
Abstract
Host genetic variants influence the susceptibility and severity of several infectious diseases, and the discovery of novel genetic associations with Covid-19 phenotypes could help developing new therapeutic strategies to reduce its burden.
Between May 2020 and February 2021, we used Covid-19 data released periodically by UK Biobank and performed over 400 Genome-Wide Association Studies (GWAS) of Covid-19 susceptibility (N=15,738 cases), hospitalization (N=1,916), severe outcomes (N=935) and death (N=828), stratified by ancestry and sex.
In coherence with previous studies, we observed 2 independent signals at the chr3p21.31 locus (rs73062389-A, OR=1.22, P=7.64×10−14 and rs13092887-A, OR=1.73, P=2.38×10−8, in Europeans) modulating susceptibility and severity, respectively, and a signal influencing susceptibility at the ABO locus (rs9411378-A, OR=1.10, P =7.36×10−10, in Europeans), which was more significant in men than in women (P=0.01). In addition, we detected 7 genome-wide significant signals in the last data release analyzed (on February 24th 2021), of which 4 were associated with susceptibility (SCRT2, LRMDA, chr15q24.2, MIR3681HG), 2 with hospitalization (ANKS1A, chr12p13.31) and 1 for severity (ADGRE1). Finally, we identified over 300 associations which increased in significance over time, and reached at least P<10−5 in the last data release analyzed. We replicated 2 of these signals in an independent dataset: a variant downstream of CCL3 (rs2011959) associated with severity in men, and a variant located in an ATP5PO intron (rs12482569) associated with hospitalization.
These results, freely available on the GRASP portal, provide new insights on the host genetic architecture of Covid-19 phenotypes.
Introduction
The severe acute respiratory syndrome – coronavirus 2 (SARS-CoV-2) is responsible for the coronavirus disease 2019 (Covid-19) which affects individuals with variable severity, ranging from asymptomatic patients to mild respiratory symptoms, hypercytokinemia, pneumonia, thrombosis and even death.1,2 Understanding the mechanisms leading to heterogeneous symptoms and susceptibility is essential in order to develop efficient treatments and improve patient care. Host genetic diversity has been shown to influence the effects of infection to several viruses,3 such as variations in CCR5 [MIM: 601373] leading to HIV resistance4 [MIM: 609423] or IRF7 [MIM: 605047] deficiency affecting Influenza susceptibility [MIM: 614680].5
In order to discover human genetic determinants to Covid-19 susceptibility and severity, several biobanks and research groups worldwide collaborated to perform Genome-Wide Association Studies (GWAS) and meta-analyses of the GWAS. In June 2020, a study involving 1,980 Covid-19 infected patients with respiratory failure was the first to reveal genome-wide significant (P < 5 × 10−8) associations at the 3p21.31 locus, encompassing SLC6A20 [MIM: 605616] and several chemokine receptors, and at the ABO [MIM: 110300] locus on chromosome 9.6 These 2 signals were later validated in independent analyses for both Covid-19 susceptibility and severity7,8 while additional significant associations were observed at loci involved in immune response or inflammation, such as IFNAR2 [MIM: 602376], DPP9 [MIM: 608258], TYK2 [MIM: 176941], CCHCR1 [MIM: 605310] and OAS1 [MIM: 164350]. Notably, these findings implicate common and uncommon variants, while studies trying to identify associations of rare variants have been unsuccessful so far.9 The largest effort is currently led by the Covid-19 host genetics initiative (Covid-19hgi),10 which completed meta-analyses of results shared by 46 studies as of January 18th 2021, and plan to release new results as additional data is made available. A major contributor to this group is the UK Biobank (UKB)11 which periodically releases the results of Covid-19 tests and related deaths, as well as health care data for its nearly 500,000 consented participants, to approved researchers.
We created a public Covid-19 GWAS results portal (https://grasp.nhlbi.nih.gov/Covid19GWASResults.aspx) in order to provide rapid deep annotation for emerging genetics results and facilitate open access to the scientific community. We contribute to this resource by performing GWAS on each Covid-19 data release from the UK Biobank, including sex-specific, ancestry-specific, and trans-ethnic Covid-19 related GWAS, along with a deep set of annotations for top variants (with P < 1 × 10−5). For each release, up to 65 GWAS have been generated including Covid-19 susceptibility, Covid-19 hospitalization, severe Covid-19 with respiratory failure, and Covid-19 death. Here we describe the results of these GWAS, 434 in total as of February 24th 2021, and report the evolution of signals associated with these Covid-19 phenotypes over the consecutive datasets released by UKB since May 2020. The latter approach, tracking the evolution of genetic signatures iteratively in UKB, suggests a valuable new analytic approach in genetic biobank studies where emerging true signals may be identified before they reach genome-wide significance based on their trajectories of significance.
Materials and Methods
UK Biobank data
Analyses are based on v.3 of the UKB imputed dataset,12 which provide genomic data for 487,320 participants from multiple ethnicities, including 459,250 of European ancestry (EUR), 7,644 of African ancestry (AFR), 9,417 of South Asian ancestry (SAS) and 11,009 of other ancestries (OTHERS). Participants were enrolled at ages ranging from 37 to 73 and are 51.16% female. UKB started to release Covid-19 test results of its participants on March 15th 2020, and periodically update this resource as new cases are reported. Furthermore, information about Covid-19 related death was made available from June 2020, while inpatient data and primary care data was first added during the summer of 2020 and are periodically updated. Details regarding the definition and selection of cases with Covid-19 susceptibility, Covid-19 hospitalization, Covid-19 severity, and Covid-19 death are available in Table S1.
Phenotype definition
Depending on the Covid-19 phenotype analyzed (susceptibility, hospitalization, severity, or death), up to 3 different subsets of participants were used as controls. For Covid-19 susceptibility, cases with positive test results were analyzed against either participants tested with negative results (labelled Tested), or against all participants without a positive test (labelled Population). For analyses of Covid-19 hospitalization, patients hospitalized due to Covid-19 were tested against non-hospitalized participants with a positive test (Positive), or non-hospitalized participants with a test (Tested), or against all non-hospitalized participants (Population). For analyses of Covid-19 severity, patients requiring invasive respiratory support or patients who died from complications were tested against non-severe participants with a positive test (Positive), or non-severe participants with a test (Tested) or all non-severe participants (Population). For analyses of Covid-19 death, patients with Covid-19 death were tested against participants with a positive test (Positive), or participants with a test (Tested) or against all participants (Population).
Analyses
Each GWAS was conducted with SAIGE v0.38,13 which controls for population stratification, relatedness and case-control imbalance, and adjusted for baseline age (at enrollment), sex and 10 genetic principal components. For the results uploaded to the GRASP portal, variants were filtered on imputation quality (r2 > 0.3), minor allele count (MAC > 2), and minor allele frequency (MAF > 0.0001). However, for the results presented in this manuscript, we applied a more stringent filter, and considered only well-imputed variants (r2 > 0.8) and common variants (MAF > 0.01). After applying this filter, the lambda (genomic control factor) ranged from 0.991 to 1.027 in all 65 analyses of the 02.24.21 data release (Table S2), indicating no systematic inflation. For analyses prior to the June 18th 2020 release, we conducted analyses on participants of European ancestry only, and started adding new analyses stratified by sex and ancestry from June 18th 2020 onward. GWAS were stratified for EUR, AFR, SAS, and OTHERS ancestries, and an additional trans-ancestry GWAS combining all participants (labelled ALL) as well as GWAS combining non-European (nEUR) participants were performed.
Associations are either reported as odd ratios (OR) and 95% confidence intervals or as beta coefficients (β) and associated standard errors (SE). Linkage disequilibrium (LD) was estimated by squared correlation (r2) using UKB EUR imputed data. To test 2 observed effects are equal, we used the Z statistic: 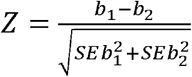 , with b1 and b2 corresponding to the observed effects and SEb1 and SEb2 the associated standard errors.
, with b1 and b2 corresponding to the observed effects and SEb1 and SEb2 the associated standard errors.
Annotation
For each analysis hosted on the portal, we provide comprehensive annotation for top results (P < 1 × 10−5) using ANNOVAR14 and the RESTful API service provided by CADD v1.6.15 We also retrieve known phenotype associations extracted from the GRASP16 and EBI GWAS catalogs,17 and known eQTLs extracted from GTeX v818 and other eQTL resources compiled from nearly 150 datasets (built upon the work of Zhang et al,19 detailed in Table S3).
Replication
The 6 Covid-19hgi meta-analyses, release version 5, excluding the UKB and 23andMe datasets, were used for our replication efforts and include results for Covid-19 susceptibility (labelled C2), hospitalization (B2) and severity (A2). The 3 meta-analyses restricted to European individuals (EUR) were used to replicate signals observed in our GWAS involving European UKB participants, while the remaining 3 trans-ancestry meta-analyses (ALL) were used to replicate signals observed either in our trans-ancestry GWAS or in non-European analyses. A signal was defined as replicated if the effect direction was concordant in the GWAS and the Covid-19hgi meta-analysis, and if the p-value in the meta-analysis was below 0.1, corresponding to a one-sided test threshold of 0.05. In addition, the Bonferroni method was applied to correct for multiple testing.
Data Availability
Permission was obtained to post UKB summary statistics under an approved application (ID 28525). The association results are available on the portal, as well as annotated top results. In addition to UKB summary statistics, results from other efforts are also hosted on the portal. Authors of Covid-19 GWAS publications have been contacted to seek approval before hosting the results of their analyses on the GRASP Covid-19 portal. Summary statistics at this time include multiple releases of the Covid-19hgi group, severe hospitalization results from Ellinghaus et al6 and the GenOMICC study,7 with all results are being re-annotated in the common framework mentioned above.
Results
UK Biobank Covid-19 demographics
Using the latest UKB data release available at this point, analyzed on February 24th 2021, we retrieved 47,413 participants with a Covid-19 diagnostic, of which 15,738 tested positive. According to inpatient care data, 1,916 positive cases were hospitalized, while 935 patients with severe Covid-19 diagnostic received respiratory support and/or died from complications (Table 1). Since May 7th 2020, we analyzed 15 UKB data releases regarding Covid-19 susceptibility, 7 data releases concerning Covid-19 related deaths, and 4 data releases for both Covid-19 related hospitalization and Covid-19 severity. Amongst Covid-19 positive participants, we observed a global increase in the percentage of female cases, starting at 45.3% at the first release analyzed, and reaching 52.47% in the last, while men were more likely to be infected (P = 9.94 × 10−7), hospitalized (P = 1.34 × 10−39), or develop severe complications (P = 3.40 × 10−29) and die from Covid-19 (P < 10−300) in the 02.24.21 data release (Table S4). There was also a decrease in the mean age of positive cases, ranging from 57.02 to 53.77, with a significant drop after the 2020 summer (Table 2), with younger individuals more likely to be infected (P < 10−300) while increase in age was associated with hospitalization (P = 1.23 × 10−73), severity (P = 1.41 × 10−93) and death (P = 3.40 < 10−300) (Table S4). Positive cases were mainly of European ancestry, representing 85.5% of all Covid-19 positive participants in the first analysis and growing to 89.3% in the last. South Asian ancestry, African ancestry, and other ancestry represent 4.4%, 3.2% and 3.1% of positive cases, respectively, in this UKB data release. However, compared to European participants, non-European participants were more likely to be infected (P = 9.34 × 10−37 for AFR, P = 6.68 × 10−82 for SAS, P = 4.31 × 10−8 for OTHERS), hospitalized (P = 2.78 × 10−28 for AFR, P = 2.23 × 10−9 for SAS, P = 1.01 × 10−6 for OTHERS), severe (P = 2.89 × 10−19 for AFR, P = 9.84 × 10−4 for SAS, P = 0.02 for OTHERS) and deceased participants (P = 2.72 × 10−10 for AFR, P = 7.04 × 10−3 for SAS) (Table S4).
Sample sizes for each analysis, as of 02.24.21
Composition of the consecutive UK Biobank Covid-19 data releases
Associations with Covid-19 susceptibility
The genome-wide significant loci in the last data release analyzed (on 02.24.21) for Covid-19 susceptibility are presented in Table 3, while all associations with P < 10−5 are available in Table S5. In addition, all signals reaching genome-wide significance in any data release analyzed are presented in Figures S1-S18.
Lead variants associated with Covid-19 susceptibility (on 02.24.21)
This project started with the analysis of Covid-19 susceptibility in participants of European ancestry, as the largest ancestry group in UKB. Consequently, it is the most repeated analysis with 15 iterations. With untested or negatively tested participants as controls (Population controls), 8 signals reached genome-wide significance (P < 5 × 10−8) at some point (Figure 1), of which only 3 remained significant in the last data release analyzed: the chr3p21.31 locus encompassing SLC6A20 and several chemokine receptors (rs73062389-A, MAF = 0.06, OR = 1.22 [1.16; 1.29], P = 7.64 × 10−14), the ABO locus on chromosome 9 (rs9411378-A, MAF = 0.22, OR = 1.10 [1.07; 1.14], P = 7.36 × 10−10) and intronic variants located in SCRT2 and SRXN1 [MIM: 617583] on chromosome 20 (rs749199237-del.G, MAF = 0.35, OR = 1.08 [1.05; 1.11], P = 1.29 × 10−8). The chr3p21.31 and ABO loci were previously reported to modulate Covid-19 susceptibility,6–8,10 while the third signal on chromosome 20 was novel.

{kind=link}
The top panel represents the evolution in significance of signals reaching genome wide significance at least once across all data releases analyzed in Europeans for Covid-19 susceptibility. The middle panel represent the mean age of cases in each data release. The bottom panel represent the number of cases in each data release.
Amongst the 5 other signals no longer significant in this data release, the APOE [MIM: 107741] variant tagging for the APOE-ε4 haplotype (rs429358-C, MAF = 0.15, OR = 1.38 [1.24; 1.53], P = 1.80 × 10−9, on 07.14.20) was the only signal previously described,20 and was notably the first report of a genetic determinant for Covid-19 susceptibility. However, this previous report was based on UKB data and this signal was not replicated in an independent dataset. This association was greatly attenuated after the summer, when the number of Covid-19 cases started to rise significantly and the mean age of infected participants decreased. The interaction between age of participants and the APOE variant was significant (P = 3.4 × 10−6) suggesting that a subset of older participant carriers of this variant was more at risk of Covid-19 infection. Remarkably, in the 02.24.21 data release, this variant is still suggestively associated with Covid-19 severity (OR = 1.42 [1.24; 1.62], P = 4.36 × 10−7 in EUR) and death (OR = 1.45 [1.25; 1.67], P = 4.62 × 10−7 in EUR) suggesting a mechanism leading to lethal complications after infection.
Using negatively tested participants as controls (Tested controls), the only signal reaching genome-wide significance in this last data release was again the chr3p21.31 locus (P = 1.85 × 10−9) with a different lead variant (rs73062394-T, MAF = 0.059, OR = 1.2 [1.13; 1.28]). This variant was in moderate linkage disequilibrium (LD) with rs73062389 (r2 = 0.59), which was the second lead variant (P = 8.46 × 10−9) for this analysis.
In other ancestry-stratified GWAS (AFR, SAS and OTHERS), no signal was found genome-wide significant in the last data release analyzed. However, in the GWAS combining all non-European participants, with Population controls, one signal was found significant at the LRMDA [MIM: 614537] locus on chromosome 10 (rs114026383-C, MAF = 0.02, OR = 2.42 [1.78; 3.29], P = 1.82 × 10−8). According to gnomAD (v2.1.1),21 this variant is mostly carried by individuals of African ancestry (MAF = 0.04) and mainly absent in other ancestries. In the GWAS of African ancestry participants, this signal is close to genome wide significance (P = 3.24 × 10−7). Interestingly, LRMDA variants have been found associated to lung function22 [MIM: 608852] and HIV viral load in an unadjusted GWAS,23 but there was no evidence of LD between these variants and rs114026383 (r2 < 0.01).
Finally, in the trans-ancestry analyses combining all UKB participants, using the Population controls, the signals at the chr3.p21.31 and ABO loci were also genome-wide significant (rs73062389-A, P = 3.99 × 10−13 and rs9411378-A, P = 1.12 × 10−9, respectively), and a third signal was observed at the MIR3681HG locus on chromosome 2 (rs112938622-G, MAF = 0.22, OR = 1.08 [1.05; 1.12], P = 4.95 × 10−8). When using Tested controls, only the chr3p21.31 signal was genome-wide significant in the last analysis (rs73062394-T, P = 1.99 × 10−8).
In sex-stratified analyses, using Population controls, the chr3p21.31 signal was significant in women (rs73062389-A, P = 1.06 × 10−8 in EUR and P = 1.35 × 10−8 in the trans-ancestry GWAS) and highly associated in men (P = 1.12 × 10−6 in EUR and P = 3.19 × 10−6 in the trans-ancestry GWAS), whereas the ABO signal was significant in men (rs9411378-A, P = 2.00 × 10−8 in EUR and P = 2.05 × 10−8 in the trans-ancestry GWAS) and moderately associated in women (P = 1.51 × 10−3 in EUR and P = 1.89 × 10−3 in the trans-ancestry GWAS). Notably, the observed effect of the ABO signal was significantly stronger in men (β = 0.135, SE = 0.023 in EUR) than in women (β = 0.068, SE = 0.021), using the Z-test for the equality of regression coefficients (P = 0.015). In addition, when using Tested controls, an intergenic variant at the chr15q24.2 locus was significant in the trans-ancestry GWAS of women (rs71401691-G, MAF = 0.07, OR = 1.23 [1.14; 1.32], P = 4.72 × 10−8), but not in men (P = 0.89). According to GTeX, the effect allele increases the transcription levels of MAN2C1 (Mannosidase Alpha Class 2C Member 1, [MIM: 154580]) in several tissues including lungs (P = 6.7 × 10−14) and esophagus-mucosa (P = 3.4 × 10−10).
Several signals were genome-wide significant in analyses using a specific set of controls, but were below the significance threshold when using another one. Notably, the LRMDA signal had a stronger effect in the Population analysis (β = 0.88, SE = 0.16) than in the Tested analysis (β = 0.62, SE = 0.16) and the intergenic variant at the chr15q24.2 locus had a stronger effect in the Tested analysis (β = 0.20, SE = 0.04) than in the Population analysis (β = 0.13, SE = 0.03), as represented in Figure S19. However, the difference in effects when using 2 different set of controls was not significant when applying the Z-test for the equality of regression coefficients (P > 0.05).
Associations with Covid-19 hospitalization, severity, and death
The genome-wide significant findings for Covid-19 hospitalization, severity and death are presented in Table 4, while all associations with P < 10−5 are available in Table S6. Similarly to the susceptibility analyses, all signals reaching genome-wide significance in any analysis are presented in Figures S20-S42. Across all analyses of hospitalization and severity, we identified 4 loci reaching genome-wide significance in the last data release analyzed, while no significant signal was observed in the GWAS of Covid-19 related death.
Lead variants associated with Covid-19 hospitalization, severity, or death (on 02.24.21)
The most recurring signal is again located at the known chr3p21.31 locus. Depending on the analysis, we observed 3 distinct leads variants at this locus: rs13071258 in the GWAS of hospitalized Europeans, rs72893671 in the trans-ancestry hospitalization GWAS, and rs13092887 in the severe Covid-19 GWAS of both Europeans and all ancestries. However, all 3 variants were in LD (r2 > 0.84), suggesting a single haplotype modulating Covid-19 hospitalization and severity. As previously reported,8 this haplotype seems distinct from the signal of Covid-19 susceptibility (Figure S43), as the 2 susceptibility variants (rs73062389 and rs73062394) are not in LD (r2 < 0.01) with the 3 severity variants.
The 3 remaining signals were not previously reported, and were identified in sex-stratified analyses. First, intronic variants in ANKS1A [MIM: 608994] were observed significantly associated in several analyses of men with Covid-19-related hospitalization, for instance in EUR using non-hospitalized participants with a positive test as controls (rs112887370-T, MAF = 0.02, OR = 3.30 [2.18; 5.00], P = 1.71 × 10−8). According to GTeX, this allele increases the transcript levels of the nearby gene C6orf106 [MIM: 612217] in some tissues (thyroid, P = 1.5 × 10−7 and testis, P = 1.7 × 10−6), while other alleles in LD also reaching genome-wide significance (e.g. rs2504165-T, OR = 2.84 [1.96; 4.12], P = 3.99 × 10−8, r2 = 0.73) can decrease the transcript levels of C6orf106 in skeletal muscles (P = 3.2 × 10−5). C6orf106 is also known as ILRUN (Inflammation and Lipid Regulator with UBA-like and NBR-1-like domains), a gene involved in the regulation of antiviral response. Second, an intergenic indel located at the chr12p13.31 locus, between CD163 [MIM: 605545] and APOBEC1 [MIM: 600130], was associated with the hospitalization of women when using non-hospitalized participants with a positive test (rs370732090-ins.ATTAT, MAF = 0.14, OR = 0.61 [0.52; 0.73], P = 3.37 × 10−8). Finally, an intronic insertion-deletion located in ADGRE1 [MIM: 600493] was associated with Covid-19 severity in European women, using participants without severe Covid-19 as controls (rs770180814-ins.GT, MAF = 0.22, OR = 1.78 [1.45; 2.19], P = 4.51 × 10−8).
Replication of novel genome-wide significant signals
The 7 novel signals identified were further investigated using the Covid-19hgi meta-analyses results, where the UKB was not included. Unfortunately, variants constituting the LRMDA and APOBEC1 signals were absent from the meta-analyses, which prevented our effort to replicate these 2 signals. Out of the 5 remaining signals, 4 did not reach nominal significance in the replication datasets: at the SCRT2 locus (rs749199237, P = 0.97 in C2 EUR), at the MIR3681HG locus (rs112938622, P = 0.27 in C2 ALL), at the chr15q24.2 locus (rs71401691, P = 0.82 in C2 ALL) and at the ADGRE1 locus (rs770180814, P = 0.55 in A2 EUR). Finally, at the ANKS1A locus, the lead variant identified in the analysis of hospitalized Europeans men did not replicate (rs112887370, P = 0.14 in B2 EUR) but another variant in LD mentioned earlier was associated at nominal significance (rs2504165-T, OR = 1.13 [1.00; 1.28], P = 0.04 in B2 EUR). Unfortunately, this association does not survive multiple-testing correction, with a threshold for significance set at P < 0.017 for 6 one-sided tests. However, the ANKS1A, ADGRE1 and chr15q24.2 signals were identified in sex-stratified analyses, and the possibility for replication in an independent sex-stratified dataset is not yet provided by Covid-19hgi.
Signals with suggestive significance trends
The significance trajectory of the most robust signals, at the chr3p21.31 and ABO loci, mostly increased after the surge of new cases following the 09.08.20 data release (Figure 1). After this date, the chr3p21.31 signal increased at each of the following 8 data release and reached genome-wide significance in the 11.03.20 release, while the ABO signal increased 5 times out of 8 and reached significance in the 01.04.21 release. In order to identify signals that may become significant in future releases, we extracted variants displaying similar positive trends in significance, meaning variants not yet genome-wide significant, but exhibiting an increase in significance since the 09.08.20 data release.
For each Covid-19 susceptibility GWAS, we extracted variants which had an increase in significance in at least 6 out of the 8 releases following the 09.08.20 data release, and reached P < 10−5 in the last data release. For other Covid-19 phenotypes, considering that fewer data releases were available, we extracted variants which increased in significance in all consecutive releases, reaching the same significance threshold of P < 10−5 in the last data release. After excluding genome-wide significant signals, 2,409 variants involved in 3,607 associations variants with suggestive trends were identified across all GWAS (Table S7).
Next, we sought to identify whether these signals with suggestive trends reached at least nominal significance (P < 0.05) in the Covid-19hgi meta-analyses. Only signals from the European and trans-ancestry GWAS were considered at this step, to match the corresponding Covid-19hgi meta-analyses. Signals with suggestive trends from Covid-19 susceptibility GWAS were sought for replication in the corresponding C2 (susceptibility) meta-analyses, while signals with trends from the hospitalization and severe Covid-19 GWAS were sought for replication in the B2 and A2 meta-analyses, respectively. In order to decrease the multiple-testing correction burden, we only extracted the lead variant at each locus, a locus being defined as a genomic region containing one or several variants with P < 10−5 separated by less than 1Mb. As a result, 329 lead variants involved in 378 associations were extracted (Table S8), of which 18 had concordant effects and reached nominal significance in the Covid-19-hgi meta-analyses (Table 5). After applying the multiple-testing Bonferroni correction, with one-sided hypothesis, only 2 signals passed the corrected significance threshold (0.05*2/378 = 2.65 × 10−4), both in trans-ancestry analyses. The first signal had a positive significance trajectory in the hospitalization GWAS (Figure S44) and is located 5Kb upstream of ATP5PO [MIM: 600828], encoding a part of the ATP synthase complex. According to GTeX, the effect allele (rs12482569-A, MAF = 0.17, OR = 1.22 [1.12; 1.33], P = 6.28 × 10−6) increases the transcript levels of ATP5PO in several tissues including the lungs (P = 4.6 × 10−18) and the esophagus-mucosa (P = 1.9 × 10−11). The second signal (rs2011959-A, MAF = 0.37, OR = 1.34 [1.18; 1.53], P = 6.50 × 10−6) had a positive trend in the GWAS of men with severe Covid-19 (Figure S45), and is located 200 bp downstream of CCL3 [MIM: 182283], a pro-inflammatory cytokine, reported by numerous studies to have unusually high levels in patients with severe Covid-19.24–26
Suggestive trends with p < 0.05 in Covid-19hgi meta-analyses
Discussion and conclusions
This project was initiated with the major aim to share results of Covid-19 host genetics analyses freely and rapidly on the GRASP portal, during a pandemic where new insights to improve patient care and develop better treatments were greatly needed. A few months after the first Covid-19 case was identified in the UK, we started to perform GWAS on each dataset released by UKB between May 2020 and February 2021, and thus far examined genetic signals associated with Covid-19 phenotypes across 15 data releases. This unique context allowed us to track the evolution of genetic associations over time, an approach rarely applied. While some works have examined the statistical properties of phased and nested case-control studies,27,28 few studies have proposed using statistical trajectories over time in genetic analyses.
As a first major observation, the majority of signals which were observed in the first stages of the project did not sustain over time. For instance, the first genome-wide significant association observed in the GWAS of Covid-19 susceptibility in Europeans with rs34338189 as lead variant is not even nominally significant in the last release (P = 0.29). Another genome-wide significant signal was observed at the APOE locus, with a significance that increased in the first 4 data releases before decreasing continually in the subsequent releases. The lead variant at the APOE locus is coding in part for the APOE-ε4 haplotype, known to increase the risk of Alzheimer’s disease [MIM: 607822], dementia, dyslipidemia and cardiovascular diseases [MIM: 617347] and is speculated to cause inflammation and cytokine storms.29 Notably, the suggestive association of the APOE signal with severe Covid-19 could support this proposition. The evolution of the APOE signal over time could also be due to an initial higher prevalence of Covid-19 in nursing homes,30 where dementia patients were at higher risk of being infected and spreading the virus due to living arrangements, and a poor understanding of transmission dynamics and appropriate safety guidelines early in the pandemic. Overall, the evolution of these signals suggests a change in the composition of cases over time, such as the diminution of age and increase of positively tested women in the later data releases, as well as the introduction of variant SARS-CoV2 strains. This change was most significant after the summer when a surge in new Covid-19 infections occurred.
The most robust findings from our study are the association of the chr3p21.31 and ABO loci with Covid-19 susceptibility and a distinct signal at the chr3p21.31 locus associated with Hospitalization and severe Covid-19. These observations corroborate several previous reports,6–8,10 although we also observed a significant difference in the effect of the ABO variant between men and women. Sex-stratified analyses also allowed us to identify a novel association between ANKS1A variants and hospitalization of men, which reached nominal significance in the Covid-19hgi hospitalization meta-analysis. Interestingly, the variants comprising this signal can also influence the expression of ILRUN, either as an upregulator or as a repressor, depending on the tissue. This gene is involved in innate immunity and has been recently shown to act as an antiviral factor in the context of SARS-CoV-2 infection, notably by downregulating the expression of ACE2 [MIM: 300335] and TMPRSS2 [MIM: 602060], the main SARS-CoV-2 entry receptors.31 This role differs from a previous report demonstrating an antiviral effect in cells infected with Hendra virus,32 which could hint at diverse roles for this gene in the context of viral infection.
We have developed an original strategy to identify associations displaying increased significance over time, and identified hundreds of loci with a significance trajectory suggesting potential future genome-wide significance. Amongst these signals, 2 of them replicated at nominal p-value in the Covid-19hgi meta-analyses, including a variant downstream of CCL3 suggestively associated with Severe Covid-19 in women, a cytokine with a well established high expression in severe Covid-19 cases.24–26 The associations we observed changing through the pandemic could reflect random effects or changes in statistical power, but some of the results suggest changes due to potential gene-environment interactions such as age, underlying health conditions (APOE) or sex makeup of cases exposed to or engaging in risk behavior. This indicates the general approach of iterative analysis and trends analyses for genetics during pandemics may have benefits in uncovering pathophysiologic clues. Additionally, other factors like predominant virus strains and changing treatment strategies through a pandemic might interact with host genetics, and be better understood by iterative analyses.
In summary, our host genomic analyses of Covid-19 have improved the comprehension of mechanisms involved in the infection and complications due to Covid-19 and we continue to perform and rapidly share these analyses with the research community. Our study has some limitations. Most importantly, our data and the work of others support large health disparities between EUR and non-EUR individuals related to COVID-19 throughout the ongoing pandemic. Despite an over-representation proportionally among cases and those with severe and fatal outcomes, the non-EUR component of UKB is a proportionally small sample limiting our statistical power to address population-specific genetic variants contributing to health outcomes. Moving forward we feel that having a diverse set of results with different phenotype definitions, sex-specific, ancestry-specific, and including external group summary statistics, all in a common genome reference and annotation framework may maximize the chance for new studies to cross-replicate or meta-analyze results as Covid-19 genetic studies continue to grow.
Data Availability
The datasets generated during this study are available at the GRASP Covid-19 portal
Description of Supplemental Data
Supplemental Data include 45 figures and 8 tables.
Declaration of Interests
The authors declare no competing interests.
Web Resources
GTeX: https://gtexportal.org/home/
CADD: https://cadd.gs.washington.edu/
EBI GWAS catalog: https://www.ebi.ac.uk/gwas/
GRASP catalog: https://grasp.nhlbi.nih.gov/Overview.aspx
Covid-19hgi meta-analyses results: https://www.covid19hg.org/results/r5/
Data and Code Availability
The datasets generated during this study are available at the GRASP Covid-19 portal: https://grasp.nhlbi.nih.gov/Covid19GWASResults.aspx
Acknowledgments
All authors were supported by NIH Intramural Research Program funds. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services. This research has been conducted using the UK Biobank Resource under Application Number 28525. UK Biobank was established by the Wellcome Trust, Medical Research Council, Department of Health, Scottish government, and Northwest Regional Development Agency. It has also had funding from the Welsh assembly government and the British Heart Foundation. All UKB analyses for this manuscript were conducted on the NIH Biowulf high performance computing cluster (https://hpc.nih.gov/). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. We also thank the NHLBI IT team for their help in keeping the GRASP portal up to date.
References




