
Abstract
Metastatic skin cutaneous melanomas that contain wild-type BRAF alleles (“BRAF WT melanomas”) remain a significant clinical challenge, primarily because of the paucity of targets for therapeutic intervention. In prior work, in silico analyses of The Cancer Genome Atlas Skin Cutaneous Melanoma (TCGA-SKCM) dataset suggested that elevated transcription of the gene that encodes the ERBB4 receptor tyrosine kinase may drive BRAF WT melanomas. Moreover, that prior work demonstrated that expression of the wild-type ERBB4 gene (WT ERBB4) stimulates clonogenic proliferation by the MEL-JUSO, MeWo, and IPC-298 human BRAF WT melanoma cell lines. Moreover, expression of a dominant-negative (K751M) ERBB4 mutant (ERBB4 DN) inhibits clonogenic proliferation by the MEL-JUSO and MeWo cell lines.
Here we have extended these findings by investigating the role of ERBB4 mutant alleles in BRAF WT melanomas. In silico analyses of the TCGA-SKCM BRAF WT melanoma dataset indicates that ERBB4 missense mutant alleles occur in a non-random manner, suggesting that melanomagenesis selects for the ERBB4 missense mutant alleles. Specifically, ERBB4 missense mutant alleles affect amino acid residues that are weakly correlated with residues conserved in the ERBB3 extracellular domains and the EGFR tyrosine kinase domain. The occurrence of ERBB4 missense mutant alleles in the TCGA-SKCM BRAF WT melanoma dataset is weakly inversely correlated with events that cause ERBB4-independent PI3K pathway signaling and is strongly correlated with events that cause elevated RAS pathway signaling. Thus, the in silico analyses suggest that ERBB4 mutant alleles stimulate PI3K signaling, which cooperates with elevated RAS signaling to drive BRAF WT melanomas. Moreover, the in silico analyses have prioritized the ERBB4 mutant alleles as candidate drivers of BRAF WT melanomas. One of the prioritized ERBB4 mutant alleles (P759L) stimulates greater clonogenic proliferation of MEL-JUSO cells than does WT ERBB4. Thus, our in silico prioritization strategy may effectively identify ERBB4 mutants that drive BRAF WT melanomas. Finally, the results of our in silico analyses suggest that ERBB4-dependent, BRAF WT melanomas may be effectively treated by a combination of a PI3K pathway inhibitor and a RAS pathway inhibitor.
Introduction
BRAF inhibitors, MEK inhibitors, and immune checkpoint inhibitors (“checkpoint inhibitors”) are transforming the treatment of advanced skin cutaneous melanomas that possess oncogenic BRAF mutations (“BRAF mutant melanomas”). The Surveillance, Epidemiology, and End Results Program of the National Cancer Institute [1] reports a 25% five-year survival of metastatic skin cutaneous melanoma patients for the period of 2010 to 2018 [2]. In contrast, a 2019 clinical trial reports 34% five-year survival of patients with advanced BRAF mutant skin cutaneous melanomas treated with BRAF and MEK inhibitors [3, 4]. A 2019 clinical trial reports 60% five-year survival of patients with advanced BRAF V600X mutant skin cutaneous melanomas (“BRAF V600X mutant melanomas”) treated with a combination of immune checkpoint inhibitors [3, 5]. Finally, combining immune checkpoint inhibitors with BRAF and MEK inhibitors will likely lead to further improvements in survival [6].
Unfortunately, approximately 50% of advanced skin cutaneous melanomas possess wild-type BRAF alleles, and contemporary treatments of advanced skin cutaneous melanomas that contain wild-type BRAF (“BRAF WT melanomas”) have yielded less impressive results. In part, these less impressive results are because of a paucity of actionable targets for the (targeted) treatment of these tumors [7]. Moreover, the five-year survival of patients with advanced BRAF WT skin cutaneous melanomas treated with immune checkpoint inhibitors is only 48%, less than the 60% experienced by patients with advanced BRAF mutant skin cutaneous melanomas in a parallel study [5]. Hence, our previous work has attempted to address this gap in treatment efficacy by evaluating the ERBB4 receptor tyrosine kinase as a candidate target in BRAF WT skin cutaneous melanomas [8].
ERBB4 (HER4) is a member of the ERBB family of receptor tyrosine kinases (RTKs), which includes the epidermal growth factor receptor (EGFR), ERBB2 (HER2/Neu), and ERBB3 (HER3). ERBB4 possesses extracellular ligand-binding domains, a single-pass hydrophobic transmembrane domain, an intracellular tyrosine kinase domain, and intracellular tyrosine residues that function as phosphorylation sites (Figure 1). Ligand binding to EGFR, ERBB3, or ERBB4 stabilizes the receptor extracellular domains in an open conformation competent for symmetrical homodimerization and heterodimerization of the receptor extracellular domains. The dimerization of the extracellular domains enables asymmetrical dimerization of the receptor cytoplasmic domains. Phosphorylation of one receptor monomer on tyrosine residues by the tyrosine kinase domain of the other receptor monomer (“cross-phosphorylation”) ensues (Figure 2). This tyrosine phosphorylation creates binding sites for effector proteins and activation of downstream signaling pathways [9].

The extracellular domains of ERBB4 exist in an equilibrium between the closed conformation that has low affinity for ligand and buried dimerization motifs and the open conformation that has high affinity for ligand and has exposed dimerization motifs. Adapted from [9].

ERBB4 ligands stimulate ERBB receptor signaling via ERBB4 homodimerization and heterodimerization. Adapted from [9].
Elevated signaling by an RTK is a hallmark of many types of cancer. Hence, RTK overexpression, ligand overexpression, and gain-of-function mutations in an RTK gene are all mechanisms for pathologic, elevated RTK signaling [10]. Indeed, EGFR and ERBB2 have been validated as targets for therapeutic intervention in numerous types of tumors; monoclonal antibodies and small molecular tyrosine kinase inhibitors have been approved to treat tumors dependent on these receptors [11-26]. It appears that ERBB3, particularly ERBB3-ERBB2 heterodimers, also drives various human tumors [27, 28].
In contrast, the role that ERBB4 plays in human tumors remains ambiguous. Part of the ambiguity reflects that an ERBB4 homodimer can function as a tumor suppressor, whereas an ERBB4-EGFR or ERBB4-ERBB2 heterodimer can drive tumor cell proliferation or aggressiveness [9]. Our previous work has demonstrated that ectopic expression of wild-type ERBB4 stimulates the proliferation of BRAF WT melanoma cell lines and ectopic expression of a dominant-negative (K751M) mutant ERBB4 allele inhibits the proliferation of BRAF WT melanoma cell lines. These data suggest that ERBB4 is a driver of BRAF WT melanomas [8].
Structural studies of ERBB family receptors and analyses of ERBB mutant alleles has revealed multiple mechanisms by which a missense mutation in an ERBB gene may cause elevated (oncogenic) signaling by that receptor protein [29]. (1) A mutation may increase the affinity of a receptor for its cognate ligands. (2) A mutation may cause a shift in the extracellular domain conformational equilibrium; the mutant may now favor the open conformation, which is competent for dimerization and signaling, over the closed conformation, which is not competent for dimerization and signaling. (3) A mutation may directly increase receptor dimerization, through the creation of novel intermolecular chemical bonds. Such activating mutations may affect dimerization of the extracellular domains, transmembrane domain, or intracellular kinase domain. (4) A mutation may destabilize the inactive conformation of the kinase domain or stabilize the active conformation of the kinase domain. (5) The substitution of a glutamate residue for a tyrosine residue at the site of tyrosine phosphorylation may mimic phosphorylation, resulting in constitutive binding of effector proteins and activation of downstream signaling events.
We have observed that ERBB4 appears to function as a driver of proliferation in multiple BRAF WT melanoma cell lines [8]. Moreover, ERBB4 mutant alleles, most of which remain largely uncharacterized, have been found in several cancers, including melanomas [9]. Thus, here we test the hypothesis that ERBB4 mutant alleles found in BRAF WT melanoma samples function as drivers in BRAF WT melanoma cell lines.
Results
A. ERBB4 non-synonymous mutations occur at a non-stochastic frequency
Our in silico analysis of ERBB4 mutant alleles is based on The Cancer Genome Atlas Skin Cutaneous Melanoma (TCGA-SKCM) dataset (470 cases) [30]. Most of the analyses reported here focus on the 227 cases that possess wild-type (WT) BRAF alleles (“BRAF WT melanomas”) [8, 30].
In the TCGA-SKCM dataset, ERBB4 missense mutant alleles are slightly more common in BRAF WT melanomas than BRAF V600X mutant melanomas (15.4% and 11.4% respectively). Therefore, we hypothesized that ERBB4 mutant alleles are selected in BRAF WT melanomas, thereby justifying the analyses reported in this work.
The ratio of non-synonymous (missense, stop gained, frame shift) to synonymous (synonymous, stop retained) mutations is an indicator of selection for mutant alleles by tumor cells. Generally speaking, if the ratio of non-synonymous to synonymous mutant alleles (N/S ratio) in a particular gene is greater than 1, it is indicative of selection for non-synonymous mutant alleles in that gene [31].
Because of the exposure of skin cells to ultraviolet (UV) light, melanomas carry numerous cytosine to thymine mutations, many of which function as passengers. This precludes using an N/S cutoff of 1 to identify genes for which mutant alleles function as drivers [32]. Thus, we have compared the N/S ratio for ERBB4 in the 227 BRAF WT melanomas of the TCGA-SKCM dataset against the N/S ratio for genes (ARMC4, CFTR, ERC2, SLIT2, and SLIT3) that are roughly the same size as ERBB4 and that from the literature do not appear to function as melanoma drivers.
The N/S ratio for ERBB4 is 5.33, which is significantly greater (p=0.003) than the average N/S ratio for the five control genes (2.44) (Table 1). Moreover, the N/S ratio for the KRAS and NRAS oncogenes and the CDKN2A, NF1, RB, and TP53 tumor suppressor genes is also greater than the average N/S ratio for the five control genes. In contrast, the N/S ratio for EGFR, ERBB2, and ERBB3 genes is not significantly greater than the average N/S ratio for the five control genes. These results suggest that mutations in ERBB4, but not mutations in EGFR, ERBB2, or ERBB3, function as drivers in BRAF WT melanomas.
The ratio of ERBB4 nonsynonymous to synonymous (N/S) mutations in the TCGA-SKCM BRAF WT melanoma dataset suggests that ERBB4 nonsynonymous mutations function as drivers of BRAF WT melanomas.
B. ERBB4 mutations are somewhat more likely to affect functionally conserved amino acid residues
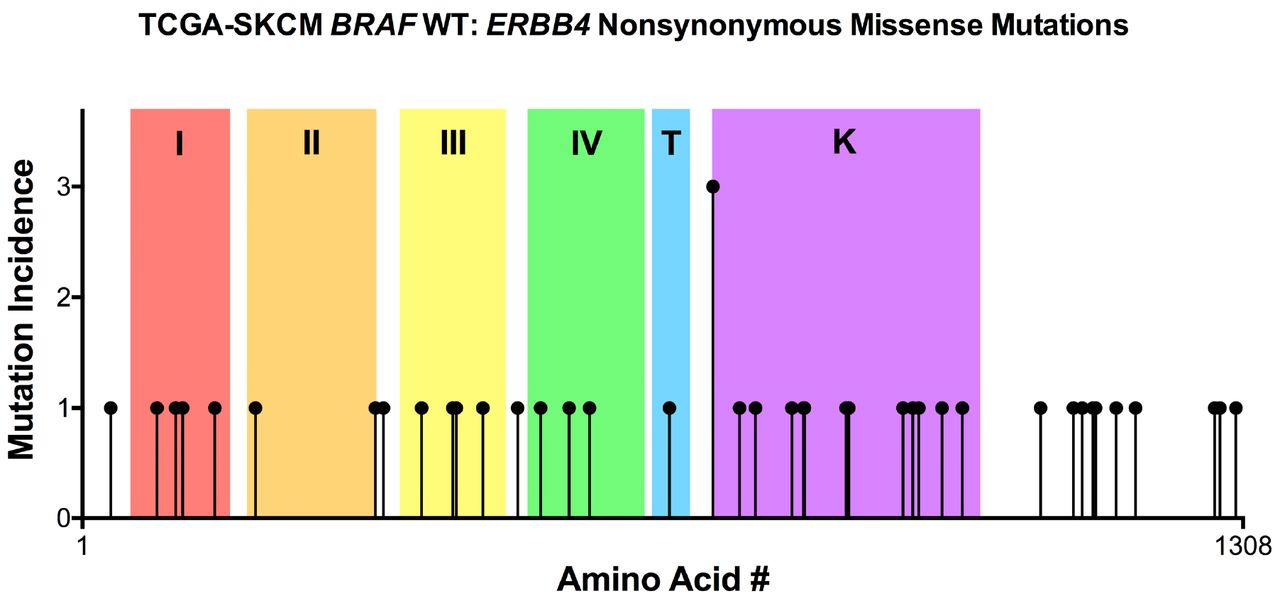
Using the TCGA-SKCM, BRAF WT melanoma dataset, we identified 40 ERBB4 missense mutations that are not coincident with an ERBB4 stop-gained mutation or splice site mutation; one of these ERBB4 mutant alleles was found in three tumor samples and the other 39 ERBB4 mutant alleles were found in a single tumor sample (Figure 3). Therefore, there is only a single, weak mutational hotspot and we must employ other approaches to identify and prioritize candidate driver mutations.

In BRAF WT melanomas of the TCGA-SKCM dataset, ERBB4 nonsynonymous missense mutant alleles are evenly distributed across the entire ERBB4 coding region.
The conservation of a particular amino acid residue across functionally related proteins suggests that residue is critical for the shared function of the related proteins. Thus, we hypothesized that ERBB4 melanoma driver mutations are more likely to affect conserved residues.
Many members of the epidermal growth factor family of peptide hormones bind to both ERBB3 and ERBB4 (Figure 2) [33]. Moreover, ERBB3 dimerization and ERBB4 dimerization are regulated by an identical mechanism (Figure 1) [9]. We have identified fifteen ERBB4 extracellular domain (ECD) I-IV amino acid residues that are affected by ERBB4 missense mutations found in the TCGA-SKCM BRAF WT melanoma data set. We hypothesized that these fifteen residues correlated with ERBB4 residues that are conserved in the ERBB3 ECDs I-IV. However, only eleven residues affected by ERBB4 BRAF WT melanoma missense mutations are conserved in the ERBB3 ECDs I-IV, which falls well short of a statistically significant correlation (p=0.3763) (Table 2a). Nonetheless, these eleven ERBB4 missense mutant alleles (G85S, R106C, R196C, P331S, G340E, A383T, S418F, T422I, E452K, R491K, and P517A) are assigned a priority point to help identify the best candidates for ERBB4 driver mutant alleles in BRAF WT melanomas.
ERBB4 missense mutations that affect residues of the extracellular domains (ECDs) are slightly correlated with residues that are conserved in the ERBB3 extracellular domains.
ERBB4 missense mutations that affect residues of the kinase domain are somewhat correlated with residues that that are conserved in the EGFR kinase domain.
Only ERBB4 and EGFR directly bind ligands and form homodimers that undergo ligand-dependent receptor cross-phosphorylation [33]. (ERBB2 does not bind ligand and ERBB3 lacks sufficient kinase activity.). We have identified thirteen ERBB4 tyrosine kinase domain amino acid residues that are affected by ERBB4 missense mutations found in the TCGA-SKCM BRAF WT melanoma data set. We hypothesized that these thirteen residues correlated with ERBB4 residues that are conserved in the EGFR tyrosine kinase domain. Indeed, all thirteen residues affected by ERBB4 BRAF WT melanoma missense mutations are conserved in the EGFR tyrosine kinase domain. However, this distribution falls just short (p=0.0565) of a statistically significant correlation (Table 2b). Nonetheless, these thirteen ERBB4 missense mutant alleles (R711C, G741E, P759L, P800L, D813N, N814T, D861N, L864P, P925S, G936E, P943S, E969K, and R992C) are assigned a priority point to help identify the best candidates for ERBB4 driver mutant alleles in BRAF WT melanomas.
Finally, three ERBB4 missense mutant alleles (R106C, G741E, L864P) found in the TCGA-SKCM BRAF WT melanoma data set correspond to gain-of-function alleles in EGFR (R108K, G735S, and L858R, respectively) or ERBB2 (L866M corresponds to ERBB4 L864P). As a result, these three ERBB4 missense alleles (Table 3) are assigned a priority point to help identify the best candidates for ERBB4 driver mutant alleles in BRAF WT melanomas.
Three ERBB4 missense mutations occur at residues that correspond to sites of oncogenic mutations in EGFR or ERBB2.
C. ERBB4 missense mutants are somewhat less common in TCGA-SKCM BRAF WT melanoma cases in which there are PI3K or PTEN alterations that are predicted to stimulate the PI3K pathway
The PI3K pathway is required for ERBB4-EGFR heterodimers to stimulate IL3-independent proliferation of BaF3 mouse lymphoid cells [34]. Likewise, an ERBB4 gain-of-function mutant allele causes increased AKT phosphorylation in a human melanoma cell line [35]. Thus, we postulated that ERBB4 driver mutants in BRAF WT melanomas stimulate PI3K pathway signaling and are inversely correlated with events (increased PIK3CA transcription, gain-of-function mutation in PIK3CA, decreased PTEN transcription, or loss-of-function mutation in PTEN) that would cause ERBB4-independent increases in PI3K pathway signaling. Twenty-nine cases in the TCGA-SKCM BRAF WT melanoma dataset contain an ERBB4 missense mutation, but no PIK3CA mutation or increase in PIK3CA transcription, nor any PTEN mutation or decrease in PTEN transcription. However, this distribution falls just short (p=0.068) of a statistically significant correlation (Table 4). Nonetheless, the 36 ERBB4 mutant alleles (E33K, G85S, R106C, R114Q, D150N, R196C, P331S, A383T, S418F, T422I, E452K, R491K, P517A, G549S, P572L, R711C, G741E, P759L, P800L, D813N, N814T, D861N, P925S, G936E, P943S, E969K, R992C, P1080L, P1117L, R1127K, R1139Q, R1142Q, P1165L, P1276S, P1282S, and P1300S) found in these 29 cases are assigned a priority point to help identify the best candidates for ERBB4 driver mutant alleles in BRAF WT melanomas.
ERBB4 missense mutants are somewhat inversely correlated with events (mutation of PIK3CA, increased PIK3CA transcription, mutation of PTEN, or decrease in PTEN transcription) predicted to result in increased PI3K pathway signaling activity.
D. ERBB4 missense mutants are significantly more common in TCGA-SKCM BRAF WT melanoma cases in which there are RAS or NF1 nonsynonymous mutations
Gain-of-function RAS gene mutations occur in about 30% of skin cutaneous melanomas, and loss-of-function mutations in NF1 occur in about 20% of skin cutaneous melanomas. Moreover, gain-of-function BRAF mutations, gain-of-function RAS gene mutations, and loss-of-function NF1 mutations are largely mutually exclusive in skin cutaneous melanomas [36].
Receptor tyrosine kinases typically stimulate RAS pathway signaling [37-43]. Hence, in the BRAF WT melanomas of the TCGA-SKCM dataset, we predicted that cases that contain an ERBB4 missense mutation would be less likely to contain a nonsynonymous mutation in a RAS gene or NF1. Surprisingly, 29 cases in the TCGA-SKCM BRAF WT melanoma dataset contain an ERBB4 missense mutation, as well as a nonsynonymous mutation in a RAS gene or NF1 (Table 5). This statistically significant correlation (p=0.0189) suggests that ERBB4 signaling cooperates with RAS signaling to drive BRAF WT melanomas. Hence, we have assigned a priority point to the 33 ERBB4 mutant alleles (E33K, G85S, R106C, D150N, P331S, A383T, S418F, T422I, E452K, R491K, P517A, G549S, P572L, F662L, R711C, P759L, P800L, D813N, D861N, P925S, G936E, E969K, R992C, P1080L, P1117L, R1127K, R1139Q, R1142Q, P1165L, E1187D, P1276S, P1282S, and P1300S) found in the 29 cases that contain a nonsynonymous mutation in a RAS gene or NF1.
ERBB4 missense mutations are significantly correlated with a RAS or NF1 nonsynonymous mutation.
E. ERBB4 missense mutations are significantly more likely in cases where there is a RAS or NF1 nonsynonymous mutation as well as no other apparent cause of increased PI3K signalling
Our in silico analyses lead us to hypothesize that gain-of-function ERBB4 mutants stimulate signaling by the PI3K pathway and that this PI3K signaling cooperates with elevated RAS signaling (caused by a gain-of-function RAS gene mutation or a loss-of-function NF1 mutation) to drive BRAF WT melanomas. Thus, we tested whether ERBB4 missense mutations are more likely to occur in cases where there is a RAS gene or NF1 mutation and NO events (increases in PIK3CA transcription, gain-of-function mutations in PIK3CA, decreases in PTEN transcription, or loss-of-function mutations in PTEN) that would cause ERBB4-independent increases in PI3K pathway signaling (Table 6). Twenty-five cases in the TCGA-SKCM BRAF WT melanoma dataset contain an ERBB4 missense mutation, as well as a nonsynonymous mutation in a RAS gene or NF1, but no event (increases in PIK3CA transcription, gain-of-function mutations in PIK3CA, decreases in PTEN transcription, or loss-of-function mutations in PTEN) that would cause ERBB4-independent increases in PI3K pathway signaling. This statistically-significant correlation (p=0.0166) supports our hypothesis. However, because the 31 ERBB4 mutant alleles found in these 25 cases received a priority point(s) through the results of Table 4 and Table 5, we have not assigned any priority points based on the data from Table 6.
ERBB4 missense mutations are significantly correlated with a RAS or NF1 mutation AND no other apparent cause of increased PI3K activity.
F. ERBB4 mutations and elevated ERBB4 transcription appear to independently drive BRAF WT melanomas
In silico analyses of the TCGA-SKCM BRAF WT melanoma dataset suggest that elevated ERBB4 transcription appears to cooperate with elevated RAS signaling (due to a gain-of-function RAS gene mutation or a loss-of-function NF1 mutation) to drive BRAF WT melanomas [8]. Thus, it is possible that ERBB4 mutant alleles require elevated ERBB4 transcription to drive BRAF WT melanomas. We have identified 129 cases in the TCGA-SKCM BRAF WT melanoma dataset that contain a non-synonymous RAS or NF1 mutation and for which ERBB4 transcription data is available. Twenty of these cases contain an ERBB4 missense mutation, eighteen cases exhibit elevated ERBB4 transcription, and three cases contain an ERBB4 missense mutation and exhibit elevated ERBB4 transcription (Figure 4). These data suggest that ERBB4 mutations and elevated ERBB4 transcription appear to independently drive BRAF WT melanomas. Hence, the 29 ERBB4 mutants (E33K, R106C, R114Q, D150N, R196C, G340E, A383T, S418F, E452K, R491K, P517A, G549S, F662L, R711C, P759L, P800L, D813N, N814T, D861N, L864P, P925S, G936E, P943S, E969K, R1142Q, P1165L, P1276S, P1282S, P1300S) found in the twenty cases that contain an ERBB4 missense allele are assigned a priority point to help identify the best candidates for ERBB4 driver mutant alleles in BRAF WT melanomas.

Within the 129 BRAF WT cases in the TCGA-SKCM dataset which harbor a RAS or NF1 nonsynonymous mutation, an ERBB4 missense mutation and elevated ERBB4 transcription appear to be largely mutually exclusive.

The non-random occurrence of ERBB4 mutations in BRAF WT tumors suggests that ERBB4 mutations drive signaling through the PI3K pathway, thereby enabling ERBB4 signaling to cooperate with RAS/NF1 mutations to drive BRAF WT tumors. Adapted from [8].
G. We have prioritized ERBB4 mutant alleles as candidate drivers of BRAF WT melanomas
Based on the in silico data presented in Table 2a, Table 2b, Table 3, Table 4, Table 5, and Figure 4, we have established and applied criteria for prioritizing ERBB4 mutant alleles as candidate drivers of BRAF WT melanomas (Table 7). Next, we will describe our efforts to determine whether prioritized ERBB4 mutant alleles function as drivers of BRAF WT melanomas.
Prioritization of candidate ERBB4 driver mutant alleles in the TCGA-SKCM BRAF WT melanoma dataset. Columns refer to tables and figures found in the manuscript and the mutations listed in the table are from the values circled in red in those tables and figures.
H. A prioritized ERBB4 mutant allele stimulates clonogenic proliferation of a BRAF WT melanoma cell line
We have shown that ectopic expression of wild-type ERBB4 (ERBB4 WT) significantly stimulates the clonogenic proliferation of MEL-JUSO, MeWo, and IPC-298 human BRAF WT melanoma cell lines [8]. Moreover, ectopic expression of the dominant-negative (K751M) ERBB4 (ERBB4 DN) mutant significantly inhibits the clonogenic proliferation of MEL-JUSO and MeWo cell lines and modestly inhibits the clonogenic proliferation of IPC-298 cells. These results indicate that ERBB4 is both sufficient and necessary to drive some BRAF WT melanoma cell lines; they also suggest that we can use the MEL-JUSO, MeWo, and IPC-298 cell lines to identify ERBB4 mutant alleles that function as bona fide drivers of BRAF WT melanomas.
We have tested whether the high priority R106C and P759L ERBB4 mutants (Table 7) stimulate a greater amount of clonogenic proliferation than ERBB4 WT. As a positive control, we tested the Y285C ERBB4 mutant, which was found in non-small cell lung carcinoma samples and stimulates ligand-dependent and -independent ERBB4 signaling and increased ERBB4 heterodimerization with ERBB2 [9, 44]. In the MEL-JUSO cells, the ERBB4 P759L and Y295C mutants caused greater clonogenic proliferation of the BRAF than ERBB4 WT. In contrast, the ERBB4 R106C and ERBB4 WT caused a similar level of clonogenic proliferation (Table 8). These data suggest that the ERBB4 P759L mutant may be a bona fide driver of BRAF WT melanomas.
In the MEL-JUSO BRAF WT melanoma cell line, the ERBB4 Y285C and P759L mutants cause greater clonogenic proliferation than ERBB4 WT.
I. A heterologous model system can be used to determine whether ERBB4 mutants that stimulate the proliferation of BRAF WT melanomas exhibit elevated ligand-dependent or ligand-independent ERBB4 signaling activity
The MEL-JUSO, MeWo, and IPC-298 cell lines exhibit endogenous expression of ERBB4 ligands [8]. Thus, ERBB4 mutants could stimulate clonogenic proliferation of these cell lines via a ligand-dependent or ligand-independent mechanism.
We and others have previously demonstrated that ERBB4 homodimers typically do not stimulate cell proliferation, whereas ERBB4-ERBB2 and ERBB4-EGFR heterodimers do stimulate cell proliferation. Indeed, ERBB4 ligands do not stimulate proliferation of BaF3 mouse lymphoid cells engineered to express ERBB4 but do stimulate proliferation of BaF3 cells engineered to express ERBB4 and EGFR or ERBB4 and ERBB2 [9]. BaF3 cells lack endogenous EGFR, ERBB2, and ERBB4 expression. But, because they endogenously express ERBB3, they are not ideal for studying the functional differences among ERBB4 homodimers, ERBB4-EGFR heterodimers, or ERBB4-ERBB2 heterodimers [45].
Thus, we have chosen to study these functional differences using derivatives of the 32D mouse lymphoid cell line, which lack endogenous expression of all four ERBB family receptors and are dependent on interleukin 3 (IL3) for their proliferation [46]. 32D cells were generated that express both ERBB4 and ERBB2, ERBB4 alone, ERBB2 alone, and the appropriate vector controls. None of these 32D cell lines exhibit proliferation in the absence of both IL3 and an ERBB4 ligand. Moreover, the ERBB4 ligand NRG1beta stimulates IL3-independent proliferation of the 32D/ERBB2+ERBB4 cells but does not stimulate IL3-independent proliferation of any of the other 32D cell lines (Table 9). Thus, 32D cell lines that co-express ERBB4 mutants and ERBB2 are ideal for studying whether these ERBB4 mutants potentiate the effects of ERBB4 ligands or enable ERBB4 coupling to proliferation independent of an ERBB4 ligand.
The ERBB4 ligand NRG1beta stimulates IL3-independent proliferation of the 32D/ERBB4+ERBB2 cell line, but not any of the control cell lines.
Materials and Methods
Analysis of the TCGA-SKCM dataset
Clinical and biospecimen data were downloaded for all 470 cases found in the TCGA-SKCM dataset [47]. All analyzed datasets are publicly available through the NIH/NCI Genomic Data Commons (GDC) portal [48]. The R statistical computing and graphics environment software [49] was used to reorganize the dataset. Statistical analyses were performed using GraphPad Prism [50].
Creating BRAF WT dataset
All BRAF WT cases were segregated from the TCGA-SKCM dataset for analysis. Cases in which there were ERBB4 mutation(s) which co-occurred with a stop gained or splice-site mutation were removed from the BRAF WT dataset for analysis of the effect of ERBB4 missense mutation in BRAF WT melanoma (Sections IIIB-F). Three mutations fell into this category (M958I, R47Q, G573D). These mutations and the stop gained mutated cases are included in the analysis for section IIIA.
Cell Lines and cell culture
Mouse C127 fibroblasts and the ψ2 and PA317 recombinant retrovirus packaging cell lines are generous gifts of Daniel DiMaio (Yale University). These cells were cultured essentially as described previously [51]. The MEL-JUSO [52] human melanoma cell line was obtained from DSMZ [53] (Braunschweig, Germany) and was cultured as recommended. The 32D mouse myeloid cell line has been described previously and was cultured as recommended [54]. Cell culture media, serum, and supplements were obtained from Cytiva [55] (Marlborough, VA). G418 was obtained from Corning [56] (Corning, NY). Genetic and mRNA expression data for the MEL-JUSO cell line were obtained from the Broad Institute Cancer Cell Line Encyclopedia (CCLE) [57].
Mutagenesis of DNA Constructs
The recombinant retroviral expression construct pLXSN-ERBB4 has been described previously [45]. We used pLXSN-ERBB4 as the parent plasmid for site-directed mutagenesis using the Q5 Site Directed Mutagenesis Kit (NEB). Mutagenesis was performed essentially as recommended by the manufacturer.
Recombinant retroviruses
Briefly, the recombinant amphotropic retroviruses LXSN, LXSN-ERBB4 (ERBB4 WT), LXSN-ERBB4 R106C, LXSN-ERBB4 Y285C, and LXSN-ERBB4 P759L were packaged using the ψ2 ecotropic retrovirus packaging cell line, and the PA317 amphotropic retrovirus packaging cell line as previously described [45, 58, 59]. 32D cells were infected with the recombinant amphotropic retroviruses LXSN, LXSN-ERBB4, LXSN-HygR, and LXSN-HygR-ERBB2 at a 0.1 multiplicity of infection. These constructs have been previously described [46, 51].
Clonogenic Proliferation Assay
C127, MEL-JUSO cells were infected with 500 and 3000 amphotropic retroviral infectious units (respectively), essentially as described earlier. After incubation with the viruses, infected cells were selected using G418 at a concentration of 800 ug/mL. The resulting colonies of G418-resistant cells were stained using Giemsa 8 and 13 days later (respectively), and colonies were counted. C127 infections served as a control for viral titer. Tissue culture plates were digitized, and clonogenic proliferation efficiency was calculated as previously described [58]. The statistical significance of differences in clonogenic proliferation was calculated using ANOVA with a p-value threshold of <0.05 (1-tailed).
Stimulation of ERBB4-ERBB2 heterodimer dependent proliferation in 32D cells
32D cells which stably express ERBB4, ERBB2, or ERBB4 and ERBB2 were treated with and without 30nM of NRG1β (Recombinant Human Heregulinβ-1, Peprotech) in the absence of IL3. After six days cells were counted using a hemacytometer to evaluate NRG1β-dependent growth.
Discussion
A. The apparent selection for ERBB4 mutant alleles in the TCGA-SKCM BRAF WT melanoma data set enables prioritization of individual ERBB4 mutant alleles
Previous in silico analyses of the TCGA-SKCM BRAF WT melanoma dataset suggest that elevated ERBB4 transcription drives the proliferation of BRAF WT melanomas [8]. In this work we show that in silico analyses of ERBB4 mutant alleles found in the TCGA-SKCM BRAF WT melanoma data set suggest that ERBB4 mutant alleles may drive the proliferation of BRAF WT melanomas.
B. An ERBB4 mutant allele that is a candidate driver of BRAF WT melanomas does in fact drive the proliferation of BRAF WT melanoma cell lines
This work has identified the ERBB4 R106C and P759L mutants as high-priority candidate drivers of BRAF WT melanomas. We have previously demonstrated that WT ERBB4 stimulates the clonogenic proliferation of the MEL-JUSO, IPC-298, and MeWo human BRAF WT melanoma cell lines [8]. Thus, we can use these cell lines to compare the biological activity of candidate BRAF WT melanoma driver mutants of ERBB4 against WT ERBB4.
The ERBB4 P759L mutant stimulates a greater amount of clonogenic proliferation of the MEL-JUSO BRAF WT melanoma cell line than WT ERBB4. In contrast, the ERBB4 R106C mutant and WT ERBB4 stimulate similar amounts of clonogenic proliferation of the MEL-JUSO BRAF WT melanoma cell line. It is possible that the ERBB4 R106C and P759L mutants will both stimulate greater clonogenic proliferation of the IPC-298 and MeWo cell lines than WT ERBB4. If so, we can use the IPC-298 and MeWo cell lines to explore the mechanisms by which the ERBB4 R106C and P759L mutants functionally differ. Those and other mechanistic insights will undoubtedly improve the in silico strategy used to identify ERBB4 mutants that are high priority candidate BRAF WT melanoma drivers.
C. 32D/ERBB4+ERBB2 and control cell lines are suitable for determining the mechanisms by which ERBB4 gain-of-function mutants stimulate cell proliferation
Here we demonstrate that ligand-induced homodimers of WT ERBB4 do not cause IL3 independence in 32D cells. In contrast, ligand-induced heterodimers of ERBB2 and WT ERBB4 do cause IL3 independence. Thus, we can use this model system to determine whether ERBB4 gain-of-function mutants cause increased ligand-dependent or -independent signaling. We can also determine whether the effects of gain-of-function ERBB4 mutants are absolutely dependent on heterodimerization or whether gain-of-function ERBB4 mutants enable ERBB4 homodimers to stimulate IL3 independence. Finally, we can use analogous 32D/ERBB4+EGFR cell lines to determine whether the biological effects of ERBB4 gain-of-function mutants are mediated by both ERBB4-ERBB2 and ERBB4-EGFR heterodimers.
D. Our in silico analyses suggest strategies for treating ERBB4-dependent, BRAF WT melanomas
Here we demonstrate that, in the TCGA-SKCM BRAF WT melanoma dataset, ERBB4 mutants are correlated with nonsynonymous mutations in a RAS gene or NF1. This surprising observation suggests that ERBB4 does not stimulate RAS signaling in BRAF WT melanomas. Instead, it appears that ERBB4 signaling cooperates with RAS signaling to drive BRAF WT melanomas.
We have previously demonstrated that the PI3K pathway is required for ERBB4-EGFR heterodimers to stimulate IL3 independent proliferation of BaF3 cells [34]. Likewise, a gain-of-function ERBB4 mutant stimulates AKT phosphorylation in a human melanoma cell line [35]. Here we demonstrate that, in the TCGA-SKCM BRAF WT melanoma dataset, ERBB4 mutations are somewhat inversely correlated with events predicted to cause ERBB4-independent activation of the PI3K pathway (mutation of PIK3CA, increased PIK3CA transcription, mutation of PTEN, or decrease in PTEN transcription). Thus, we have postulated that, in BRAF WT melanomas, ERBB4 gain-of-function mutants stimulate PI3K signaling, which cooperates with elevated RAS signaling to drive the proliferation of these tumor cells.
Hence, we postulate that a suitable treatment for ERBB4-dependent, BRAF WT tumors would consist of a combination of a RAS pathway inhibitor and a PI3K pathway inhibitor. We believe that the most suitable RAS pathway inhibitor would be a MEK inhibitor (trametinib, binimetinib, selumetinib, or cobimetinib). The toxicity of the PI3K and mTOR inhibitors (alpelisib, sirolimus, everolimus, temsirolimus) may preclude the use of these drugs in this application. However, no specific ERBB4 inhibitors have been approved by the FDA. However, because ERBB4 mutants are likely to drive BRAF WT melanomas through signaling by ERBB4-EGFR or ERBB4-ERBB2 heterodimers, anti-EGFR drugs (gefitinib or cetuximab) or anti-ERBB2 drugs (lapatinib or pertuxumab) may be suitable. Future work is needed to evaluate these agents.
Data Availability
All data produced in the present study are available upon reasonable request to the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}