
Abstract
The COVID-19 pandemic has revealed the importance of virus genome sequencing to guide public health interventions to control virus transmission and understand SARS-CoV-2 evolution. As of July 20th, 2021, >2 million SARS-CoV-2 genomes have been submitted to GISAID, 94% from high income and 6% from low and middle income countries. Here, we analyse the spatial and temporal heterogeneity in SARS-CoV-2 global genomic surveillance efforts. We report a comprehensive analysis of virus lineage diversity and genomic surveillance strategies adopted globally, and investigate their impact on the detection of known SARS-CoV-2 virus lineages and variants of concern. Our study provides a perspective on the global disparities surrounding SARS-CoV-2 genomic surveillance, their causes and consequences, and possible solutions to maximize the impact of pathogen genome sequencing for efforts on public health.
One-Sentence Summary The causes, consequences and possible solutions for disparities in genomic surveillance observed in the COVID-19 pandemic.
The importance of genomic surveillance
RNA viruses accumulate genetic changes at high evolutionary rates, some of which allow adaptations to selective pressures induced by antivirals, vaccines, and host immunity (1). More than 20 months after the emergence of SARS-CoV-2, many countries continue to face large outbreaks of COVID-19 (2), recently driven by novel viral variants with constellations of amino acid changes, some acquired by convergent evolution (3). Variants of concern (VOCs) – such as Alpha/B.1.1.7; Beta/B.1.351; Gamma/P.1; and Delta/B.1.617.2 (and its descendent AY lineages) – have genotypic and phenotypic traits that pose increased risks to global public health, since they may affect diagnostics or therapeutics, confer higher transmissibility, lead to higher disease severity, and/or immune escape from natural infections and/or vaccines (4, 5). Variants of interest (VOI) – including Eta/B.1.525; Iota/B.1.526; Kappa/B.1.617.1 and; Lambda/C.37 – share some genetic traits with VOCs, but further evidence is needed to determine their risks to public health (6). To allow timely public health responses to emerging variants, it is essential to keep track of SARS-CoV-2 genetic diversity, preferably in real time (4, 7, 8). Following the evolution of VOCs/VOIs many countries have initiated or scaled up genomic surveillance, leading to an unprecedented number of viral genomes in publicly accessible databases, with >2,400,000 consensus genome sequences deposited in GISAID (9), >916,000 high-throughput sequencing datasets and >969,500 consensus sequences in NCBI (10) as of July 20th, 2021. However, there are striking differences in the spatial and temporal intensity of genomic surveillance worldwide. Here we investigate global SARS-CoV-2 genomic surveillance during the first 15 months of COVID-19 pandemic, highlighting causes and consequences of surveillance disparities, and identifying key aspects for timely variant detection.
Global disparities in the genomic surveillance of SARS-CoV-2
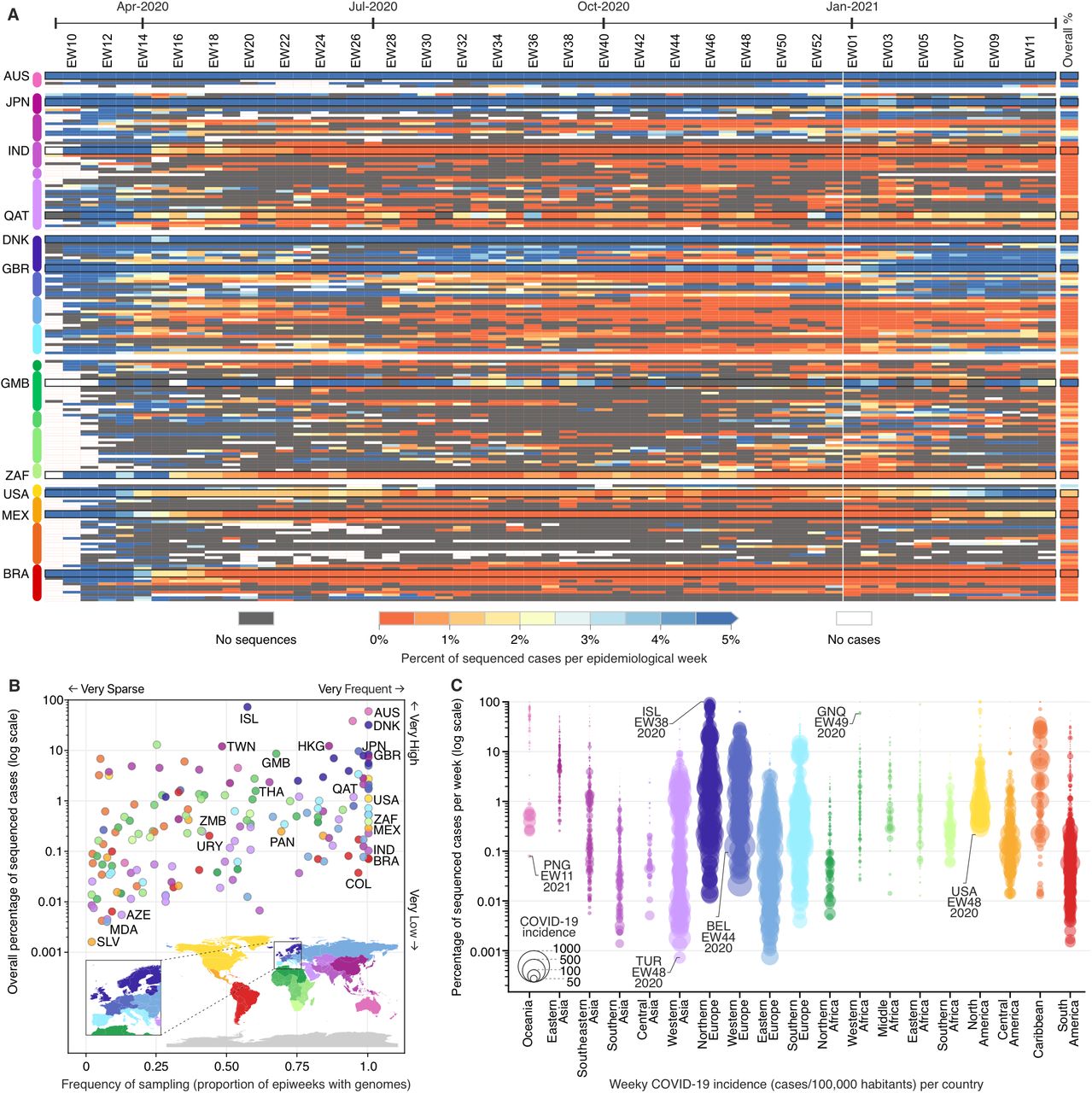
The impact and responses to control the COVID-19 pandemic differ greatly across geographic regions (11). In the early stages of the pandemic, high-income countries (HIC) relied on well-resourced laboratories to perform molecular testing and sequencing (12, 13), while low-and middle-income countries (LMIC) faced challenges in molecular diagnosis and SARS-CoV-2 sequencing (13–15). To investigate spatial and temporal heterogeneity in sequencing efforts, we analysed the percentage of COVID-19 cases that were sequenced from each country between February 2020 to March 2021 (Fig. 1A). We observed that 100 out of 167 countries sequenced <0.5% of confirmed cases (Fig. 1B), and only 16 countries were able to sequence >5% of their overall confirmed cases. While HICs and LMICs reported similar numbers of cases (65.3 and 61.2 million, respectively), they respectively sequenced 1.81% and 0.11% of their cases (Table S1). We found a moderate negative correlation between weekly sequencing percentages and reported COVID-19 incidence (cases/100K pop., r² = −0.52; p-value < 0.001), suggesting that countries that kept incidence at low levels (Fig. 1C; Fig. S1) generally had the means or opportunity to sequence a high proportion of cases, as observed in Hong Kong (12%), Taiwan (12%), New Zealand (38%), Australia (59%) and Iceland (73% sequenced cases). Only 20 out of 167 countries included in this study were able to sequence more than 5% in weeks where COVID-19 incidence was high (>100 cases per 100,000 pop.), mainly high-income countries in Northern Europe, Western Europe, and Southern Europe (Fig. 1; Fig. S1). For example, despite facing high weekly COVID-19 incidence after October 2020, Denmark and the UK were still able to keep their sequencing efforts above 10% in most weeks, and attain an overall proportion of 32% and 8% sequenced cases, respectively (Fig. 1A-B; Fig. S1).

(A) Percentage of sequenced COVID-19 cases per country per epidemiological week (EW), between February 23rd, 2020 and March 27th, 2021 (based on metadata submitted to GISAID up to May 30th, 2021). (B) Frequency and overall percentage of sequenced cases per country. This plot summarizes the data shown in (A), where the x-axis shows the percentage of epidemiological weeks with sequenced cases, and the y-axis displays the overall percentage of cases shown in the rightmost column of panel (A). (C) Percentage of cases sequenced per EW per country, per geographic region. Each circle represents an epidemiological week with at least one sequenced case, and their diameters highlight the incidence (cases per 100,000 habitants) in each country (e.g. “ISL-EW38-2020” shows data from week 38 in 2020, in Iceland). Country codes (ISO 3166-1): AUS = Australia; AZE = Azerbaijan; BEL = Belgium; BRA = Brazil; COL = Colombia; DNK = Denmark; GBR = United Kingdom; GMB = Gambia; GNQ = Equatorial Guinea; HKG = Hong Kong; IND = India; ISL = Iceland; JPN = Japan; MDA = Moldova; MEX = Mexico; PAN = Panama; PNG = Papua New Guinea; QAT = Qatar; SLV = El Salvador; THA = Thailand; TUR = Turkey; TWN = Taiwan; URY = Uruguay; USA = United States; ZAF = South Africa; and ZMB = Zambia.
Many LMICs or countries with low levels of country-wide genomic surveillance were only able to sequence >5% when weekly incidence was low (<10 cases/100,000 pop.). For example, Gambia, which reported a low cumulative incidence of 226 cases/100,000 pop., sequenced nearly 8% of all cases up to mid-March 2021 (Fig. 1)(16). However, most countries in Africa and Asia, despite experiencing low incidences, were not able to scale up genomic surveillance like Gambia, Japan, Hong Kong, New Zealand and Australia, which experienced similar COVID-19 incidences (Fig. 1B-C; Fig. S1). In most Latin American countries, sequencing >1% of cases has proven to be a difficult task, particularly during periods of high incidence. Despite the low percentages of sequenced cases, surveillance in Latin America has been consistent, with heavily affected countries such as Brazil, Mexico, Chile, Colombia and Peru generating genomes nearly every week (Fig. 1B-C). This suggests that sequencing high or even moderate percentages of cases (0.1% to 1%) each week is still not feasible for most LMICs. Our study reveals another concerning fact: more than 20 LMICs, especially in Africa, do not have openly available genomes, or are only represented in the global genomic surveillance due to cases associated with travel from those locations being sequenced abroad (Fig. S2). Overall, these results show that with the worsening of the pandemic, few countries were able to maintain thorough genomic surveillance, especially LMICs, who generated few (red shades in Fig. 1A) or no sequences (dark grey) for many weeks (Table S2). European countries constitute exceptions, sequencing high or very high percentages of cases, nearly on a weekly basis (Fig. 1; Fig. S2).
Sequencing regularity and turnaround time
The rapid public sharing of data is essential for genomic surveillance (17). In 2020, the turnaround time between sample collection and genome submission varied greatly across geographic regions (Fig. 3; Fig. S3; see also (18)). Some countries have been performing surveillance mainly in near real-time, with a median turnaround time below 21 days (Fig. S3), as observed in Northern Europe (median turnaround time = 19 days). When cases started to rise in the second wave in Europe, in October 2020, countries in the region began to focus on sequencing more recent cases, shortening the median time from 43 to 19 days (see last epidemiological weeks in Northern Europe, Fig. 2). This marked change in early October coincides with, an}d could have happened in response to, the emergence and spread of B.1.1.7 (23). Similar trends were also observed in other regions, likely in an attempt to capture early introductions of B.1.1.7 (24).

Delays between sample collection and genome submission across epidemiological weeks (turnaround time) in different regions, between February 23rd, 2020 and March 27th, 2021, based on metadata submitted to GISAID up to May 30th, 2021.

Covariates that show the highest correlation with the overall percentage of COVID-19 sequenced cases (along the period shown in Fig. 1A). (A) Expenditure on R&D per capita; (B) GDP per capita; (C) Socio-demographic index; (D) Overall percentage of Influenza sequenced cases in 2019 (HA segment). The colour scheme of geographic regions is the same as in Figures 1 and 2. Solid line shows the linear fit. *PPP = purchasing power parity, USD = US dollar 2005.
Much longer turnaround times were observed in countries in eastern and central Africa, where sequencing was mainly retrospective (median turnaround time = 78 days, Fig. S2). Longer turnaround times could be a result of sequencing projects to investigate reinfections (19), vaccine escape (20), or to understand past epidemic dynamics (21, 22), types of research that are slower than public health surveillance. But longer turnaround times in a context of surveillance can be caused by delays in tasks that go from ‘sample to sequence’ and/or from ‘sequence to database’. Delays from ‘sample to sequence’ may occur as a result of insufficient lab personnel, delays in shipment of samples and reagents, and as a result of poor coordination, which leads to missing or incomplete metadata connected to samples, such as date and location of collection (7, 14, 15, 25, 26). Likewise, the lack of experienced professionals to quickly and accurately perform bioinformatics tasks (genome assembly, data collation and submission, etc) may extend the ‘sequence to database’ phase, and hamper timely responses (25). Delays may also come from concerns of having findings scooped and published by other researchers (27), revealing that the matter of data ownership, including genomic data, should be resolved in consultations with data providers, database managers, and publishers, to properly acknowledge these efforts, and facilitate rapid data sharing for the benefit of public health (7, 27, 28).
Factors associated with genomic surveillance capacity
Disparities in national wealth, in investment in research and development (R&D), and in the extent of national coordinated sequencing efforts impact the ability of countries to perform genomic surveillance (7, 13, 26). To investigate the impact of socioeconomic factors on SARS-CoV-2 genomic surveillance preparedness around the world, we explored how a list of country-level covariates are correlated with the percentage of sequenced COVID-19 cases in each country (Table S2). The strongest correlations with the log10-transformed percentage of sequenced cases are shown by expenditure on R&D per capita (r2 = 0.47), GDP per capita (0.37), socio-demographic index (0.31), established influenza genomic surveillance capacity (0.30) and fraction of out-of-pocket health expenditure out of total health expenditure (−0.35) (Fig. 3, Table S2). Using the same set of covariates, we also explored their correlations with the log-transformed mean turnaround time (Supplementary Table S3). The strongest correlations of the log-transformed mean turnaround time are with universal health coverage (r2 = −0.45), healthcare access and quality index (−0.44), socio-demographic index (−0.42) and health expenditure per capita (−0.4) (Fig. S4, Table S3).
These results reveal that socioeconomic factors represent important obstacles. Efforts must be made to improve the genomic capacity in LMIC countries to prevent the unnoticed emergence and spread of variants (13). To start, diagnostic capacity needs to be enhanced, as case underreporting directly impacts the ability of countries to detect variants and their frequency changes.
Sampling strategies for rapid variant detection
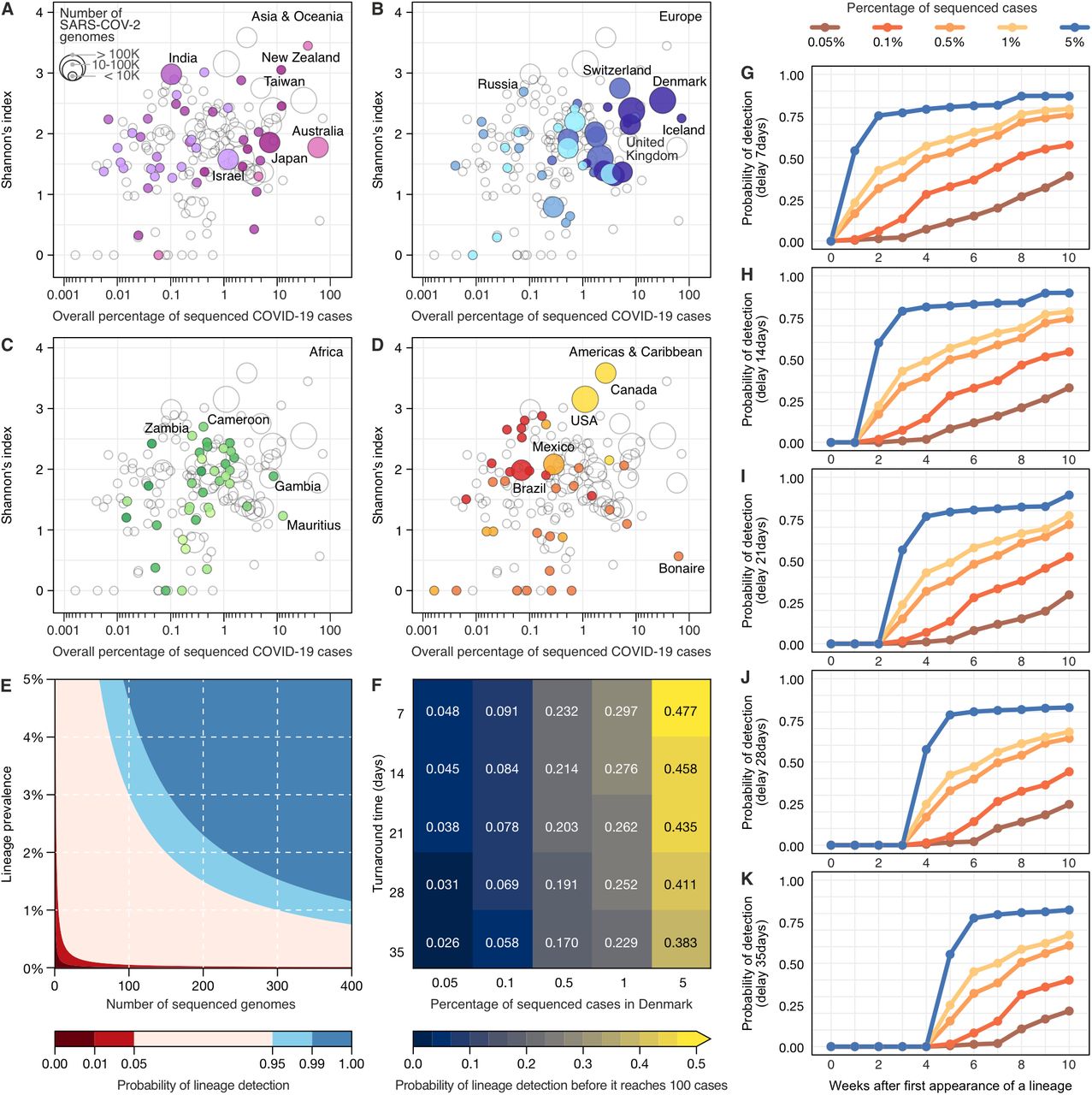
Since the initial detection and emergence of the VOC B.1.1.7/Alpha in the UK, countries across the world have sought to intensify genomic surveillance. As shown above, genomic surveillance is mainly characterized by three key aspects towards detecting previously-identified variants in a timely manner: the percentage of sequenced cases (Fig. 1), the frequency of genome sampling (Fig. 1B and Fig. 2), and the turnaround time (Fig. 2). Since resources are limited and socioeconomic factors affect the ability of countries to perform surveillance (Fig. 3 and Fig. S4), in a quantitative manner we evaluated how sequencing percentage and turnaround time affects a country’s ability to detect a previously-identified variant (Fig. 4). Initially, by exploring the diversity of lineages detected by each country in 2020-2021, we found that sequencing high percentages of cases may not necessarily lead to detection of more viral lineages (Fig. 4A-D, Fig. S5). High numbers of infections may favour the emergence of new variants (29), and with high global connectivity, some countries are more likely to import new lineages from abroad (24, 30, 31), factors that explain differences in viral lineage diversity detected in the countries. However, as expected, countries sequencing low percentages of cases and few genomes tend to detect less lineage diversity (Fig. S6 and S7). We also estimated the probability of first detection of previously identified lineages (across all lineages), under different combinations of turnaround time and sequencing percentages, assuming a scenario of random and uniform sampling at national level. To begin, we looked at the landscape of detection using binomial confidence intervals, to specifically highlight what combinations of ‘number of cases’ and ‘sequencing percentages’ can confidently say that a lineage not encountered in sequence data is also not very common. As an example, this approach allowed us to infer that when the prevalence of a rare lineage is 2%, a surveillance program would need to sequence 300 representative cases to detect at least one genome of that lineage with 95% probability (Fig. 4E). On a different look at the empirical data (Fig. 4F), we evaluated how different sequencing efforts are able to detect a lineage before it reaches 100 cases, and our analysis revealed that the percentage of sequenced cases have a larger role than turnaround time. By looking at this effect over time (Fig. 4G-K), we found that sequencing higher percentages of cases enable rapid detection of lineages, even with turnaround time delays.
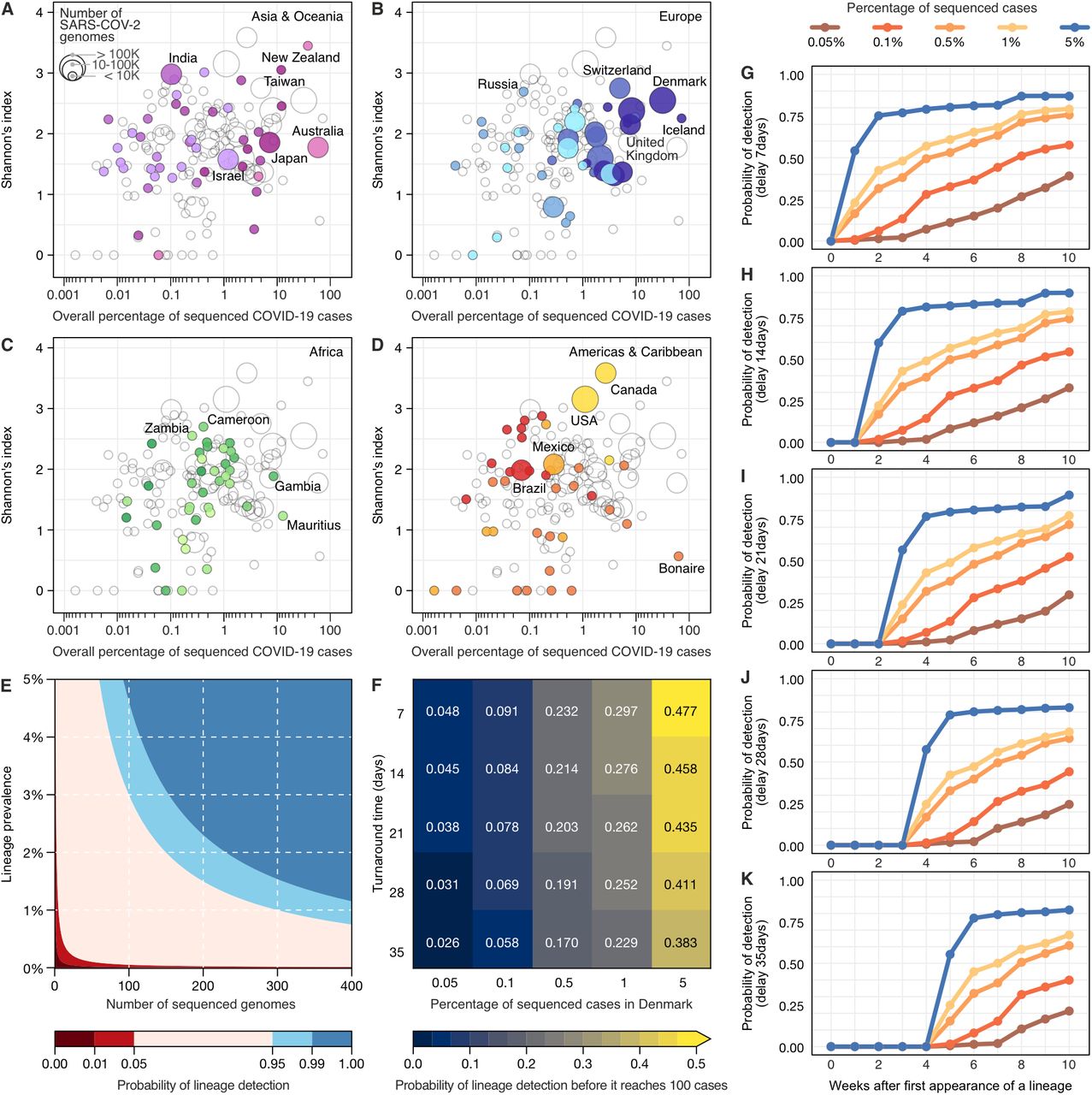
(A-D) Shannon index of lineage diversity reported in each country adopting different sequencing efforts. The colour scheme of geographic regions is the same used in Figures 1, 2 and 3. (E) The probability of detecting at least one genome of a rare lineage under different sequencing regimes. (F) Relative importance of decreasing genome sequencing turnaround time versus increasing sequencing percentage measured as probability that a lineage found in simulated datasets was detected before it had reached 100 cases in the ground truth dataset (described in Fig. S8). (G-K) Probability of lineage detection considering delays of 7, 14, 21, 28 and 35 days between sample collection and genome submission (turnaround time).
To track a virus such as SARS-CoV-2, which accumulates 2-3 substitutions per month, sampling on a weekly basis is recommended (32). Throughout this pandemic, despite differences in diagnostic capacity, weekly incidences as high as 100 cases/100,000 pop. were reported in many countries (Fig. 1C; Fig. S1). Considering the findings presented above, it is important that local public health labs improve their capacity to be able to sequence at least 0.5% of the cases during peak incidence, always adopting strategies to obtain random and representative sampling (in terms of age, sex, clinical spectrum, and geographical distribution) (33). For example, if a location (country, state, city) with 10 million habitants is reporting a weekly incidence of 100 cases/100,000 pop., a 0.5% threshold could be achieved by sequencing 1 genome for every 200,000 habitants, which we propose as a reasonable benchmark. Based on empirical data (Fig. 1; Fig. S1) and our statistical analysis (Fig. 4E-K), if public health labs worldwide use such a benchmark to set their minimal operational limits to sequence at least 0.5% of the cases at high incidence (100 cases/100,000 pop.), with quick turnaround time (<21 days), it would greatly improve our global capacity to detect new variants and track changes in variant prevalence.
Conclusion
We provide a comprehensive overview of SARS-CoV-2 genomic surveillance patterns observed worldwide, highlighting disparities in surveillance capacity in different geographic regions, in terms of percentage of sequenced cases, frequency of sampling (Fig. 1), and turnaround time (Fig. 2). Differences in socioeconomic (Fig. 3), epidemiological (Fig. S1), and political factors (26) are associated with genomic surveillance capacity and timeliness. Consequently genomic surveillance in most countries can not provide rapid responses (quick turnaround, below 21 days), perform mainly sparse surveillance (less than 75% of the weeks are sampled), and are unable to achieve even the minimum percentage of sequenced cases we propose here as a benchmark (at least 0.5% of reported cases in high incidence weeks). Infectious diseases represent a global threat, and as such, require international, coordinated efforts to allow the rapid detection of emerging pathogens (4, 26). Since the identification of cases is an essential step that enables genomic surveillance, it is essential to enhance diagnostic capacity within countries, beyond the metropolitan areas. Further, in order to maintain constant and rapid genome sequencing, local coordination, adequate staffing and training, and appropriate analytical tools are essential for enabling rapid responses to emerging infectious disease threats to public health. To that end, we need to implement better protocols for performing representative sampling (see 28, 33), so that affordable, impactful and cost-effective genomic surveillance strategies can be adopted. Finally, efforts must be made to provide funds, training, and logistic support for LMICs to improve their local genomic surveillance capacity, to allow public health decision making in regions where resources may be scarce.
Data Availability
Data used in this study can be found in this GitHub repository: https://github.com/andersonbrito/paper_2021_metasurveillance
Funding
Internal Fondsen KU Leuven/Internal Funds KU Leuven, Grant No. C14/18/094 (GB)
Research Foundation – Flanders, “Fonds voor Wetenschappelijk Onderzoek - Vlaanderen” G0E1420N, (GB)
Research Foundation – Flanders, “Fonds voor Wetenschappelijk Onderzoek - Vlaanderen”, G098321N (GB)
National Institutes of Health F31 AI154824 (GWH, MAS)
National Institutes of Health R01 AI153044 (MAS)
National Institutes of Health U19 AI135995 (MAS)
Oxford Martin School, EUH2020 project MOOD (MUGK)
Branco Weiss Fellowship (MUGK)
Rockefeller Foundation (MUGK)
Google.org (MUGK)
Fast Grant from Emergent Ventures at the Mercatus Center at George Mason University (NDG)
Centers for Disease Control and Prevention (CDC) Contract # 75D30120C09570 (NDG)
Oxford Martin School (OGP)
Wellcome Trust (NRF)
Royal Society Sir Henry Dale Fellowship 204311/Z/16/Z (NRF)
Medical Research Council-São Paulo Research Foundation (FAPESP) CADDE partnership award (MR/S0195/1 and FAPESP 18/14389-0) (http://caddecentre.org/) (NRF)
Author Contributions
Conceptualization: AFB, NDG, NRF; Data curation: Danish Covid-19 Genome Consortium; Formal analysis: AFB, ES, GD, GWH, CCK, SB, SF, MAS, GB; Validation: AFB, ES, GD, GWH, MUGK, SCH, OGP, CD, SB, SF, NDG, GB, NRF; Methodology: AFB, GD, ES. Visualization: AFB, GD, ES. Writing – original draft: AFB, ES, GD, CCK, GB, NRF; Writing – review & editing: AFB, ES, GD, GWH, MUGK, SCH, ECS, OGP, CD, MAS, NDG, GB, NRF; Funding acquisition: NDG.
Conflicts of Interest
NDG is an infectious diseases consultant for Tempus Labs. MAS receives grants and contracts from the National Institutes of Health, the US Food & Drug Administration, the US Department of Veterans Affairs and Janssen Research & Development. OGP has undertaken work for AstraZeneca on SARS-CoV-2 classification and genetic lineage nomenclature.
Data and materials availability
Data used in this study can be found in this GitHub repository: https://github.com/andersonbrito/paper_2021_metasurveillance
Acknowledgements
We gratefully acknowledge the authors from the Originating laboratories responsible for obtaining the specimens, as well as the Submitting laboratories where the genomic data were generated and shared via GISAID, on which this research is based. An acknowledgement table can be found in Table S5. GD acknowledges Joshua Batson, whose work shared on Twitter (@thebasepoint) inspired the creation of Figure 4E.
Footnotes
↵‡ Co-senior authors





{kind=link}
{kind=link}
{kind=link}
{kind=link}