
ABSTRACT
Understanding the evolution of SARS-CoV-2 virus in various regions of the world during the Covid19 pandemic is of the utmost importance to help mitigate the effects of this devastating disease. We describe the phylogenomic and population genetic patterns of the virus in Mexico during the pre-vaccination stage, including asymptomatic carriers. A RT-qPCR screening and phylogenomics reconstructions directed a sequence/structure analysis of the Spike glycoprotein, revealing mutation of concern E484K in genomes from central Mexico, in addition to the nationwide prevalence of the imported variant 20C/S:452R (B.1.427/9). Overall, the detected variants in Mexico show mutations in the N-terminal domain (i.e., R190M), in the receptor-binding motif (i.e., T478K, E484K), within the S1-S2 subdomains (i.e., P681R/H, T732A), and at the basis of the protein, V1176F, raising concerns about the lack of phenotypic and clinical data available for the postulated variants of interest (VOIs) 20B/478K.V1 and P.3. Moreover, the population patterns of Single Nucleotide Variants (SNVs) from symptomatic and asymptomatic carriers sampled with a self-sampling scheme, revealed a bimodal distribution of polymorphisms in all three sampled localities from central Mexico, and confirmed the presence of several fixed variants, mostly from the 241T-3037T-14408T-23403G haplotype common in Asia. Despite gene flow among Mexican localities, we found differences in both the allelic frequencies among localities and the allelic imbalance of the mutation S194L of the nucleocapsid protein between symptomatic and asymptomatic carriers. Our results highlight the dual role of Spike and Nucleocapsid proteins in adaptive evolution of SARS-CoV-2 to their hosts and provide a baseline for specific follow-up of mutations of concern during the vaccination stage in Mexico.
IMPACT STATEMENT Following self-sampling, screening of mutations of concern, and a combined phylogenomic and population genetics pipeline, we reveal the occurrence of two variants of interest with mutations in the Spike protein, P.3 (B.1.1.28.3) and 20B/478K.V1 (B.1.1.222, recently named B.1.1.519) in Mexico during the pre-vaccination stage, plus a mutation in the Nucleocapsid protein, S194L, that associates only with symptomatic patients versus asymptomatic carriers in the population investigated. Our research can aid epidemiological genomics efforts during the vaccination stage in Mexico by contributing with a combined analytical platform and information about variants within different genetic backgrounds.
DATA SUMMARY The Mexstrain-Nextstrain data can be found at: http://www.ira.cinvestav.mx/ncov.evol.mex.aspx.
Data generated and used in this study is provided as supplementary Tables:
Table S1. Self-sampling performance validation data generated in 58 volunteers.
Table S2. Metadata of SARS-CoV-2 genomes generated and investigated.
Table S3. Complete dataset of the population genomics analysis of SARS-CoV-2.
INTRODUCTION
Mutation D614G of SARS-CoV-2 Spike protein was the first mutation implicated in increased transmission and a more efficient viral replication in human cells (Korber et al. 2020). It emerged as a consequence of natural selection in the S1 region of the Spike protein as the virus colonised human populations outside of Asia to Europe and to the rest of the world (Zhang et al. 2020). Shortly after the appearance of D614G, other mutations in the Receptor Binding Domain (RBD) of the Spike protein appeared, including but not limited to K417N, L452R, E484K and N501Y. Their increasingly high frequency in recent months, their potential for increased transmission coupled with immunological escape (Greaney et al. 2021; Li et al. 2020, 2021; Zhou et al. 2021; Garcia-Beltran et al. 2021), and the likely increased mortality (at least of the B.1.1.7 variant; Davies et al. 2021) have alerted the medical and scientific community to follow emerging and prevalent variants as Variants of Interest (VOI) and Variants of Concern (VOC) (“WHO | SARS-CoV-2 Variants” 2021; Scientific Advisory Group for Emergencies 2021). VOIs and VOCs are both defined as highly occurring mutations and/or providing an increased fitness to the virus with a concomitant phenotypic change that results in an increased threat to human health (Resende et al. 2021), but VOIs require experimental evidence of the suspected phenotypic dangerous trait to become VOCs.
Irrespective of their threat level to humans, emerging variants can be detected by their phylogenetic position provided by the Nextstrain (Hadfield et al. 2018) and Pangolin platforms, the latter being the primary source of classification and nomenclature of SARS-CoV-2 (Rambaut et al. 2020). Mutations included in VOIs and VOCs can also co-occur with other mutations, both within the variable N-terminal domain (NTD) region and/or the flexible RBD of the Spike protein or throughout the SARS-CoV-2 genome. Such co-occurring mutations, including indels, are expected given the virus’ current evolutionary trajectory. Of the sixteen emerging lineages identified during the summer of 2020, four have evolved into lineages with VOCs bearing signature shared mutations: 20I/501Y.V1 (United Kingdom, B.1.1.7), 20H/501Y.V2 (The Republic of South Africa, B.1.351), 20J/501Y.V3 (Brazil, P.1 or B.1.1.28.1), and 20C/S:452R (California USA, B.1.427/9). Together with the P.1 closely related VOI P.2 or B.1.1.28.2, these VOCs often share mutations E484K and/or N501Y. Mutations defining VOCs, e.g. E484K, are increasingly common worldwide as they are imported from their regions of origin into naive populations; but also as independent evolution converges to the same point mutations, for instance, in the recently reported VOIs 20A/S.484K and 20C/S.484K (B.1.525/526, Lasek-Nesselquist et al. 2021; Annavajhala et al. 2021).
The aforementioned evolutionary emerging scenarios are worrisome for their potential to extend the duration of the pandemic, as these lineages emerge and spread concomitantly to vaccinated populations. The convergence of K417N, E484K and N501Y in different geographical regions with independent evolutionary trajectories suggest that during the “pre-vaccination stage” of the pandemic, similar selection pressures took place to increase SARS-CoV-2’s fitness (McCormick et al. 2021). For instance, 20J/501Y.V3 or P.1 evolved in parallel with the 20B/S.484K or P.2 variant in Brazil, and they share the Spike protein mutations E484K and V1176F, but not N501Y. Likewise, although geographically unrelated, it can be argued that the places of origin of VOCs 20I/501Y.V1, 20H/501Y.V2, 20J/501Y.V3 and 20C/S:452R have in large common populations with worrying recorded epidemiological statistics (e.g. deaths, cases, R0, positivity, etc.). Cases where natural or unintended immunological pressures, such as the use of convalescent plasma (Kemp et al. 2021) or high prevalence of the disease leading to a high immunological burden (Resende et al. 2021), can help us understand what to expect during the vaccination stage of the pandemic, but further population genomics and epidemiological data is needed.
The finer-scale population genetic patterns of the virus as it adapts to local conditions, and its interaction in diverse human genetic backgrounds have just started to be investigated. SARS-CoV-2 has a high-fidelity transcription and replication process (Ma et al. 2015) compared to other single-stranded RNA viruses, resulting in generally lower genetic diversity (Rausch et al. 2020). Yet the large SARS-CoV genomes allow efficient exploration of the sequence space (Forni et al. 2017) so the array of possible variants during SARS-CoV-2’s trajectory as it adapts to humans’ large populations and even crosses over to other species (Oude Munnink et al. 2021), perhaps even in the absence of the Angiotensin-Converting Enzyme-2 or ACE2 receptor (Montagutelli et al. 2021), is yet to be described in full. One of the few previous population-level studies in SARS-CoV-2 showed that haplotype diversity and allele frequencies in infection clusters are likely caused by natural selection (Liu et al. 2020). Additionally, in early 2020 there were two significant lineages identified by population-level analyses (Tang et al. 2020); in late 2020, there were six viral subtypes based on the estimation of increased nucleotide diversity (Morais et al. 2020; Yang et al. 2020); and in 2021, there were four sub-strains in the US alone detected by population-level analyses (Wang et al. 2021).
Here, we provide a baseline analysis for the evolution of SARS-CoV-2 in Mexico between March 2020, when the first case of Covid19 was registered in the country, and February 2021, when large-scale vaccination started. Based on such analysis, we postulate the occurrence of two VOIs: P.3, which evolved from 20B/P.2, and 20B/478K.V1 or B.1.1.222, emerging within Mexico. We use publicly available genomic data for Mexico, plus 85 genomes generated by us. Our data include sequences from asymptomatic carriers identified using a novel self-sampling strategy developed to increase success rate in sampling these individuals without uncertainty. Two specific SARS-CoV-2 epidemiological scenarios (the prevalence of mutations of concern and population-level viral allelic variation) are investigated to better understand the transition from the pre-vaccination stage to the vaccination stage, and to provide an ad hoc bioinformatics pipeline for real-time analysis of SARS-CoV-2 genomes in Mexico. Our report is timely and of interest as the Mexican national vaccination program involves diverse vaccine technologies and coincides with the launch in March 2021 of a national epidemiological genomics program that aims at providing a thousand genomes per month from hospital samples throughout the country.
METHODS
Self-sampling for identification of asymptomatic SARS-CoV-2 carriers and sample preparation
We sampled asymptomatic SARS-CoV-2 carriers from private organizations represented by a Human Resources (HR) in the City of Irapuato, State of Guanajuato. The HR contact person would report on suspected cases of Covid19 with symptoms, which were sent to the clinic for an assessment and diagnosis by health care professionals. At this point, all contacts of the suspected Covid19 patient were identified through contact tracing, including their family members, and provided with a self-sampling kit with instructions for collection of nasopharyngeal swabs (Figure S1). Validation in 58 volunteers was done by self-sampling and assisted sampling by a qualified healthcare professional, followed by quantification of nucleic acids with Qubit™ Flex Fluorometer (Thermo Fisher), and in some cases, by qPCR amplification of the human marker RNAse P as control (Table S1).
Additive manufacturing technology became an essential player as supply shortages affected swab and viral transport media (VTM) production globally. Our group used 3D printing for emergency manufacturing devices, including face shields and swabs. For this, we partnered with Prothesia (https://www.prothesia.com/covid19), a Mexico based company dedicated to creating software algorithms to manufacture custom orthotic devices. FDA-cleared 3D-printed NP swabs designed by USF-Northwell and manufactured by Prothesia were used for self-sampling. Manufacturing was done using methacrylate monomers, urethane dimethacrylate and a photoinitiator as reagents, with stereolithography technology on Formlabs printers, curing and washing stations. The tip of the swab features a biomimetic design based on the “cattail” (Typha) plant, and it has a rounded nose to maximise comfort and lateral alternating nubs to maximize surface area for sample collection.
Samples were transported and saved in 700 µl of VTM (Atila Biosystems Inc) and/or DNA/RNA Shield (Zymo Research). The DNA was saved for future investigations. This provided enough material for two extractions of RNA, each of 300 µl, done with chemagic™ Viral DNA/RNA 300 Kit H96 from PerkinElmer, Quick-RNA Viral Kit (Zymo Research) or QIAmp Viral RNA Mini Kit (Qiagen); and one extraction of DNA of the entire 700 µl, done with ZymoBiomics DNA Kit, saved for future investigations. Around 31.5 ng/µl of RNA and 73.3 ng/µl of DNA were obtained per extraction in a total volume of circa 50 µl. The RNA material was used for SARS-CoV-2 diagnostics using suitable thermocyclers for RT-qPCR, with kits validated by InDRE and provided by Genes2Life, Mexico, to the academic diagnostics laboratories of Guanajuato (Wov19) and Jalisco (Decov triplex). Detection of San Luis Potosí SARS-CoV2 positive samples was done with the GeneFinder COVID-19 PLUS RealAmp Kit.
Identification of mutations of concern
Screening for E484K and N501Y mutations was done by RT-qPCR, using module 2 of the MUT-SARS-CoV-2 kit (Genes2Life SAPI de CV, Mexico) simultaneously in the same reaction using RNA previously identified as positive for SARS-CoV-2. The reactions were carried out using a QuantStudio 5 RT System thermal cycler from Applied Biosystem. This assay uses four fluorescent probes that specifically hybridize with the target sequences to discriminate each base/mutation. Thus, the detection and discrimination are carried out in a fourplex assay, being the channels for the FAM and HEX dyes dedicated to detection of wildtype background; and the Cal Fluor Red 610 and Quasar 670 dyes channels used for detection of the E484K and N501Y mutations, respectively. A total of 1560 RNA samples from the States of San Luis Potosí (170), Jalisco (1330) and Mexico City (60), which were previously identified as positive for SARS-CoV-2, were screened with this assay.
Genome assembly and sequencing of SARS-CoV-2
Two different approaches were used to produce sequencing libraries. A total of 78 cDNA samples from Guanajuato (45) and San Luis Potosi (33) were prepared using the Illumina/IDbyDNA Respiratory Pathogen ID/AMR Enrichment Kit following the vendor’s protocol for library preparation. In a nutshell, libraries were prepared with Illumina RNA Prep with Enrichment (Illumina, Catalog no. 20040536) and IDT for Illumina DNA/RNA UD Indexes (Illumina, Catalog no. 20027213). Illumina RNA Prep with Enrichment consists in a On-Bead Tagmentation followed by a single hybridization step to generate enriched DNA and RNA libraries. After amplification, libraries were enriched as 3-plex reactions using the Illumina Respiratory Pathogen ID/AMR Panel to detect several viral (RNA) and bacterial (DNA) pathogens, including SARS-CoV-2, via probe capture. The DNA bacterial data will be reported elsewhere. Libraries were sequenced using the Illumina NextSeq 500 platform using a configuration for 75bp paired-end. Further 24 sequencing libraries from Jalisco (9) and San Luis Potosí (15) were constructed using the QIAGEN library preparation kit QIAseq_SARSCov2_Primer_Panel, and sequenced in a shotgun fashion using Illumina MiSeq 2×150 v3. SARS-CoV-2 genome sequences were produced by mapping the sequencing reads to the NC_045512.2 reference genome and constructing a consensus sequence using the Explify pipeline at Illumina BaseSpace hub site or with Illumina DRAGEN Bio-IT Platform. Of the 102 genomic libraries generated only 35 (GISAID quality), 82 in total, passed the criteria for their use within our phylogenomics and population genomics analyses, respectively. Details on these genome sequences are provided in supplementary Table S2.
Phylogenomics of SARS-CoV-2 mutations of concern
All complete and high-coverage SARS-CoV-2 genome sequences from Mexico (from samples obtained up to February 2021) were downloaded from GISAID on March 2021 (https://www.gisaid.org/). These were 1554 genomes. We also downloaded from GISAID all genome sequences represented in Nextstrain SARS-CoV-2 global analysis (https://nextstrain.org/ncov/global, date: March 2021). We joined the above set of sequences (and their associated metadata) into a single set for phylogenomic analyses by using in-house Perl scripts (https://github.com/luisdelaye/Mexstrain), giving place to our ad hoc Mexstrain platform (http://www.ira.cinvestav.mx/ncov.evol.mex.aspx) for phylogenomic analyses using a local version of Nextstrain (Hadfield et al. 2018). We used a ‘global’ sampling scheme grouping 10 sequences per ‘country year month’ and modified the defaults/include.txt file to include all Mexican sequences. For the rest of the parameters we used those provided by the ncov Nextstrain build. All configuration files are available upon request. The obtained phylogenomics were used to manually investigate mutations of concern according to Pangolin nomenclature (Rambaut et al. 2020).
Incidences and relationships between SARS-CoV-2 mutations of concern
We focused on the gene coding for the Spike protein. All 1554 Mexican SARS-CoV-2 sequences were compared against the reference genome sequence MN908947 used by Nextstrain (the sequence MN908947 used by Nextstrain version 3.0.3 is identical to NC_045512.2). We identified 337 combinations of mutations and 315 unique mutations from 1552 (two sequences were filtered out because of quality issues) sequences using in-house Perl and Python scripts. To identify homologous positions between Mexican and reference sequences, we used the multiple sequence alignment generated by our local Nextstrain installation. We transformed the output file to study the incidence for 315 mutations, we grouped them in 11 clades, and we studied their covariances between one another, applying in-house scripts with R packages: tidyverse (Wickham et al. 2019), circlize (Gu et al. 2014), and Python modules: NumPy (Harris et al. 2020), Pandas (McKinney et al. 2010), matplotlib (Hunter, 2007), seaborn (Waskom et al. 2017) - R version 4.0.4 (Ripley 2001), R Studio 1.4.1106 (“RStudio”), Python 3.8 (Van Rossum & Drake 2009), and JupyterNotebook 6.1.4 (Kluyver et al. 2016). Scripts will be available upon publication at github.com/plissonf/Phylogenomics_SARS-CoV-2_Mexico.
Population genomics of SARS-CoV-2
Nucleotide variants and small indels allow the assessment of allele frequencies and the intra-host imbalance of ancestral (known Wuhan reference) and derived (alternative) alleles that can inform the appearance of new variants and their evolution. Due to the possibility of having SNPs due to RNA modification rather than RNA replication errors, we did not separate allele frequencies (i.e. A–G and C–T mismatches) that could potentially be due to deamination events (Li et al. 2020), so we refer to nucleotide variation at the population level as Single Nucleotide Variants (SNVs) more generally. We designed a mapping-based approach to call SNVs and indels directly from sequencing reads rather than assembled genomes. Raw reads from a total number of 102 pair-ended libraries were processed by first removing adapters, low quality bases and short reads using the fastP program (Chen et al. 2018) using default parameters. They were then mapped to the NC_045512.2 version of the SARS-CoV-2 reference genome using BWA (Li & Durbin 2009) with default parameters. Sam alignments were then converted to bam files and sorted using samtools (Li et al. 2009). Bam files were then used to call variants (SNVs + indels) via the freebayes program (Garrison & Marth 2012). VCF files were then concatenated and parsed to retain only biallelic SNVs supported by at least 20 sequencing reads and alignment qualities above 30 using in-house R scripts. Allelic frequencies were estimated on the basis of 85 samples that passed our filters. Data visualization and statistical analysis were performed using the tidyverse (Wickham et al. 2019), ggridges (Wilke 2021) and rstatix (Kassambara 2021) R libraries. Additionally, we used the ANGSD (Korneliussen et al. 2014) program to construct a genetic covariance matrix between samples using genotype likelihoods directly from the bam files. The genetic covariance matrix was then used for a Principal Components Analysis using the prcomp function of R.
Positions of mutations of concern in the glycoprotein Spike of SARS-CoV-2
We extracted the Electron Microscopy structure (7A94, Benton et al. 2020) of the trimeric spike (S) glycoprotein of SARS-CoV-2 bound to one Angiotensin-Converting Enzyme-2 (ACE2, yellow). We depicted different domains and subdomains of S in various colors and textures (cartoon, surfaces) using MacPyMOL v.1.7 (Schrodinger LLC, http://www.pymol.org). We rendered 3D representations with ray before proceeding to screen captures. We marked the important mutations S477N, T478K, E484K, D614G, P681H/R, and T732A.
Ethical committee clearance
Ethical committee clearance. All the RNA samples that are reported in this study were obtained during a surveillance program part of a research protocol compliant with El Comite de Bioetica para la Investigacion en Seres Humanos del Centro de Investigacion y Estudios Avanzados del IPN (COBISH) led by Dr. Betzabe Quintanilla Vega, who approved research for all SARS-CoV-2 carriers in the State of Guanajuato (069/2020). The samples from the State of San Luis Potosi were obtained through the clinical diagnostic service provided by the Centro de Investigacion en Ciencias de la Salud y Biomedicina of the Universidad Autonoma de San Luis Potosi as part of a research protocol compliant with the corresponding local authorities, represented by Dra. Liliana Rangel Martinez according to the Ley Estatal de Salud and NOM-012-SSA3-2012, who approved obtaining and using the samples for this paper (SLP/09-2020). For samples from Jalisco the Laboratory for the Diagnosis of Emerging and Reemerging Diseases (LaDEER), the University of Guadalajara, was compliant with local ethical authorities. Obtaining and using research samples from Jalisco were approved by the Comites de Investigacion, Etica en Investigacion y Bioseguridad de la Universidad de Guadalajara led by Dra. Barbara Vizmanos Lamonte CI-00821 and CI-00721). Moreover, all samples were treated and contained by international standards of Interim Laboratory Biosafety Guidelines for Handling and Processing Specimens Associated with Coronavirus Disease 2019 (COVID-19), and all positive diagnostics were informed to patients and the corresponding Health local authorities between 24 and 48 hours of sampling. An informed written consent for the use of surveillance samples was obtained from all patients.
RESULTS
Sampling of asymptomatic SARS-CoV-2 carriers
It was relatively straightforward to have access to samples from Covid19 patients approaching the diagnostics laboratory or the hospital, which was the case for the samples from San Luis Potosi and Jalisco States, but it was not the case for asymptomatic carriers of the SARS-CoV-2 virus from Guanajuato. We solved this problem by tracing back contacts of patients with symptoms and by using a de novo self-sampling scheme to collect samples. We developed a highly efficient 3D polycarbonate swab that ensured capture of larger amounts of nucleic acids than what is obtained with traditional cotton swabs (or other materials), greatly reducing the risk of false negatives. The performance of the swab and self-sampling method was validated with 58 volunteers subject to nasopharyngeal sampling by a medical professional and self-sampling following simple instructions provided by us (see Methods and supplementary Figure S1).
The self-sampling approach yielded a minimum amount of 20 ng/µl per extraction, carried out in duplicates (supplementary Table S2). We obtained sufficient nucleic acids to perform several RT-qPCRs for separate diagnostic purposes or mutant screening, as well as the entire sequencing of their genomes (see Methods). By following this approach, we also reduced the risk among members of the potential clusters investigated and health professionals were under less viral exposure. For the State of Guanajuato, a total of 23 asymptomatic SARS-CoV-2 carriers, plus a total of 37 patients that eventually developed symptoms, could be identified after a screening of 484 individuals. Related metadata and genomic SARS-CoV-2 IDs are provided in Supplementary Table S2.
Targeted RT-qPCR identification of variants of concern in Mexico
We adopted an RT-qPCR approach to identify mutations of concern previously detected in VOCs circulating worldwide. We screened for the Spike protein mutations E484K, N501Y and 69-70 deletion. A total of 1560 SARS-CoV-2 positive samples from patients coming from Mexico City (60) and the States of San Luis Potosi (170) and Jalisco (1330) from the pre-vaccination stage were characterized. This effort allowed us to identify only one sample during January 2021 (frequency of 0.59%, 1/170) from the State of San Luis Potosi (supplementary Figure S2-B). The genome sequence of the associated SARS-CoV-2 virus was obtained by Mexican authorities (Mexico/SLP-InDRE_454/2021, GISAID EPI ISL 1219714), confirming the mutation E484K; but also, independently by us in this study, as the raw reads were required for our population genomics analysis. Likewise, following the same approach, a total of nine independent samples containing the E484K mutant could be detected in samples from the State of Jalisco (supplementary Figure S2-A). The genome sequences of three of these samples were generated by Mexican authorities (Mexico/JAL-InDRE_371, 372, 373/2021,GISAID EPI ISL 1093145,1093146,1093147), and us, including the remaining six samples needed for our population genomics analyses (Mexico/JAL-LaDEER-145365, 145340, 139093, E39931, 133706, 147248/2021,GISAID EPI ISL_1360409, 1360408, 1360412, 1360411, 1360407, 1360410). Sample collection of the Jalisco sequences (frequency of 0.69%, 9/1330) occurred during January 2021 (Supplementary Table S1).
Phylogenomics reveals evolution and spread of mutations of concern in Mexico
To further identify potential mutations of concern and characterize their phylogenetic associations, as well as their co-occurrence and frequency at the clade level, we constructed an ad hoc database for Mexican genome sequences, called Mexstrain (see Methods), which includes selected references to allow for the correct establishment of phylogenetic relationships in the Nextstrain platform. The Mexstrain version used for our analyses consists of 4242 genome sequences (1554 from Mexico), and includes our newly generated high-quality genome sequences from asymptomatic carriers (5) and symptomatic patients (30) from Central Mexico (Guanajuato, San Luis Potosi and Jalisco States). As such, Mexstrain provides the universe of currently available genomic data for the pre-vaccination stage (March 2020 - February 2021) of the Covid19 Pandemia in Mexico, allowing us to identify the presence of additional Spike protein mutations of concern in at least two sequences, at positions 13 (S → I), 18 (L → F), 20 (T → I), 26 (P → S), 144 (144→X), 152 (W → C), 190 (R → M), 417 (K → N), 452 (L → R), 477 (S → N), 614 (D → G), 677 (Q → H), 681 (P → R) and 1176 (V → F), in samples distributed throughout different clades and/or emerging lineages (Table 1).
Mutations of concern with multiple occurrences in Mexico during the pre-vaccination stage (see supplementary Figure S4).
More specifically, the genome data generated by Mexican authorities and other laboratories include sequences with the mutation E484K in the context of known VOCs or VOIs. This include (i) one single sequence associated with 20J/501Y.V3 or P.1 (Mexico/JAL-InDRE_245/2021, GISAID EPI ISL 1008714); (ii) as mentioned in previous section, three sequences from Jalisco annotated as P.2 variants (Mexico/JAL-InDRE_371, 372, 373/2021), first identified by us and sequenced in parallel together with further six sequences also identified by us (Mexico/JAL-LaDEER-133706, 139093, 145340, 145365, 147248, E39931/2021; and (iii) two sequences from the State of San Luis Potosí (Mexico/SLP-InDRE_192/2020 and Mexico/SLP-InDRE_454/2021). The latter sequences belong to Nextstrain clade 20A (pangolin lineages B.1 and B.1.396 respectively), which makes them closer to the recently identified E484K-containing lineage 20A/S.484K (or B.1.525) than to P.2.
The above-mentioned scenarios (ii) and (iii), together with further E484K-containing variants identified by RT-qPCR screening (supplementary Figure S2), places the States of San Luis Potosi and Jalisco as epicenters of E484K-containing SARS-CoV-2 VOIs in Mexico. P.1 and P.2 share mutations E484K, D614G and V1176F (the latter appearing to be specific to these closely related emerging lineages), but not N501Y. As expected from independently evolving VOCs, mutation V1176F is absent from the two sequences from the State of San Luis Potosi. In contrast, all samples from Jalisco State (scenario ii) are situated in the same lineage as P.2 or B.1.1.28.2. The sequence Mexico/JAL-InDRE_373/2021 includes the mutation R190M, an alternative version of mutation R190S present in the VOC P.1 or B.1.1.28.1, which can be early signs of evolution in Mexico. Indeed, all Mexican sequences form a distinctive clade supported by the mutations ORF1a: V1071A, P1810L, S3149F (Figure 1C & Supplementary Figure S3). Based on these observations we propose to re-name the Jalisco P.2 variant as P.3 or B.1.1.28.3, raising awareness of the need to trace these E484K-containing variants.

A) The Mexstrain phylogenomic tree showing VOI/VOC identified during the pre-vaccination stage. B) Geographic distribution of VOI/VOC in Mexico. C) VOI 20B/P.3 or 20B/B.1.1.28.3 evolving in Jalisco, Mexico, from P.2. ORF1a and Spike amino acid mutations (ORF1a: V1071A, P1810L, S3149F; S: R190M) are indicated with green stars.
Our phylogenomics analyses also revealed a different 484 mutation involving another amino acid substitution, namely, E484Q in the sequence Mexico/OAX-InDRE-61/2020 (August 2020, GISAID EPI ISL 576264). Interestingly, this substitution involves a basic residue, glutamine (Q), and therefore it is tempting to speculate that a similar effect to that generated by the basic residue lysine (K) may be in place. Irrespective of the codon usage of SARS-CoV-2 (Sheikh et al. 2020), the mutations from E to K (G → A) and from E to Q (G → C) involve single and non-sequential nucleotide point mutations, and thus they may offer alternative solutions to the same phenotype. A similar scenario with multiple combinations at the codon level, within different lineages in Mexico, can be seen in the mutation of concern S477N/I (Singh et al. 2021). These mutations happen in seven sequences from different States distributed throughout different lineages (B.1.404 and B.1, for instance), and they involve mutations G → A and G → U in the second letter, respectively, which can be generated from two different sets of codons. As both mutations, N and I, are known to be of concern, this is important. The case of mutation 681 from P to R/H, present in the Mexican VOI 20B/478K.V1 discussed next, also provides an important example of the SARS-CoV-2 genomic plasticity. These cases, and others included in Table 1 and supplementary Figure S4, raise the importance of the characterization of allelic variation de novo as we do in the population genomics section.
Mutagenesis of concern does not occur solely within the RBD of the Spike glycoprotein. For instance, mutation L18F has been identified in the N-terminal domain (NTD), which is also part of the globular head of the Spike protein and is rich in glycosylation sites (Watanabe et al. 2020). This mutation happens in variants 20H/501Y.V2 or B.1.351 and 20J/501Y.V3 or P.1, and thus it has been implicated in antibody scape. Within the period of analysis, this mutation occurs in Mexico in two forms: (i) in the single case associated with 20J/501Y.V3 or P.1 previously mentioned; and (ii) in two different lineages within several clades, accounting for at least six different backgrounds in a total of eight samples. A similar scenario for the NTD deletion 144X can be found in four samples from the North East of Mexico, Nuevo Leon and Tamaulipas States, associated with 20I/501Y.V1 or B.1.1.7, and more importantly, in seven samples identified in two different lineages, 20A/B.1.189 and 20B/B.1.1.222. These examples of convergent evolution of mutations of concern, together with many others, are also included in Table 1 and supplementary Figure S4.
In agreement with public announcements of Mexican Health authorities, a genomic lineage belonging to 20B/B.1.1.222 and characterized by mutations T478K, P681H/R and T732A, was confirmed. More than one third of available sequences with a nationwide distribution could be identified (Figure 1 & 2 and supplementary Figure S4). Although occurrence in Mexico City seems higher, the earliest sequence with these three mutations (lineage B.1.1.29) is from the South East State of Oaxaca, recorded as early as July 2020 (Mexico/OAX-InDRE_258/2020, GISAID EPI ISL 1054990). Moreover, as enough genomic data is available, including some of the genome sequences released here, we investigate overall mutation’s frequency and co-occurrence systematically. For this, we first identified 315 mutations of concern in the Spike protein that occur in at least one genome sequence during the period of analysis. We then analyzed their incidences and their covariances across the 1552 genome sequences in which these mutations occur. The results of this analysis are presented in Figures 2A & B and supplementary Figure S5. Figure 2A highlights the incidences of the 51 most frequent mutations of concern within the 11 clades used to organize this data.
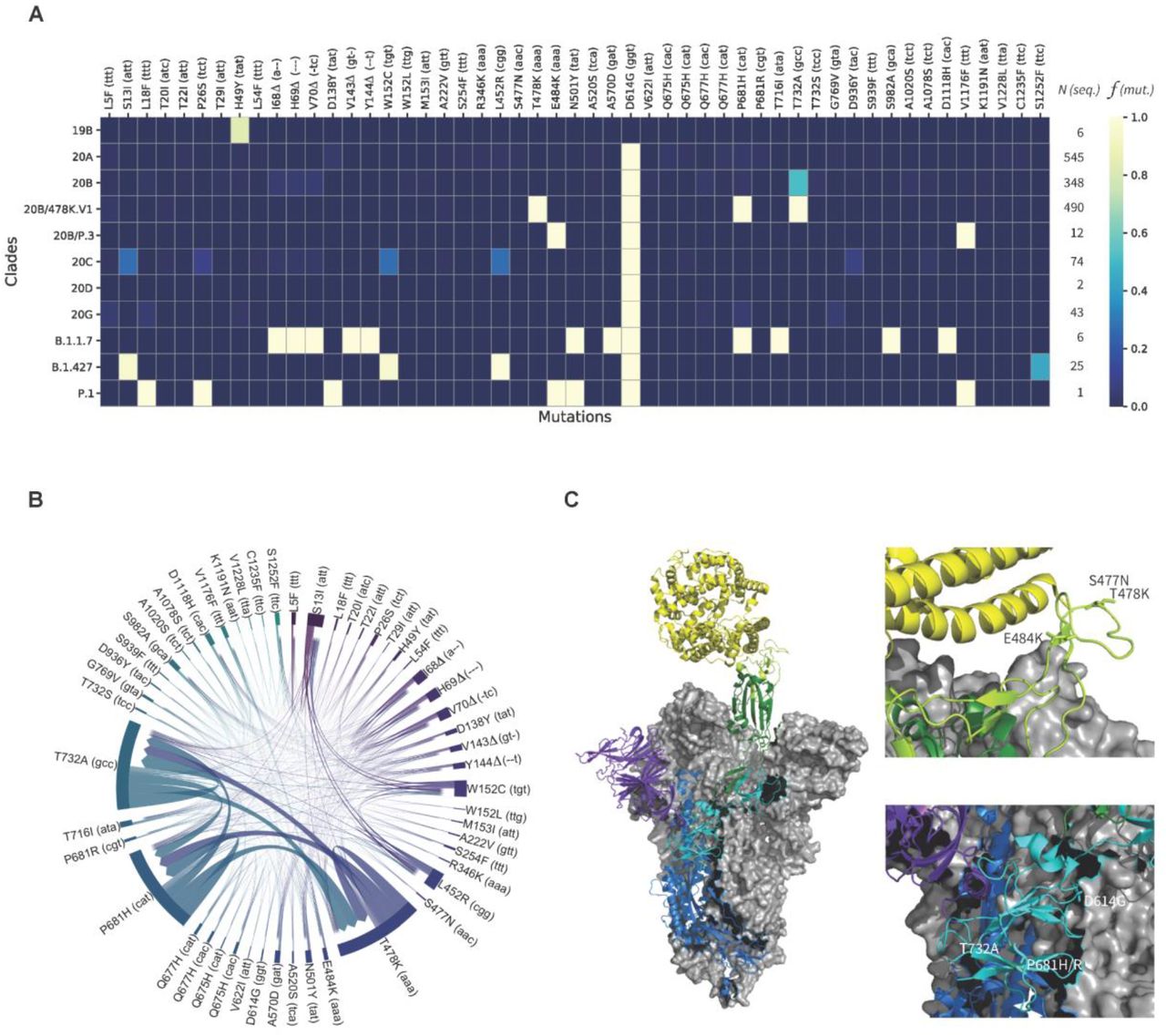
A) The heatmap highlights relative frequencies f for 51 important mutations (out of 315) across 1552 viral genomes arranged in 11 clades (19B, 20A-D, 20B/478K.V1, 20B/P.3, 20G, B.1.1.7, B.1.427, P.1). N indicates the number of available genome sequences per clade. See supplementary Figure S5 for a heatmap including all mutations. B) The chord diagram displays the covariances between the 51 most frequent mutations. The thickness of the arrows denotes the strength of a covariance. C) Left - Electron Microscopy structure (PDB:7A94 ref) of the trimeric Spike (S) glycoprotein of SARS-CoV-2 bound to one Angiotensin-Converting Enzyme-2 (ACE2, yellow). Two chains of the viral protein are shown in gray surfaces. The third chain comprises the N-terminal domain (purple), the S1 (cyan) and S2 (blue) protein subdomains, the Receptor-Binding Domain (RBD, green), and the Receptor-Binding Motif (RBM, light green). Right - Positions of the important mutations S477N, T478K, E484K, D614G, P681H/R, and T732A in two different regions.
We then selected the 51 mutations that were identified in at least five genome sequences overall, indicating the number of available genome sequences (N) per clade and the incidence of each mutation expressed as its relative frequency (f), also per clade. The highest incidences are depicted in yellow (close to 1.0), while low relative frequencies or inexistent mutations shown in shades of blue. For example, roughly 80% of the clade 19B sequences display the Spike protein mutation H49Y, which appears to be unique to that clade. The insertions or deletions of the other mutations of concern illustrate the evolutionary relationships between subsequent clades, starting with clade 20A. Besides the conserved D614G mutation, sequences from the newly detected VOIs, 20B/478K.V1 and 20B/P.3, share mutations with VOCs B.1.1.7 (P681H) and P.1 (E484K, V1176F), and present novel mutations such as T478K and T732A. Figure 2B further validates the co-occurring mutations of concern mentioned above, particularly between T478K, P681H, and T732A, from the dominant clades 20B and 20B/478K.V1. With only 25 sequences available, this analysis also allowed noting for weaker covariances between S13I, L452R, and W152C associated with VOI B.1.427/9.
Among these results, although not at a high frequency, cases of T478K co-occurring with other mutations of concern could be detected, including: (i) K417N, which is present in 20H/501Y.V2 or B.1.351, identified in one sample (February 2021, GISAID EPI ISL 1137473); (ii) L18F, which is present in 20J/501Y.V3 or P.1, identified in one sequence from the State of Oaxaca, in the parental lineage B.1 (January 2021, GISAID EPI ISL 1168605); and (iii) the deletion of amino acid in position 144, which occurs in 20I/501Y.V1 or B.1.1.7 and 20A/S.484K or B.1.525, happened in one sample from Mexico City (February 2020, GISAID EPI ISL 1181713). Similar to convergent evolution of E484K leading to VOIs from the State of San Luis Potosi, mutation T478K is not only present in the entire country, but the sequence Mexico/OAX-InDRE_535/2021 (January 2021, GISAID EPI ISL 1168605) has it, and belongs to 20B/B.1. It is also interesting to note that despite the fact that mutations T478I/R have been identified as dangerous (Li et al. 2020; Muecksch et al. 2021), these mutations could not be found in Mexico during the period of analysis.
3D structural analysis of selected mutations of concern occuring in Mexico
To predict the alleged roles the detected mutations of concern may take in the transmission and/or viral replication, we localized their positions onto the glycoprotein Spike of SARS-CoV-2. For this, we focused on S477N, E484K and D614G as previously identified mutations of concern; and in mutations T478K, P681H/R and T732A identified in the VOI 478K.V1. We used the electron microscopy trimeric structure (PDB:7A94) published by Benton and co-workers (Benton et al. 2020). These authors reported several structures free or bound to one or several Angiotensin-Converting Enzyme-2 (ACE2), of which we picked the example bound to a single ACE2 for simplicity, as shown in Figure 2C. In that figure, two of the three chains within the glycoprotein S are depicted in gray structures to keep our attention to the single-chain interacting with ACE2 (yellow). The third chain includes the N-terminal domain (purple), the S1 (cyan) and S2 (blue) protein subdomains, the Receptor-Binding Domain (RBD, green), and the Receptor-Binding Motif (RBM, light green). Three mutations of concern S477N, T478K, and E484K, are in direct vicinity with the human receptor ACE2, located within the RBM. The mutations D614G and P681H/R are found in the S1 protein subdomain near T732A, taking part in S2. Overall, these structural projections emphasize the expected risks associated with these mutations, in particular for the VOI 478K.V1 emerging in Mexico.
Population genomics of SARS-CoV-2 in Central Mexico
Our reference-based (NC_045512.2) approach yielded high-quality read alignments for 85 of the 102 sequence libraries with a median value of uniquely mapped reads of 177,688 and 1st and 3rd quartiles of 16,292 and 1,751,201, respectively (supplementary Table S3). They were distributed in 43 libraries from Guanajuato, 31 from San Luis Potosí and 9 from Jalisco. Once polymorphic sites were filtered by sequencing depth, quality of the alignments and population frequency, 82 (97%) of the 85 initial samples were retained. From a total of 500 polymorphic sites across these samples, 59 high-quality SNVs/indels were retained, spanning the ∼30 Kb of the SARS-CoV-2 reference genome. These polymorphic sites include 53 SNV and six indels ranging from two up to seven nucleotides in length. This result shows that the use of raw reads from multiple genomes increased the predictive power of genomic variation, in particular for low-frequency variants that could be overseen in lower sample size experiments (Y. Wang et al. 2021). The landscape of genetic variation in our samples suggests that there is enough within- and among-population genetic variation of the virus to give rise and permanence of variants (nearly fixed in the population), likely by convergent evolution due to selection pressure as the virus adapts to the human host.
We found that the majority of the genomic variants (SNVs + indels) occur in low frequencies, with the exception of a few variants, such as P323L in the RdRp protein; E484k, D614G and V1176F spanning the Spike protein region, and R203K and S194L in the Nucleocapsid region (Figure 3A & 3B). We also found the mutation T478K in 4 of the 31 San Luis Potosi genomes and nowhere else, but it was filtered out of the population genetic analyses due to its low population frequency (< 5%).

A) The genome-wide population frequency of iSNVs is plotted as lollipops. Darker colors reflect the per-site median imbalance of the alternative (derived) allele to the reference (ancestral) allele. B) Heatmap showing the relative allele frequencies of the most common variants in the population in a per-location fashion. C) Density plots showing the distributions of the allele frequencies of the 59 high-quality polymorphic sites for each location. D) The three principal components of genetic covariance among hosts are shown as violin plots for each location, suggesting that hosts from Guanajuato (GTO) have a genetically distinct composition from those of Jalisco (JAL) and San Luis Potosi (SLP). E) The distributions of the per-variant intra-host allelic imbalance are shown as density plots for each location. F) The violin plots depict the distributions of allelic imbalance for symptomatic and asymptomatic hosts within Guanajuato.
Our results are consistent with previous reports that highlight the dual role of Spike and Nucleocapsid proteins in adaptive evolution of SARS-CoV-2 to their hosts (Wang et al. 2021). The mismatch distribution of the per-site population frequencies (Figure 3C) suggests that there is indeed selection on multiple mutations that result in several high frequency variants in the population. To test for overall gene flow patterns that could explain our results (e.g. substructure due to clustered sampling or demographic factors) we used the 82 samples with high-quality polymorphic sites to generate a genetic covariance matrix representing the relatedness among samples, which in turn was used to perform a PCA. The principal components PC1, PC2 and PC3 explained the 52%, 21% and 15% of variance genetic differences, respectively. The PC1 suggests overall gene flow among the three locations with the exception of a small portion of the samples from Guanajuato separated from those from San Luis Potosi and Jalisco (Figure 3D). This suggests that even if multiple lineages of the virus are circulating within these three regions, they are mostly admixed. By assessing the allelic imbalance between the ancestral and the derived alleles, we found that for the majority of the testable polymorphic sites, the derived allele is found in higher proportions (Figure 3E), mostly in Jalisco, which is likely explained by the pre-screening of the E484K in this locality. Surprisingly, in some polymorphic sites that were extracted from non-screened genomes, namely in Guanajuato, the ancestral allele dominates the sequencing reads in the sample.
The Guanajuato samples include asymptomatic patients, which could help explain the smaller cluster that we found (Figure 3D & 3E). To further investigate the role of viral genetic composition in the presence or absence of symptoms in the Guanajuato cluster, we compared the median values of allelic imbalance between symptomatic patients and asymptomatic carriers, and found that overall, symptomatic patients show higher proportions of the derived allele (Wilcoxon test; Effect size = 0.12, T = 17,808, p = 0.00692) (Figure 3F). By comparing the site-wise allelic imbalance between symptomatic and asymptomatic carriers from Guanajuato, we were able to detect the C28854T; S194L SNV in the Nucleocapsid region showing a significative higher (ANOVA; F = 20.5; p = 0.003) proportion of the derived T:L allele in symptomatic patients, with a population frequency of 30% in our dataset (Figure 3A). The mutation S194L was recently reported to be associated with mortality (Joshi et al. 2021), and not surprisingly the 65% of patients bearing S194L in our dataset developed symptoms. This nucleocapsid mutation S194L (C28854T) is an interesting find in this population-level context, never reported explicitly for Mexico to our knowledge.
Motivated by this result, we analyzed the prevalence of the N:S194L mutation in the extended Nexstrain phylogeny, and found two major clades with high prevalence of the associated variant: (i) the clade where most of the Mexican genomes are placed, including around one fifth of all sequences available for the period of analysis; and (ii) a mostly Asian clade related to the emerging lineage of the VOI 20A/N.194L. The phylogenetic placement of the S194L variant when we include all Mexican genomes is surprising, as it seems to be highly prevalent in the Mexican population, and as frequent as the Asian clade (Figure 4). The phylogeny reconstructed is more congruent with convergent evolution than a common origin, and although many Pangolin lineages are annotated within the 20B clade, our discovery of the association of this mutation with the development of symptoms, suggest that these lineages should be better defined for Mexico.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Occurrence of N:S194L mutation in Mexican sequences (1554) included in MexStrain, showing an emerging lineage (red asterisk) plus a handful of sequences elsewhere in the phylogeny. B) Same analysis including all sequences contained in MexStrain (all countries), showing appearance of the 20A/N.194L lineage (blue asterisk). C) Zoom-in of emerging N:S194L Mexican lineage (red asterisk), showing many Pangolin associations within the 20B clade. D) Same analysis as in A. and B. but using NextStrain in extent (web version: https://nextstrain.org/ncov/global), showing lineage 20A/N:S194L (yellow, blue asterisk) reported first in India and the Mexican 20B/N:S194L lineage (blue, red asterisk).
DISCUSSION
Our analyses revealed two Spike protein VOIs emerging in Mexico during the pre-vaccination stage: 20B/478K.V1 or B.1.1.222 (discussed below) and P.3 or B.1.1.28.3, which contains the E484K mutation (Figure 1). On one hand, variants featuring the E484K mutation were detected in a lineage in San Luis Potosi different to the closely related and previously reported bona fide P.1, P.2, and now, P.3. P.3 sequences from Jalisco, identified after targeted RT-qPCR screening (and the mutation confirmed after genome sequencing), still appear at a low frequency (0.68%) but emphasizes the advantage of directed screening to anticipate potential epidemiological hotspots. With current data, however, it can only be concluded that P.3 is the result of an early P.2 entry followed by local evolution, warranting the follow-up of the behavior of E484K-containing variants in this region and throughout Mexico. In agreement with this, data available for Mexico and the rest of the world for the pre-vaccination stage suggests convergent evolution and spread of the virus containing mutation E484K (Lasek-Nesselquist, Lapierre, et al. 2021; (Annavajhala et al. 2021); (Ferrareze et al. 2021), suggesting a major role of this amino acid change in higher transmission and/or viral replication and increased viral fitness. On the other hand, 20B/T478K.V1 can be undoubtedly classified as a VOI, with all the potential elements to become a VOC in Mexico and worldwide. This has been suggested as well by other authors (Di Giacomo et al. 2021; Lasek-Nesselquist, Pata, et al. 2021), which report the occurrence of this variant in Mexico, USA and Europe, with an emphasis in mutations T478K and P681H. Importantly, during the pre-vaccination stage this VOI was ascribed to B.1.1.222, which at time of submission appeared more often as B.1.1.519. Beyond nomenclature, the phenotypic implications of this VOI, including potential clinical outcomes and impact upon diagnostics, represent a pressing issue that remains to be addressed.
Our 3D structural analysis of the Spike protein hints towards two regional hotspots for viral mutagenesis. The first one is in the flexible loop of the RBD, and includes S477N, T478K, and E484K. These mutations may affect the interactions between the Spike protein and the ACE2 receptor (Singh et al. 2021; Muecksch et al. 2021). The other hotspot is within S1-S2 subdomains, which includes the furin-like protease cleavage site (Xia et al. 2020; Starr et al. 2020), and contains D614G, P681H/R, and T732A. Within this structural context, mutations T478K and S477N, involving changes from similar hydroxylated side-chains (Thr and Ser) into positively charged basic amino acids (Lys and Asn) may have analogous functional roles reinforcing the ability of the virus to bind to the human ACE2 receptor (Starr et al. 2020). Interestingly, S477 has been identified as the most flexible amino acid within the RBM and with the largest number of mutations (Singh et al. 2021), which might be stabilized by mutation T478K. A case exemplifying a similar scenario in which physically close mutations contribute towards an analogous structural solution has been suggested for the Spike protein mutation Q613H, recently suggested to take a similar role of the well-studied and broadly distributed D614G in an emerging lineage, A.23.1, in Uganda, Africa (Bugembe et al. 2021).
The prevalence of local haplotypes or increased allelic variation within asymptomatic carriers and/or symptomatic patients can be signs that the virus is evolving through natural selection to favor its own fitness, even at an increased cost to the host. Recent studies in SARS-CoV-2 have indeed highlighted the importance of population-level studies (Yang et al. 2020; Mishra et al. 2020; Wang et al. 2021; Graudenzi et al. 2021). Haplotypes in higher frequencies with co-occurring allelic variants at the population level are likely increasing the fitness of the SARS-CoV-2 virus (Yang et al. 2020). We found variants that include known haplotypes and recognized VOCs, with varying degrees of frequencies. Specifically, our samples contain high frequencies of most of the SNVs for one of the early strain types (VI) that spread out of China that includes C241T, C3037T, C14408T and A23403G mutations, and that has the haplotype of allelic associations 241T-3037T-14408T-23403G, also with high frequency detected worldwide in December 2020 (Korber et al. 2020).
The bimodal distribution of allelic frequencies is typical of evolutionary scenarios that result from diversifying selection, which is more likely given the difference in the derived allele frequency among symptomatic and non-symptomatic patients within Guanajuato. Previous population genetics in the Ebola virus show that its strains have co-occurring mutations that have functional implications related to the recovery process of the patients (Liu et al. 2019) and of essence to viral evolution and its adaptation to its host. They also showed that intra-host SNVs were shared by more than two patients, suggesting gene flow among them in addition to recurrent mutations. Along these lines, we found that the Nucleocapsid S194L mutation could play a role in developing symptoms, although this would have to be confirmed with clinical and epidemiological studies in the Mexican population. The S194L variant was recently reported to be associated with higher mortality in Gujarat, India. Joshi et al (2021) found in March 2021 that this variant had an allele frequency of 47.62 and 7.25% in deceased patients, compared to 35.16 and 3.20% of the same variant in recovered patients, from the Gujarat and global datasets, respectively (although the difference was only significant in the global dataset). Surprisingly, this mutation occurs at a very high frequency (89.9%, N=1270) in lineage B.1.1.289 (Plante et al., 2021), the variant isolated in Denmark and shown to co-infect humans and minks (Oude Munnink et al. 2021). Although the connection of B.1.1.289 to our dataset is unclear, it is possible that since our initial sampling this strain has become even more common in Mexican viral genomes.
A clear advantage of the population-level approach as we do here, is that even though the overall quality of an assembled genome could fail to meet the requirements for commonly used platforms such as Nextstrain, we can still recover a good number of high-quality reads with a minimum depth to identify SNVs from fragmented genomes. This calls for an additional global repository of less complete genomes that can be used for population-level studies. As was previously mentioned by Yang et al (2020), SNVs may become an important consideration in SARS-CoV-2 classification, surveillance and tracking as they travel through the world and accumulate additional mutations by convergent evolution. In the meantime, our finding of the mutation N:S194L contributes to an additional site for RT-qPCR screening of samples outside of the Spike region.
It is reasonable to believe that at the moment of emergence of these variants and mutations a massive or global immune pressure elicited by vaccination or antiviral treatments was not present. Also, it has been reported that reinfections of SARS-CoV-2 can occur (Tomassini et al. 2021; Zhang et al. 2020; Kellam & Barclay 2020; Lu et al. 2020). This type of evolution has been observed in other pathogens like respiratory syncytial virus (RSV). In 2009, during the influenza A/H1N1 pandemic, a 72-nucleotide duplication in the G gene of the RSV-A occurred. During the following three years multiple independent duplication events occurred around the world (Comas-García et al., 2018). In fact, multiple and independent events of convergent evolution of the gene G in RSV-A and RSV-B have been reported (Muñoz-Escalante et al. 2019). The mechanism that drives RSV and SARS-CoV-2 convergent evolution may be similar. That could be explained by the lack of a specific antiviral treatment, the lack of vaccines (SARS-CoV-2 convergent events occurred before the distribution of vaccines), a similar R0 of both pathogens, a global high transmission rate and the capacity to escape from the humoral immune response.
Understanding the role of intra-host selection pressures and their subsequent transmission as vaccination continues is critical. For instance, we do not know if new variants occur mostly as the result of admixture, or the mixtures of different variants within the same person, nor their relationship to various vaccines or mixed vaccination in a diverse human background. As different vaccines are used within and across regions, and varying timing of vaccination programs across countries takes place, genome surveillance will still be of essence to monitor and identify emerging phylogenetic lineages that could become VOCs. Foremost, the recent appearance of various sub-strains that result from prolonged individual infections and subsequent transmission, as well as regional surges and episodic outbreaks (Wang et al. 2021) prompt more studies of phylogenomics coupled with population level analyses that can help provide detailed recommendations to decision-makers. In follow-up studies with broader geographic sampling and additional samples in Mexico and elsewhere, we will be able to estimate intra-host SNVs, their effective viral population size, and at a larger scale, to identify super-dispersers and intermediate steps in human transmission that help reconstruct the virus’ microevolution. For now, epidemiological genomics efforts targeting the identified mutations herein are an important first step to control and assess the impact of the mentioned variants of interest and concern, upon the Mexican population during the vaccination stage.
Data Availability
All data is available in public databases.
http://www.ira.cinvestav.mx/ncov.evol.mex.aspx
CONFLICT OF INTEREST
Authors declare no conflicts of interests other than OGG and MDS from GrupoT and FV from Prothesia, who declare commercial interests.
ACKNOWLEDGEMENTS
We would like to acknowledge colleagues and members of the public who provided samples, as well as all authors who make SARS-CoV-2 sequence data available via the GISAID initiative. We are in debt for technical support, access to diagnostics services and/or critical discussions with Francisco de Alba, Carlos Angulo, Guillermo Corona, Eloisa Díaz-Francés, Fernando Fontove, Rafael Franco, Ámbar Gómez, Bruno Gómez-Gil, Ileana Gutiérrez, Gabriel Hernández, Luis Hernández, Alfredo Herrera-Estrella, Silvia Jerez, Beatriz Jiménez, Alejandro Ledesma, Enrique López, Miguel Nakamura, Arturo Nieto, Mizraim Olivares, Hilda Ramos, Victor Rivero, Nelly Selem, Xavier Soberón and Dulce Valdivia. We also thank Ricardo Grande, Francisco Pulido and Gloria Vazquez from the Unidad de Secuenciación Masiva y Bioinformática of the “Laboratorio Nacional de Apoyo Tecnológico a las Ciencias Genómicas (CONACyT no. 260481) for their support in sequencing services. This work was funded by Conacyt, grant No. 313075 to ACJ, and own resources of GrupoT (OGG). FBG is recipient of a Newton Advanced Fellowship, Royal Society, UK (NAF\R2\180631). FP is supported by a Cátedras CONACYT fellowship. Work in the ACG laboratory at UASLP was funded by the income generated by the Clinical Laboratory Diagnostic Unit.
REFERENCES




