
Summary
Background E. coli is a highly diverse species that generates a huge global burden of antimicrobial-resistant infections. Although it is one of the most well-studied model organisms, we lack a synthesis of the genomic distribution of antimicrobial resistance (AMR) genes according to phylogenetic and ecological variables.
Methods We implemented Mash-based phylogrouping on ∼16,000 RefSeq E. coli genomes, and statistically modelled the link between phylogroups, host species categories, geographic subregions, climate, and AMR genes. We predict the burden of AMR across these categories using several metrics including overall counts of resistance genes, multidrug- and extensively-drug resistant (MDR and XDR) classifications, and frequencies of extended-spectrum β-lactamase and carbapenemase genes.
Findings AMR burden was highly variable across all measures between phylogroups, host species, geographic locations and climates. Phylogroups spanning the commensal-pathogen spectrum were found with high levels of AMR. MDR genomes were highly prevalent in North Africa and South-Eastern Asia, while XDR isolates were more common in Eastern Asia. Wild birds were associated with the highest levels of XDR and carbapenemase-encoding isolates. Carbapenemases, MDR genomes and most blaCTX-M groups were positively associated with temperature.
Interpretation This study synthesises data on clinical, ecological and anthropogenic variables impacting AMR in E. coli on an unprecedented scale, modelling its burden across clinically-relevant measures. It highlights key findings with direct relevance to managing the global spread of AMR and understanding its ecology. Finally, it emphasizes knowledge gaps in public databases that, if investigated, would further develop our understanding of AMR in this important pathogen and the risk posed across contexts.
Funding EU Horizon 2020 Programme
Biotechnology and Biological Sciences Research Council (BBSRC)
Introduction
Escherichia coli is a remarkably diverse species both genetically and ecologically. It occupies various niches, ranging from a commensal of many warm-blooded organisms, to a globally devastating pathogen, to a free-living environmental bacterium.1 It is a leading cause of mortality associated with drug-resistant infections and is the pathogen responsible for the most deaths attributed to AMR in 2019.2 In addition, carbapenem-resistant and extended-spectrum β-lactamase (ESBL)-producing E. coli are World Health Organization priority pathogens, for which development of new antibiotics are urgently needed.3 In light of the ongoing AMR crisis, it is vital to understand how resistance genes are distributed across E. coli genomes isolated from different host species, geographic locations and climates, as well as across the E. coli phylogeny.
Previous studies have investigated associations between AMR and phylogroup in E. coli in specific populations, typically restricted to smaller sample sizes (e.g. 4,5). The importance and increasing feasibility of larger studies is being recognised, with a recent study analysing the distribution of resistance genes across phylogroups in a large E. coli dataset.6 Most research focuses on AMR in human-associated and pathogenic isolates.7 To address this, another study focusing on ∼1,000 isolates selected to represent the diversity of E. coli, mostly including isolates from non-clinical origins, found AMR genes were associated with humans or domesticated animals, independent of phylogroup.8 Therapeutic and growth promoter usage of antibiotics in livestock is also well-documented, and has been linked to high rates of AMR in livestock-associated isolates.9 Resistance may also spill over into wild animal populations, and there is increasing evidence of wildlife as potential reservoirs of AMR.10
The overall prevalence of AMR also varies geographically, with associations between specific AMR genes and regions. Particular clinically-important antibiotic resistance genes may also become endemic in certain areas whilst being comparatively rare elsewhere, such as OXA-producing Enterobacteriaceae in Turkey.11 Meanwhile, the blaCTX-M enzymes make up the largest group of extended-spectrum β-lactamases (ESBLs), having become globally disseminated and displacing other enzymes of this class.12 Whilst antibiotics are used most in high-resource settings, the burden of AMR is estimated to disproportionately affect lower and middle income countries, despite the scarcity of data in these regions.2 In addition to anthropogenic factors impacting AMR incidence geographically, climate may also influence its development and spread.13
While previous studies have individually shown their importance, a large-scale study synthesising the broader genetic, ecological and anthropogenic factors that are associated with AMR occurrence in E. coli is lacking. Focusing on these factors collectively by taking a One Health approach is needed, as their combined contributions are vital for understanding the ecological distribution and drivers of AMR, and informing policy towards limiting its spread. Using the wealth of genomic data and metadata from different hosts and geographic locations, as well as expanding on previous phylogenetic analyses with recently-developed high-resolution phylogrouping approaches14, we sought to provide such an overview. In a final dataset of over 16,000 genomes, we used powerful statistical modelling approaches to estimate overall counts of AMR genes according to geographic subregion, host category and Mash-based phylogroup. Next, we analysed the distribution of multidrug-resistant (MDR) and extremely drug-resistant (XDR) bacteria, a measure of the number of antibiotic classes an isolate is resistant to. Finally, we focused on clinically-important β-lactamase genes, namely blaCTX-M extended-spectrum β-lactamases and carbapenemases, to demonstrate the power and utility of our approach. We provide the data generation scripts for each stage of this analysis as reproducible bioinformatics pipelines to enable researchers to expand upon this work in different datasets and contexts.
Methods
Genomes
An initial dataset of 26,883 E. coli genomes was retrieved from the National Center for Biotechnology Information (NCBI) RefSeq database in February 2022 in nucleotide fasta format using ncbi-genome-download v0.3.0 (https://github.com/kblin/ncbi-genome-download).
Mash-based phylogrouping
Genome clustering was performed using methods from Abram et al.14 In brief, Mash sketches were produced for each genome with a sketch size of 10,000 and k-mer size of 21.15
Subsequently, pairwise distances between all genomes were calculated using the mash dist function in order to construct a distance matrix. Values were converted to Pearson’s correlation coefficients and then subtracted from 1 to obtain a distance measure, and clustered using the hclust function in R with the ward.D2 method. As defined in Abram et al.14, cluster height was set as the maximum height present in the hclust dendrogram multiplied by 1.25 × 10−2, and clusters used to produce dendrograms. We performed quality control by removing genomes with a mean mash distance to all other genomes >0.04. To confirm that results were congruent with phylogrouping schemes used in the literature, this approach was complemented with traditional phylogrouping16, using the EzClermont command-line tool.17
Detection of AMR genes and retrieval of metadata
NCBI AMRFinderPlus v3.10.21 was used to predict the identity of acquired AMR genes.18 We removed efflux and blaEC β-lactamase genes from the dataset, due to their ubiquity. As we could not use phenotypic resistance categories, we used the classes included in NCBI AMRFinderPlus output as a proxy for categorising genomes as multidrug-resistant (MDR; AMR genes conferring resistance to ≥ 3/20 antibiotic classes) and extensively drug-resistant (XDR; AMR genes conferring resistance to ≥ 10/20 antibiotic classes).19
Host and location metadata were retrieved and categorised using the Bio.Entrez utilities from Biopython v1.77 (Supplementary material). We filtered genomes for those with complete metadata for host species and geographic location, which excluded 10,600 genomes, resulting in a final dataset of 16,192 genomes. Geographic locations were split into 20 subregions (Fig. S1), and sample sizes across all metadata categories are shown in Tables S1-S3. Minimum monthly mean temperatures (MMMTs) were taken from monthly mean temperatures for the period 1961-1999, retrieved from the World Bank (https://datacatalog.worldbank.org/search/dataset/0040276, accessed June 2022).
Statistical analysis, computational resources and code availability
Snakemake v6.2.1 was used to generate reproducible pipelines to perform the data generation for each step of this manuscript. Univariate models were used throughout, and model specifications and selection protocols are provided in the supplementary material. The University of Exeter’s Advanced Research Computing facilities were used to carry out this work.
Results
Mash-based phylogrouping of E. coli isolates
To characterise AMR spread across the genetic diversity of E. coli, we used Mash distance calculation followed by hierarchical clustering to group the full E. coli genomic dataset into more high-resolution phylogenetic clusters. We identified 14 clusters of E. coli (an additional 6 compared to PCR-based phylogrouping), with a fourth D cluster and a second F cluster compared to the previous study (Fig. S2). By comparing with in silico PCR-based phylogrouping and results from the previous study14, we determined the final phylogroup assignment for each cluster (A, B1, B2-1, B2-2, C, D1, D2, D3, D4, E1, E2, F1, F2 and G). We also identified a 15th cluster, within which the majority of genomes were typed as cryptic using traditional phylogrouping, and this cluster was removed due to its size (n=14).
Modelling AMR gene counts across phylogroups, geographic regions, host species and climates
To provide an overview of the total burden of AMR genes across ecological and genetic categories, we modelled the total counts of AMR against phylogroup, host species, MMMT (in country of isolation) and geographic subregion using data from 16,192 genomes (Figure 1; raw data in Fig. S3). The E2 and D4 phylogroups were associated with far higher counts of AMR genes than their closest relatives E1 and D3, respectively (Fig. 1A). In addition, groups A, F1 and F2 were associated with high counts of AMR. We found a moderate positive association between minimum temperature and counts of AMR genes, with a 5.2% increase across the predicted range (−30°C–30°C) (Fig. 1B). Geographic areas associated with high AMR gene counts included South-Eastern, Eastern, Western, and Southern Asia, Northern Africa, and South America (Fig. 1C, Table S10). Finally, genomes isolated from wild birds, humans and agricultural or domestic animals were all associated with higher counts of AMR than wild animals (Fig. 1D).

Estimated means with 95% confidence intervals for total counts of AMR genes for a given genome from models of A) phylogroup with approximate phylogeny, B) minimum mean monthly temperature of country, C) geographic subregion (95% confidence intervals presented in Table S10) and D) host species category. Each plot is based on a separate statistical model.
Distribution of multidrug- and extensively drug-resistant E. coli
To provide increased clinical relevance to our AMR predictions, we categorised genomes as MDR (≥ 1 AMR genes conferring resistance to ≥ 3 classes) and XDR (≥ 1 AMR genes conferring resistance to ≥ 10 classes) following detection of 477 unique resistance genes conferring predicted resistance to 20 classes. MDR genomes were highly prevalent across most regions, reflecting trends for overall AMR gene counts, with high probabilities in South-Eastern Asia and Northern Africa (Fig. 2A, Table S11). XDR genomes were far less common, and most prevalent in Eastern Asia. In addition to occurring in regions with high MDR rates, XDR genomes were also relatively common in Western Asia, and Eastern and Western Europe (Fig. 2B, Table S12). There was a positive association between the probability of a genome being MDR and MMMT of a country (Fig. 2C), with a 22.4% increase in MDR probability from −30°C–30°C. There was a negative association between XDR probability and temperature (Fig. 2D), with a smaller effect size (12.5% decrease in XDR probability across the same temperature range). To investigate the latter trend, we determined which countries had the highest proportions of XDR isolates, finding by far the most in China (MMMT = −8.49°C) followed by other countries with relatively low temperatures such as France (3.11°C), Germany (−0.48°C) and Russian Federation (−27°C) (Fig. S4).

Estimated probabilities that a given genome will be A) MDR and B) XDR according to subregion (95% confidence intervals presented in Tables S11-S12). Estimated probabilities with 95% confidence intervals that a given genome will be C) MDR and D) XDR across a range of mean annual temperatures. Each plot is based on a separate statistical model. MDR, multidrug-resistant; XDR, extensively drug-resistant.
Across phylogroups, MDR probability was high (above 0.25 for all groups except E1; Fig. 3A). MDR probability was also above 0.5 for 9/14 of the phylogroups, and was highest in D4, F2 and E2. There was a low probability of a genome being XDR across phylogroups, with no XDR genomes found in group E1, and all other probabilities below 0.1 (Fig. 3B). The highest XDR probabilities were found in groups A and F1. Predicted MDR probabilities were above 0.4 in all host species, highest in humans and lowest in wild birds and animals (Fig. 3C). However, XDR genomes were far more common in wild birds than any other host category (Fig. 3D). All other hosts had XDR probabilities ≤ 0.05.
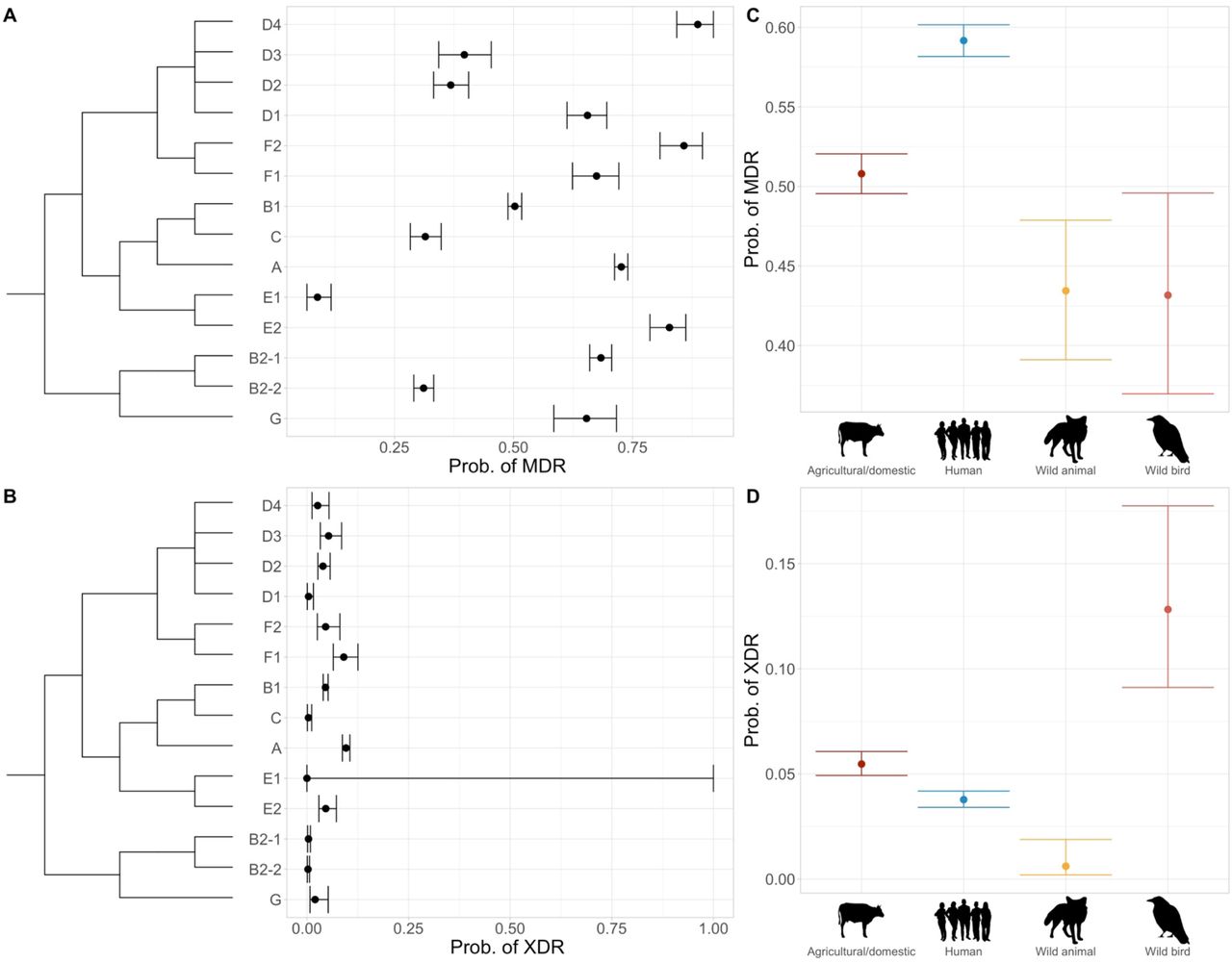
Estimated probabilities with 95% confidence intervals that a given genome will be A) MDR and B) XDR according to phylogroup (with approximate phylogeny shown). Estimated probabilities and 95% confidence intervals that a given genome will be C) MDR and D) XDR according to phylogroup. Each plot is based on a separate statistical model. Where confidence intervals span the whole range of probabilities for a phylogroup, there were too few or zero genomes to estimate them with, but the group has been included to show the whole phylogeny. MDR, multidrug-resistant; XDR, extensively drug-resistant.
blaCTX-M genes and carbapenemases
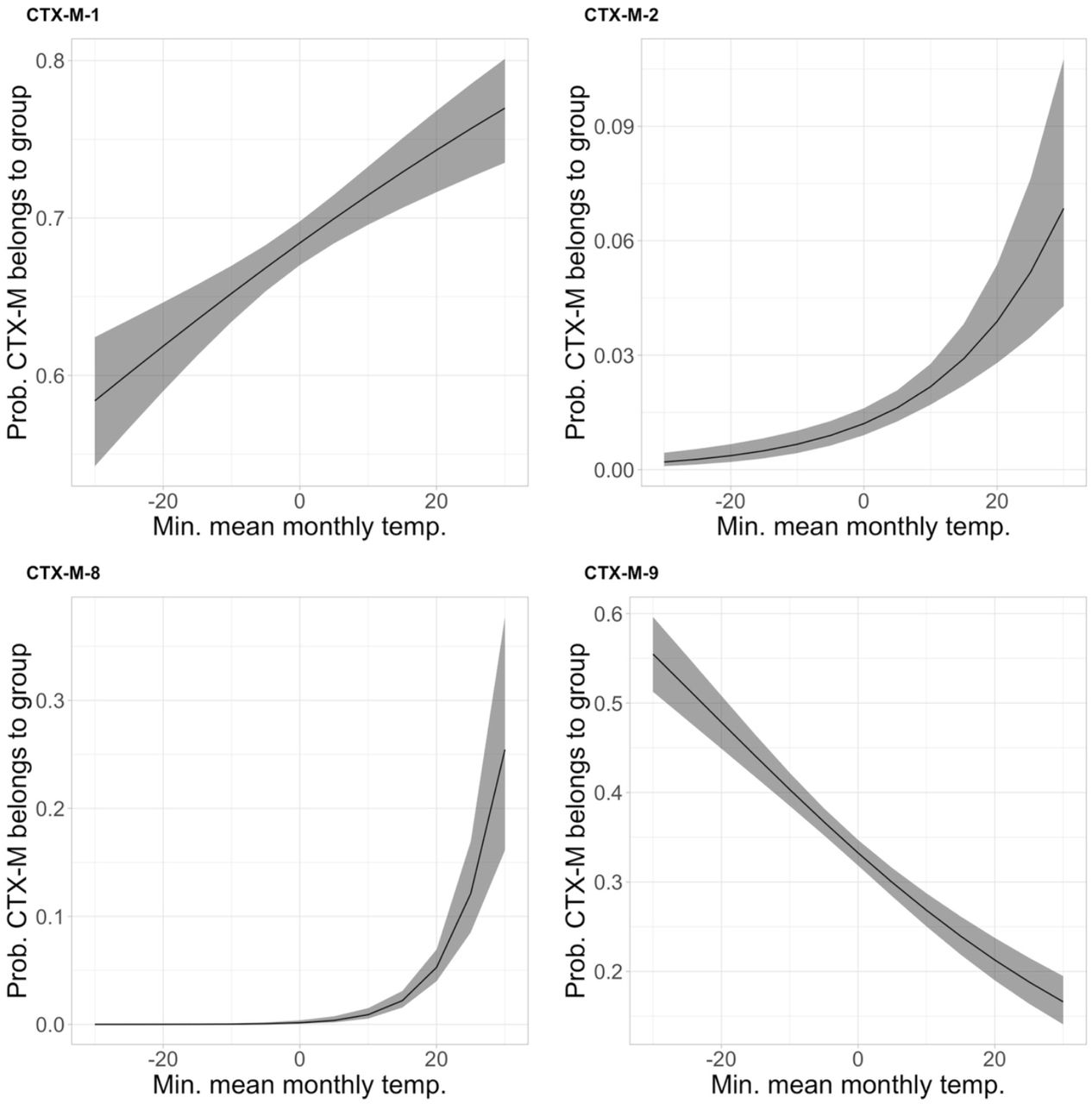
To demonstrate the power of our approach on clinically-important AMR genes, as well as current resistance gene pandemics, we next focused on blaCTX-M and carbapenemase genes. We detected 40 unique blaCTX-M genes across the final dataset, most commonly blaCTX-M −15 (n=2,536), blaCTX-M-14 (n=1,063) and blaCTX-M −55 (n=903). blaCTX-M genes were frequent in some phylogroups, at around or above 0.6 probability a genome would possess at least one in the D4, F2, E2 and B2-1 groups (Fig. 4A). However, across the majority of phylogroups, probabilities of having a blaCTX-M gene were far lower. Genomes from human hosts had the highest probability of blaCTX-M presence (Fig. 4B). There was a 4.4% decrease in the probability of a genome encoding a blaCTX-M gene across the temperature range (−30°C–30°C) (Fig. 4C). When analysing blaCTX-M-encoding genomes by evolutionary cluster, we found a strong negative association with temperature only remained for the blaCTX-M −9 group (containing blaCTX-M-14), while genes from the blaCTX-M-1 (containing blaCTX-M-55 and blaCTX-M-15), −2 and −8 groups were more probable at higher temperatures (Fig. 5). High probabilities of possessing a blaCTX-M gene were predicted in South-Eastern Asia, Northern Africa and Eastern Europe (Fig. 4D, Table S13). All other regions had < 0.45 probability of blaCTX-M gene presence.
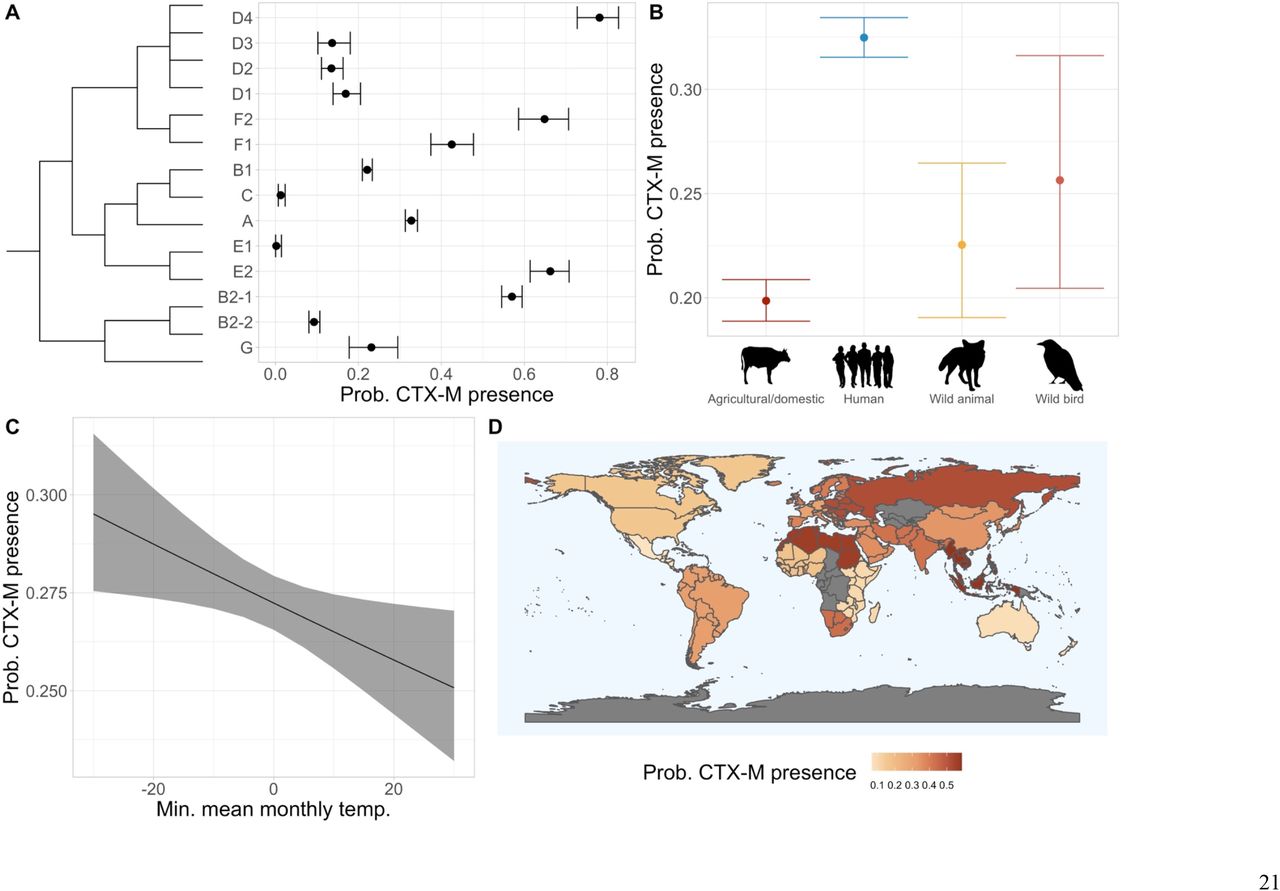
Estimated probabilities with 95% confidence intervals that a given genome will possess a blaCTX-M gene from models of A) phylogroup with approximate phylogeny, B) host species category, C) minimum mean monthly temperature and D) geographic subregion (95% confidence intervals presented in Table S13).

For genomes that possess blaCTX-M genes, estimated probabilities with 95% confidence intervals that blaCTX-M genes will belong to each class according to minimum mean monthly temperature. Each plot is derived from a separate statistical model.
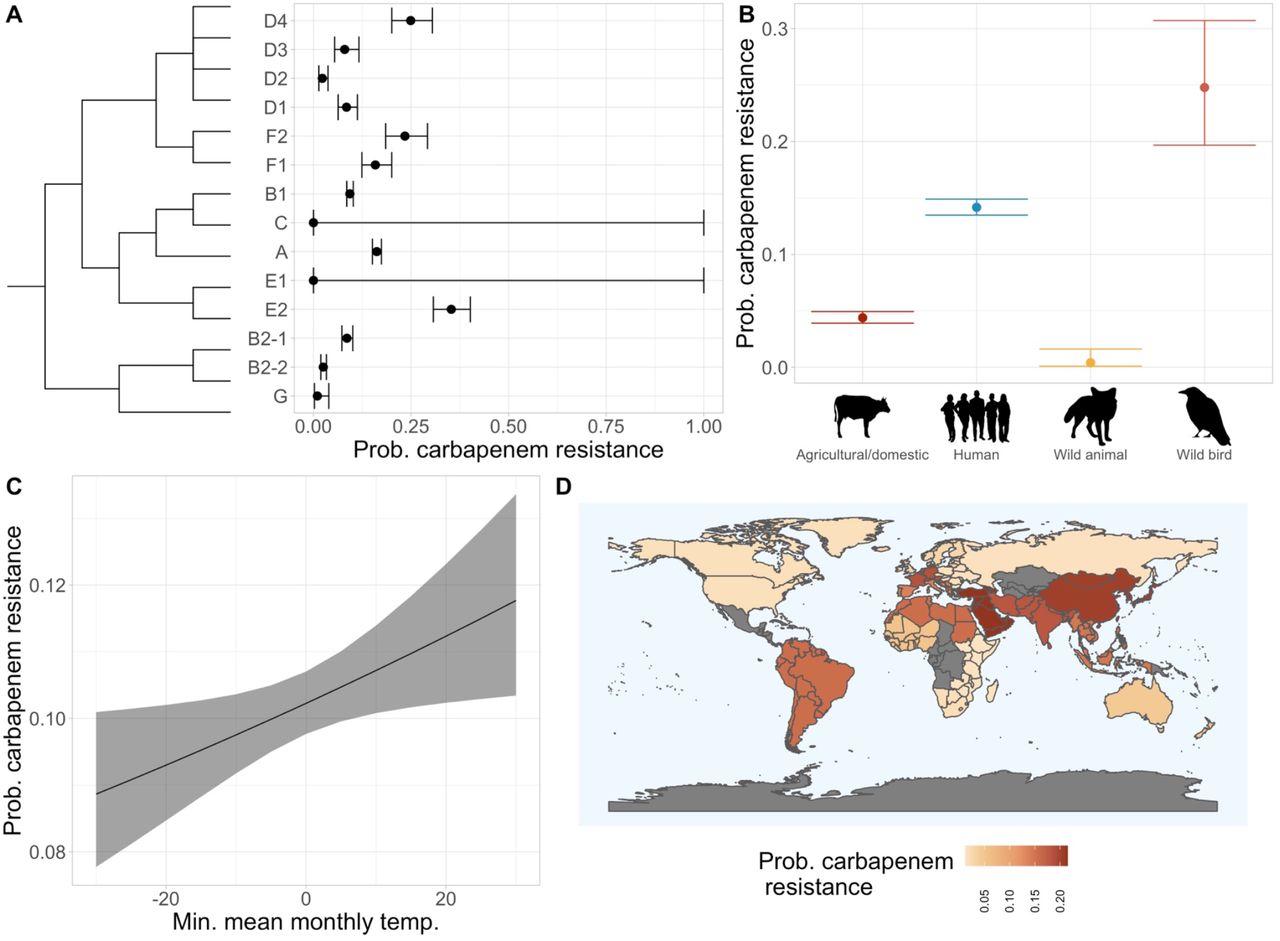
A total of 46 different carbapenemases from the NDM, KPC, OXA, IMP, VIM, IMI and GES classes were identified, the most frequent being NDM-5 (n=893) and KPC-2 (n=264). Carbapenemases were less frequent than blaCTX-M genes, with < 0.20 predicted probability of occurrence in genomes from all phylogroups except for E2, D4 and F2 (Fig. 6A). No carbapenemases were found in genomes from the E1 and C phylogroups, and they were far more common in genomes from wild birds compared to all other hosts, with ∼10% higher predicted probability of possessing this gene type than genomes from humans (Fig. 6B). These genes were uncommon in genomes from livestock, domestic animals and other wild animal hosts. The probability of a genome possessing a carbapenemase encoding gene increased with MMMT by 2.9% from −30°C to 30°C (Fig. 6C). Finally, carbapenem resistance genes had the highest predicted probabilities of occurring in genomes from Western and Eastern Asia, and Western Europe (Fig. 6D, Table S14).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Estimated probabilities with 95% confidence intervals that a given genome will possess a carbapenemase gene from models of A) phylogroup with approximate phylogeny, B) host species category, C) minimum mean monthly temperature and D) geographic subregion (95% confidence intervals presented in Table S14). Where confidence intervals span the whole range of probabilities for a phylogroup, there were too few or zero genomes to estimate them with, but the group has been included to show the whole phylogeny.
Discussion
We mapped the distribution of AMR in E. coli across phylogenetic groups, host categories, geographic subregions and temperatures, in an unprecedented synthesis of current genomic data and its associated metadata. As well as providing an informative predictive overview of different AMR metrics across these categories, we highlight some key findings relevant to managing the global spread of AMR and understanding its ecology.
Phylogroups associated with commensal lineages have high rates of AMR
Current knowledge of the phylogenetic distribution of AMR in E. coli derives predominantly from smaller-scale studies in specific regions, hosts or sample types. Though we found high rates of AMR across metrics in known pathogenic lineages, such as the O157:H7-dominated E2 phylogroup, we found a high incidence of XDR genomes in phylogroup A, which is typically associated with commensal lifestyles.20 Additionally, Phylogroup F, which we found consistently associated with greater AMR counts and MDR and XDR categories, is also associated with fewer virulence traits than closely related groups.21 Our comprehensive analysis of the distribution of blaCTX-M and carbapenemase genes by phylogroup confirms and validates previously observed trends, such as the spread of blaCTX-M-15 in isolates from group B222, as well as highlighting previously unknown CTX-M-enriched lineages like F2 and D4. Taken together, these results suggest AMR genes may be accumulating most in lineages associated with long-term carriage in humans, which may represent reservoirs for subsequent transfer to more pathogenic lineages.
Wild birds may be key AMR reservoirs
We found high counts of AMR genes and high XDR probabilities in isolates from wild birds. Previous research finds varying prevalence of AMR genes in wild bird populations, suggesting that they may be important disseminators of resistance.23–25 In addition, we predicted a 25% chance of an E. coli genome from a wild bird possessing a carbapenemase gene – around 10% higher than isolates from humans. Previous work has found high rates of carbapenemases in Enterobacteriaceae from individual bird populations: for example, 16-19% of gulls from Australia and Europe carried isolates possessing carbapenemases.26 This highlights a need for further study of E. coli in wild birds, to further determine their role in the spread of AMR genes as well as investigate their genetic diversity and potential pathogenicity. Though the sample size of the wild bird group in our study was relatively low (n=230), its high load of AMR genes compared to isolates from humans and livestock or domestic animals, groups known to be enriched for these genes, is notable. This group was also geographically and taxonomically diverse, including isolates from corvids, gulls, wildfowl, waders and birds of prey from different continents. There are very few studies which assess E. coli in wild animals in general.27 The high levels of AMR we found may be due to the relatively high mobility of birds compared to other taxa, and an abundance of generalist host species in our dataset which utilise diverse foraging habitats, both of which increase potential exposure to anthropogenic sources of resistant bacteria.28,29
Different regions are associated with MDR and XDR E. coli, and clinically-important β-lactamases
Particular geographic areas are known to be associated with a high prevalence of AMR: for example, ESBL-producing E. coli are most commonly found in the Indian subcontinent, China and Southeast Asia, the Middle East, Northern Africa and Central and South America.30 Global surveillance programmes are key for monitoring AMR prevalence internationally, but due to global disparities in reporting, many regions are missing systematic data.31 Therefore, the global AMR burden is challenging to estimate, particularly across different types of resistance genes. Our models showed that North Africa and South-Eastern Asia were associated with the highest counts of AMR and MDR probabilities. Meanwhile, by far the highest XDR proportions were found in Eastern Asia, particularly China, with relatively high probabilities also found in Western Asia, and Eastern and Western Europe.
blaCTX-M genes occurred at the highest frequencies in South-Eastern Asia and Northern Africa, as well as Eastern Europe in our study. Previous studies reported high rates of ESBL genes in Enterobacteriaceae from Northern Africa, at between 30-40% in hospital settings, with the highest rates at ∼80% in hospitals in Tunisia.32 We found carbapenemase genes were most likely to occur in genomes from Western and Eastern Asia (∼20%) and Western Europe (18%). These enzymes are globally distributed with local variations in the type of carbapenemase found. For example, KPC-type carbapenemases are endemic in the US, Italy and Greece, NDMs are endemic in the Indian subcontinent, and OXA-48-like enzyme are endemic in parts of North Africa and the Middle East as well as the Indian subcontinent.33 This global spread was reflected in our data, with probabilities of carbapenemase genes above 0.15 across distant regions.
AMR is associated with increasing temperature
A factor that may partly explain the geographic variations in AMR is climate. We found that temperature was positively associated with counts of AMR genes and probability of isolates being MDR. Conversely, XDR genomes were negatively associated with temperature, likely because of the high XDR incidence in China, a country with a relatively low minimum temperature (−8.7°C). Antibiotic usage data is not freely available for many countries, particularly lower and middle income countries, and from non-clinical settings, such as agriculture. Therefore, we were unable to control for this potential confound, meaning that trends for temperature in our study may encompass broader socio-economic trends in the transition from global North to South. Previous work has found positive associations between temperature and AMR in the USA and Europe34,35, as well as between AMR and other socio-economic factors such as infrastructure and governance.36 These reports suggested mechanisms including higher temperatures facilitating increased bacterial growth and raising rates of horizontal gene transfer, which spreads resistance determinants, with important implications in the context of climate change.34
We found opposing trends for temperature, with positive and negative associations for carbapenemases and blaCTX-Ms, respectively. As blaCTX-Ms are a highly diverse type of resistance gene, we focused on its different evolutionary clusters. We investigated whether, for an isolate that possesses a blaCTX-M gene, the class of that gene predicts differing temperature associations. Three of the four blaCTX-M groups we identified had a positive association with temperature, with only enzymes from the blaCTX-M-9 group being relatively more likely at colder temperatures. The blaCTX-M-9 group contains the blaCTX-M-14 enzyme, one of the most globally dominant blaCTX-Ms, which was first described in China, as well as the blaCTX-M-27 gene, which is becoming common in Japan, China, South-East Asia, North America and Europe.37 Therefore, its negative association with temperature may represent particular regional lineages of E. coli carrying this group of genes.
Limitations and future work
This study it is one of the first to directly characterise AMR genes in a large genomic dataset and analyse their frequencies according to different metadata categories, as opposed to levels of phenotypic resistance identified through systematic surveillance. Though powerful, such surveillance programmes are restricted to regions with sufficient resources to run them, and focus on human populations, overlooking AMR prevalence in livestock, wildlife and the wider environment. Our methods may help to predict resistance prevalence in regions with lower surveillance coverage, and we demonstrate their utility in predicting the spread of known problem genes such as blaCTX-M ESBLs and carbapenemases. However, the nature of genomic data from human pathogens means our findings are derived from a dataset which is biased towards isolates from human and livestock host species and high-income countries.38 In fact, our work directly quantifies the disparities in sampling between host species, locations and phylogroups in publicly-available genomes. Conflicting results from genomic studies on E. coli may frequently be driven by small sample sizes being used to draw wider inference about this hugely diverse species. Our study goes some way to addressing this issue by pooling and statistically modelling trends from thousands of genomes across several decades. Future work based on systematic sampling efforts will be necessary to plug the knowledge gaps we identify here.
Contributors
EP, SVH and ERW conceptualised the study. WHG refined the directions of the analysis and methodology. EP performed all data and statistical analysis and wrote the first manuscript draft. All authors contributed to interpretation of the results and editing subsequent versions of the manuscript. TD independently verified the data underlying the conclusions of the manuscript. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication after obtaining approval from all coauthors.
Data Availability
All pipelines, analysis and modelling scripts, as well as data used in this study are available at https://github.com/elliekpursey/AMR-phylogroups. Genomic data is available from the NCBI.
Data sharing
All pipelines, analysis and modelling scripts, as well as data used in this study are available at https://github.com/elliekpursey/AMR-phylogroups. Genomic data is freely available from the NCBI.
Declaration of Interests
We declare no competing interests.
Acknowledgements
EP is supported by a PhD studentship equally funded by a grant from the European Research Council under the European Union’s Horizon 2020 research and innovation programme (ERC-STG-2016-714478 to ERW) and the College of Life and Environmental Sciences, University of Exeter, UK. EP thanks Liam Patrick Langley for statistical modelling advice and constructive feedback on the manuscript. TD is supported by European Research Council (ERC-2017-ADG-788405 to ERW). SVH acknowledges BBSRC for funding (BB/R010781/1 and BB/S017674/1).
References




