazil. We anticipated that successful SWGA reactions would be positively correlated with parasite burden, therefore, our screen involves first prioritizing samples for SWGA using a parasite-specific qPCR [45](Figure 4A). Based on qPCR results, 66 patient samples with the highest parasite burden were selected for SWGA (Supplementary Table 2). SWGA reactions were then arrayed in 96-well plates using different SWGA primer sets (Figure 4B). Since PS2 and PS3 performed more poorly on synthetic samples (Figure 1C), we chose to only use these primer sets in second-round SWGA reactions that had first undergone an initial round of SWGA with PS1 or PS4. These ‘nested’ SWGA reactions aim to amplify greater breadth of the parasite genome. Following SWGA, sequencing libraries were prepared, pooled, and subjected to shallow sequencing (Figure 4C). For each patient sample, all SWGA reactions yielding ≥20% reads mapping to the parasite from a shallow sequencing run were considered successful. The corresponding libraries were pooled (Figure 4D) and subjected to resequencing (Figure 4E). This screen of 66 patient samples yielded parasite genomes from 18 patients (27% success rate) with a median percentage of the parasite genome covered at ≥10x of 86.6% and a median genome coverage of 38x (Table 1). Given broad coverage across each of the 18 parasite genomes generated by SWGA, we next sought to call single nucleotide polymorphisms (SNPs) and insertions/deletions (INDELs) against the reference L. braziliensis genome. Across all 18 SWGA-generated genomes we observed a median of 85,032 SNPs and 17,857 INDELs, a finding that is consistent with the number of SNPs/INDELs previously reported in genome sequences from cultured isolates of L. braziliensis [7].

(A) qPCR is used to prioritize samples that have the highest parasite burden and, therefore, the greatest likelihood of success for SWGA. (B) SWGA is carried out in 96-well plates using multiple primer sets and primer set combinations (plate rows) for each patient (plate columns). (C) Shallow sequencing is used to determine which samples showed the best amplification by SWGA. (D) All successful SWGA reactions are pooled for each patient and (E) subjected to deep sequencing.
Selective whole genome amplification of L. braziliensis from primary patient samples.
Integrating SWGA and WGS genomes for population genomics of L. braziliensis in South America
Several L. braziliensis genomes have been generated from cultured parasite isolates, which prompted us to ask whether SWGA generates genomes of sufficient quality to compare with isolate data for large-scale population genomic studies. We carried out an integrated analysis of our 18 L. braziliensis SWGA genomes together with 41 publicly available L. braziliensis genomes generated from cultured isolates, including 4 from Bahia, Brazil [46], 10 from Pernambuco, Brazil [7], 1 from Rondônia, Brazil [47], 18 from Peru [47], 6 from Colombia [8], and 2 from Bolivia [8,47]. Collectively, these 59 genomes span a wide geographic range (Figure 5A), with our SWGA samples contributing genomes from areas of Bahia, Brazil that were not previously covered by other studies (Figure 5B). Principal component analysis (PCA) of SNP data from these genomes shows clear separation by geographic location (Figure 5C), with L. braziliensis genomes from Brazil clustering tightly together (Figure 5C, upper right) but distinct from Colombian, Peruvian, and Bolivian isolates. Two genomes from a forested region of Brazil appear distinct from other Brazil samples [7], while a single genome from Rondônia in Western Brazil – bordering Bolivia – clustered with the Peru/Bolivia/Colombia isolates (Figure 5C, lower right). These data support the hypothesis that geography influences population genetic structure in L. braziliensis. Upon closer examination of the dense cluster of highly similar genome sequences from Northeastern Brazil (Figure 5C, inset), we observed a separation between SWGA sequences from Bahia (inset; triangles) and those from Pernambuco (inset; circles). To confirm that this separation was not an artifact of using SWGA, we included two control samples in which genome sequence data was generated from the same cultured laboratory clone of L. braziliensis from Brazil by either traditional WGS (Figure 5C, inset; black circle) or SWGA (white triangle). These two data points are indistinguishable from each other on PCA and cluster with other genomes from Brazil, demonstrating that the SWGA method itself is not likely to be a significant contributor to the variation observed in this analysis.

(A) Map showing all 59 samples, from this study and four previously published reports, included in the analysis. (B) Zoomed view of Bahia, Brazil showing region covered by samples from this study. White point indicates position of field hospital where patients were seen. (C-D) Principal component analysis of SNP data from 59 genomes, colored by country of origin. (E) Maximum likelihood tree constructed using 994393 variants from 59 L. braziliensis genomes and the L. guyanensis outgroup, compared to the L. braziliensis reference. Branch length of outgroup was shortened for figure preparation. Tree is rooted using the L. guyanensis outgroup.
To view the genomic variation for these 59 genomes with more clarity, we plotted the first four principal components – which collectively account for over 38% of the total variance – separately, allowing us to see how each sample contributes to each principal component (Figure 5D). When viewed in this way, PC1 clearly separates two of the Colombia isolates from all other genomes, consistent with a high number of SNPs previously described for these samples[8]. PC2 separates Brazil samples from all other samples, regardless of whether they are from SWGA or WGS of cultured isolates. PC3 separates the two WGS samples from Paudalho, Pernambuco, Brazil, from all others, while PC4 separates samples originating from Colombia versus Peru. Collectively, these data point to country and, to a much smaller extent regional differences, as being associated with genetic variation in L. braziliensis. Our data show that integrating these data opens the door to comparing SWGA data in the context of a growing number of WGS datasets for L. braziliensis.
Phylogenetic analysis supports the hypothesis that both the forested Pernambuco, Brazil samples and two samples from Colombia are quite unique (Figure 5E). Similar to the PCA, the tree shows that the sample from Western Brazil is more similar to samples from Peru and Bolivia. The SWGA samples are most closely related to the public data also from Bahia, Brazil followed by the non-forest Pernambuco, Brazil samples. The phylogenetic tree further supports the conclusion that SWGA and WGS genomes can be compared as our cultured laboratory clone of L. braziliensis falls within the same clade with a high bootstrap value.
Identifying variants unique to Northeast Brazil where treatment failure rates are high
Treatment failure rates are high in Northeastern (NE) Brazil [48] and our SWGA genomes cover a region in NE Brazil not well represented by previous WGS studies. This, together with the fact that many of our SWGA genomes (15/18) came from patients who failed therapy with pentavalent antimony (Supplementary Table 2), prompted us to ask whether our data could be used to identify parasite variants unique to NE Brazil, and therefore, potentially linked to treatment failure. Toward this end, we carried out a systematic identification and annotation of genomic variants from all 59 L. braziliensis genomes available, yielding over 600,000 high-quality variants, including nearly 110,000 missense and 634 frame-shift variants (Figure 6A, column 1). Over 120,000 of these variants were present in our SWGA genomes (Figure 6A, column 2), and our data identified 5,812 novel variants not previously observed in other studies including 1,204 missense and 277 frame-shift variants (Figure 6A, column 3). Notably, nearly half of the total L. braziliensis frame-shift mutations were contributed by our SWGA data.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Table showing variants identified by integrated analysis of WGS and SWGA genomes (top), and studies included (+) or excluded (-) from the analysis (bottom). Venn diagrams indicate how each of the five studies (labeled a-d) were used in the integrated analysis to generate the variants shown in table column above. (B) Bubble chart showing results of Gene Ontology (GO) enrichment for Molecular Function terms associated with 149 genes containing frame-shift variants (left) or 152 genes identified with high-frequency missense mutations in Northeast (NE) Brazil (right). All terms shown were associated with ≥ 5 genes. FC = fold change; FDR = false discovery rate (Benjamini-Hochberg correction). (C) Four representative parasite genes that were enriched for high-frequency missense mutations in genomes from Northeast Brazil.
Next, we focused our analysis on two types of variants, frame-shift and missense mutations, since they have a high potential for impacting protein sequence. In particular, we were interested in these variants when present in NE Brazil – including our 18 SWGA genomes and 14 genomes from two other studies [7,46] – but absent from genomes collected from Colombia, Bolivia, Western Brazil, and Peru (Figure 6A, column 4). 316 frame-shift mutations were found to be specific to NE Brazil and occurred in 303 genes, of which 51% (154) were annotated as conserved hypothetical genes (Supplementary Table 3). Gene ontology enrichment analysis of the remaining 149 genes revealed enrichment of functional terms associated with post-translational modifications, including protein phosphorylation (2.7 fold enrichment; FDR = 0.03) (Figure 6B, left). In addition, we identified 13,831 missense mutations specific to NE Brazil. We reasoned that many of these variants were likely observed at low frequency (only found in one or a few samples), thus we further refined this list by selecting for variants that were observed at high frequency in NE Brazil but not elsewhere (see methods). This analysis yielded 1916 variants. To focus on genes with the potential to be most impacted by these mutations, we selected only genes that had ≥ 2 of these missense mutations, resulting in a list of 347 genes, of which 52% (195) were conserved hypothetical proteins (Supplementary Table 3). GO analysis of the remaining 152 genes showed significant enrichment of ubiquitin transferase activity (> 6 fold enrichment; FDR = 0.01) (Figure 6B, right). Included amongst this list were 12 genes with putative kinase domains, 4 SPRY-domain/HECT-domain-containing (ubiquitin-transferase) proteins (LbrM.32.2.004170, LbrM.13.2.001230, LbrM.07.2.000290, and LbrM.35.2.006640), one ubiquitin carboxyl-terminal hydrolase (LbrM.16.2.000720), one putative E1 ubiquitin-activating enzyme (LbrM.34.2.002970), and two putative cullin protein neddylation domain-containing proteins (LbrM.16.2.001260 and LbrM.25.2.001240) (Supplementary Table 3).
Five RNA binding proteins were also identified in this analysis (LbrM.18.2.000200, LbrM.18.2.001450, LbrM.24.2.001860, LbrM.29.2.001510, LbrM.30.2.001230, and LbrM.33.2.001710). In some cases, these genes had high frequency variants both within and outside of NE Brazil, but present at different locations in the gene (Figure 6C and 6D). For other genes, high frequency missense mutations were only observed in NE Brazil (Figure 6E and 6F). Collectively, these results raise the possibility that L. braziliensis strains circulating in NE Brazil may undergo unique post-transcriptional or post-translational modifications that could, in turn, increase resistance to chemotherapy.
DISCUSSION
The slow growth of L. braziliensis, together with low parasite burden present at the site of the lesion and relative scarcity of modern laboratory infrastructure in areas endemic for CL, have made it difficult to isolate, culture, and sequence a diverse range of parasite strains for population genomic studies. One recent strategy for addressing these challenges in L. donovani used custom biotinylated ‘bait’ sequences and streptavidin-conjugated beads (Agilent SureSelect technology) to enrich for parasite DNA in samples from visceral leishmaniasis patients [15]. However, the relatively high cost of this assay coupled with the need to redesign new baits for different species of Leishmania limit more widespread adoption of this approach. The data presented here show that simple pools consisting of ten 8-mer primers can be used to selectively amplify L. braziliensis genomes – and likely L. major – from complex primary patient samples. Aside from these oligonucleotide primers, only the Phi29 polymerase is needed and the SWGA proceeds as an isothermal room-temperature reaction, bypassing the need for a thermocycler. Furthermore, since SWGA is an amplification-based protocol, only small amounts of total DNA are needed. Taken together, our data show that SWGA is a low-cost method to generate high-quality genomes even in resource-limited areas.
Although we successfully amplified 18 parasite genomes from primary patient samples, this represented only a 27% success rate from the 66 samples we attempted to amplify with SWGA. One open question is how the efficiency of the SWGA method can be improved so that a higher number of patient samples yield parasite genomes. Host-specific restriction enzymes [20,49] may offer one appealing solution for Leishmania, particularly since L. donovani reportedly lacks C-5 DNA methylation, potentially opening the doors to using methylation-sensitive restriction enzymes to preferentially degrade host DNA [50]. Based on our data from SWGA of synthetic controls (Figure 2), primer sets 1 and 4 yielded the greatest percent of reads aligning to L. braziliensis, while primer sets 2 and 3 performed more poorly. Interestingly, sets 1 and 4 share more primers in common with each other, than they do with sets 2 and 3. Thus, we could use the sequences in sets 1 and 4 to refine the SWGA algorithm to identify new primers that may demonstrate improved performance. Despite these limitations, SWGA offers several exciting potential uses for Leishmania genetics. The Phi29 polymerase used in SWGA is highly processive and can produce amplicons up to 100kb or more in length, potentially allowing long-read sequencing of SWGA reactions to resolve complex regions in the parasite genome. We expect that SWGA will make capturing genomes of Leishmania parasites from sympatric mammalian hosts (e.g. human and canine) and insect vectors all from the same geographic area relatively straightforward, thus empowering the design of sophisticated population genetic studies.
Our 18 SWGA genomes included 15 from patients who failed treatment after a single round of chemotherapy with antimony (Supplementary Table 1). This bias in favor of successful SWGA of parasite genomes from patients who fail therapy is likely due to the higher parasite burden observed in these patients [10], thus putting the total amount of parasite DNA above a threshold for successful SWGA. Understanding why some patients have higher parasite load than others, prior to initiating chemotherapy, may help identify the root causes of treatment failure in this disease. There are many possible explanations, including variable parasite load in the insect vector, variability in host immunity, differential host immune evasion by the parasite, differing parasite replication rates, and more. All of these potential explanations could involve parasite strain genetics, yet prior to this study little was known about how L. braziliensis strains in NE Brazil – where failure rates are high – compared to those observed elsewhere in South America. Future studies to formally identify parasite variants associated with treatment outcome will require some consideration for how to successfully obtain genomes from patients who cure and, therefore, have the lowest parasite load prior to treatment. One potential solution would be to perform SWGA on skin biopsies collected from patients early in the course of disease, before the development of an ulcer. Previous studies have shown that this early stage of the disease is when parasite burden and failure rates are highest [51]. Notably, our screening approach (Figure 3), is scalable and could be used to tackle this challenge by rapidly testing many different samples and patients to identify the optimal setting to generate genomes from very low burden infections.
By integrating our SWGA genomes with public WGS data, we were able to carry out a population genetic study of L. braziliensis that spanned four S. American countries. The high-frequency variants we identified in NE Brazil were enriched in protein kinases, RNA-binding proteins, and ubiquitin-transferases. We hypothesize that these mutations may impact RNA or protein stability in the parasites. Interestingly, Leishmania and Trypanosoma parasites lack traditional promoter-based gene regulation and thus rely heavily on post-transcriptional and post-translational mechanisms for modulating gene expression in the face of environmental stressors and cues [52,53]. For example, RNA binding proteins in Trypanosomes are critical for differentiation of the parasite through its lifecycle [54,55]. Post-translational modifications have also been linked to drug resistance in Leishmania, as evidenced by the discovery that calcium-dependent protein kinase 1 (CDPK1) is important in resistance to paramomycin [56]. In addition, overexpression of translational machinery, such as ribosomal proteins, is sufficient to reduce sensitivity of L. donovani to multiple drugs, including antimony [57]. Collectively, our data constitute a high priority set of variants and genes that could be experimentally explored in future studies to identify mechanisms of drug resistance in Leishmania braziliensis.
MATERIALS AND METHODS
Data and code availability
Raw reads for all 18 SWGA genomes are available on the Sequence Read Archive (SRA) under accession number PRJNA875085. All code used for analysis of depth and breadth of coverage in SWGA samples, and annotation, analysis, and visualization of variants is available as a fully reproducible dockerized code “capsule” archived on Code Ocean (DOI pending).
Human and mouse sample collection
Skin lesion biopsies were obtained with informed consent from cutaneous leishmaniasis (CL) patients seen at the Health Post of Corte de Pedra, Bahia, Brazil, in accordance with local ethical guidelines (Ethical Committee of the Federal University of Bahia Medical School, Salvador, Bahia, Brazil and the University of Pennsylvania Institutional Review Board). All samples were collected prior to initiating treatment. A 4-mm diagnostic punch biopsy was collected from the border of the lesion of CL patients and DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega). CL diagnosis was determined by a positive skin lesion PCR for L. braziliensis and a positive intradermal skin test with Leishmania antigen. These diagnostic DNA samples were the same ones used in this study. For some patients, an additional biopsy was collected and stored in RNAlater (Thermo Fisher Scientific) for shipment. Biopsies were homogenized, and DNA was extracted using the MP Bio FastPrep Tissue Homogenizer and Qiagen Blood and Tissue kit according to the manufacturer’s instructions.
For mouse experiments, L. braziliensis (MHOM/BR/01/BA788 strain) and L. major (Friedlin strain) parasites were grown in Schneider’s insect medium (GIBCO) supplemented with 20% heat-inactivated fetal bovine serum (Atlanta Biologicals) and 2 mM glutamine (Sigma). Metacyclic promastigotes were enriched from stationary-phase parasite cultures by density gradient centrifugation before infection as previously described [58]. Briefly, parasites were suspended in PBS and layered on a step gradient of 40% and 12% Ficoll 400 (Sigma) before centrifuging at 2400 rpm for 10 minutes. C57BL/6 mice were infected intradermally in the ear with 1×106 L. braziliensis or L. major. At the peak of ear swelling (∼4-6 weeks post-infection), mice were humanely euthanized, ears were collected, homogenized, and DNA extracted as described above for human samples. All animal work was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee, University of Pennsylvania.
SWGA primer design and validation
We used the program swga [16] to generate a list of 172 candidate primers that preferentially bind to the Leishmania braziliensis reference genome (MHOM/BR/75/M2904 2019) over a complex background genome that consisted of human (GCA_000001405.28), Staphylococcus aureus (GCA_000746505.1), and Streptococcus pyogenes (GCA_000006785.2). We scored these candidate primers and designed primer sets using an updated machine-learning-guided and thermodynamically-principled version of the SWGA algorithm, swga2.0 [31](software available at https://anaconda.org/janedwivedi/soapswga). Overall, 23 unique 8-mer primers with the highest evaluation scores calculated from swga2.0 were generated (Integrated DNA Technologies). The last two bases of the primers were phosphorothioated, which prevents primer degradation by phi29 polymerase [21]. In silico validation was carried out by counting exact matches for each SWGA primer against a range of target and background genomes using the Unix grep command, and hits per Mbp and the fold difference in predicted binding sites were calculated and visualized using Prism 9. The target genomes included L. braziliensis (see above), L. major (TriTrypDB-55_LmajorFriedlin), L. donovani (TriTrypDB-46_LdonovaniBPK282A1), L. infantum (TriTrypDB-56_LinfantumJPCM5), and L. amazonensis (TriTrypDB-56_LamazonensisMHOMBR71973M2269). Background genomes included human (Homo_sapiens.GRCh38), Mus musculus (GCF_000001635.27_GRCm39), and Canis lupus familiaris (Canis_lupus_familiaris.CanFam3.1). The human, mouse, and canine reference genomes were filtered to only include the autosomal chromosomes, sex chromosomes, and mitochondrial DNA for the analysis. Primers were grouped into four sets of 10 primers each (Supplementary Table 1). Genomic DNA extracted from human foreskin fibroblasts (HFF) cells and an axenic culture of L. braziliensis promastigotes using the DNeasy Blood and Tissue kit (Qiagen) were mixed to generate 1% and 0.1% L. braziliensis:human DNA (w/w).
SWGA on primary patient and mouse samples
DNA from human or mouse samples was quantified using a Qubit 3.0 fluorometer. qPCR was performed on a ViiA 7 machine (Applied Biosciences) using SsoAdvanced Universal Probes Supermix (BioRad) for both Leishmania kinetoplast DNA [45] and the human 18S rRNA gene (Biomeme Inc). Ct values for Leishmania were normalized using the human 18S rRNA gene to prioritize lesions with the highest parasite burden for SWGA. All qPCR reactions were carried out in duplicate. SWGA was performed by combining ∼50 ng of the sample DNA, 3.5mM of an SWGA primer set, 1x phi29 buffer, 30 U of phi29 polymerase enzyme (New England Biolabs), 4mM dNTPs (Thermo Fisher Scientific), 1% bovine serum albumin and nuclease-free water in a total volume of 50μL. Thermocycler cycling conditions included a 1 hr ramp down step (35°C to 30°C; 10 min per degree), 16 hr amplification step at 30°C, 10 min denaturing step at 65°C and hold at 4°C. For second-round SWGA reactions, ∼50 ng of first-round SWGA product was subjected to a second round of SWGA with a different primer set. Ten ng of first-round or second-round SWGA product was used to generate libraries using the Hackflex [59] protocol and subjected to shallow sequencing on an Illumina NextSeq 500 or NextSeq 2000 to produce 1-4 million 75 or 150 single-end reads per SWGA reaction. Reads were trimmed with Trimmomatic [60], aligned to the appropriate Leishmania reference genome using Bowtie2 [61], and summarized with MultiQC [62]. All SWGA reactions that showed >20% reads aligning to L. braziliensis were pooled by patient and subsequently resequenced to generate ≥100 million paired-end 150 bp reads. Genome coverage was estimated based on the median gene coverage, excluding genes with outlier coverage, removed with iterative Grubbs’ test.
Variant calling, phylogeny, and somy analysis
Sequencing data from different SWGA primer sets were combined for each sample using the Unix cat command. In addition to data from the 18 SWGA samples, publicly available raw sequence reads were also obtained for 41 L. braziliensis cultured isolates from Colombia, Bolivia, Brazil, and Peru [7,8,46,47] that were subjected to whole genome sequencing (WGS). Reads were trimmed with Trimmomatic [60] (filtering parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36) and mapped to the L. braziliensis MHOM/BR/75/M2904 2019 reference genome using bwa-mem v.0.7.17 [63]. Alignments were reported in bam files, which were sorted, and indexed with SAMtools [64], and reads were tagged with a sample ID using Picard Tools AddOrReplaceReadGroups [65] similar to previously described [66]. Genome coverage was estimated using BEDtools genomecov command with 100 bp windows [67]. The percent of the L. braziliensis genome covered at ≥1x, 5x, and 10x was calculated from the resulting bed file. SNPs and indels were called using The Genome Analysis Toolkit (GATK) v.4.1.0.0 [68] HaplotypeCaller and Freebayes v.1.3.2 [69] in ‘discovery’ mode, with a minimum alternative allele count set to ≥5. Only variants found by both methods were retained for downstream analysis. The SWGA and WGS data were merged and sorted with BCFtools v.1.9 [70] and regenotyped using Freebayes. A bed file that contained only regions with ≥10x coverage in at least 14 out of the 18 SWGA samples was used to filter the SWGA and public WGS data for population and phylogenetic analysis.
For phylogenetic analysis, biallelic sites were selected with BCFtools [70], and variant calls were filtered by quality (QUAL>500) with VCFtools [71] and by linkage disequilibrium with Plink v.1.9 [72] (parameters used: r2=0.5, step size=1, window size=10kb). Principal component analysis was carried out with Plink v.1.9. For phylogenetic tree generation, L. guyanensis MCAN/CO/1985/CL-085 (ERR205773) was mapped to the L. braziliensis reference as above to be used as an outgroup to root the tree. Sequences were extracted from the merged SWGA, public, and outgroup variant call format (VCF) file with vcf2phylip v2.8 [73] and a maximum likelihood phylogenetic analysis was performed using IQ-TREE v.2.0.6 [74] (parameters used: ModelFinder Plus, and 10000 bootstrap replicates for SH-aLRT). The resulting tree and geospatial data were visualized with Microreact [75]. Genomic variants were annotated with snpEff [76], which was configured using a custom database prepared from the L. braziliensis genome fasta file, coding sequence (CDS) fasta file, Gene Transfer Format (GFF) file, and codon usage data, all of which were obtained from TriTypDB.org (release 58) [77,78]. Filtering of variants by quality and type was carried out using SnpSift [79], and comparisons of variants between any two sets of samples were carried out using the isec function from BCFtools [70]. For high-frequency variants, snpSift was used to identify only missense mutations with an allele count greater than the number of samples in the group (n=32 for NE Brazil, n=27 for non-NE Brazil). For example, since Leishmania is diploid, an allele count of 32 in a group of 32 samples could be achieved if all samples were heterozygous for a mutant allele or if half of the samples were homozygous. Data visualization was carried out using R/Bioconductor [80,81], the vcfR package [82], ggplot2 [83], DataGraph v4.7.1, Prism 9, and Sketch v91. Chromosomal somy estimation was based on the proportion of reads in the alternate allele in biallelic heterozygous positions. VCF files were imported in R using vcfR and only biallelic positions with at least 5 reads in each allele were kept. For each chromosome, the proportion of reads corresponding to the alternate allele in each SNP position was obtained and their distribution was used to infer the chromosomal somy.
Data Availability
Raw reads for all 18 SWGA genomes are available on the Sequence Read Archive (SRA) under accession number PRJNA875085.
ACKNOWLEDGEMENTS
This study was funded in part by grants from the National Institute of Allergy and Infectious Diseases (5R01AI143790, 5T32AI007532-24, and 5R01AI149456-03). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
BIBLIOGRAPHY
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.
- 15.↵
- 16.↵
- 17.
- 18.
- 19.
- 20.↵
- 21.↵
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.
- 36.↵
- 37.↵
- 38.
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵




