
ABSTRACT
BACKGROUND Sarilumab (anti-interleukin-6 receptor-α monoclonal antibody) may attenuate the inflammatory response in Covid-19.
METHODS We performed an adaptive, phase 2/3, randomized, double-blind, placebo-controlled trial of intravenous sarilumab 200 mg or 400 mg in adults hospitalized with Covid-19. The phase 3 primary analysis population (cohort 1) was patients with critical Covid-19 receiving mechanical ventilation (MV) randomized to sarilumab 400 mg or placebo. The primary end point for phase 3 was the proportion of patients with ≥1-point improvement in clinical status from baseline to day 22.
RESULTS Four-hundred fifty-seven (457) and 1365 patients were randomized and treated in phases 2 and 3, respectively. Among phase 3 critical patients receiving MV (n=289; 34.3% on corticosteroids), the proportion with ≥1-point improvement in clinical status (alive not receiving MV) at day 22 was 43.2% in sarilumab 400 mg and 35.5% in placebo (risk difference [RD] +7.5%; 95% confidence interval [CI], –7.4 to 21.3; P=0.3261), representing a relative risk improvement of 21.7%. Day 29 all-cause mortality was 36.4% in sarilumab 400 mg versus 41.9% in placebo (RD –5.5%; 95% CI, –20.2 to 8.7; relative risk reduction 13.3%). In post hoc analyses pooling phase 2 and 3 critical patients receiving MV, the hazard ratio (HR) for death in sarilumab 400 mg compared with placebo was 0.76 (95% CI, 0.51 to 1.13) overall, improving to 0.49 (95% CI, 0.25 to 0.94) in patients receiving corticosteroids at baseline.
CONCLUSION In hospitalized patients with Covid-19 receiving MV, numerical benefits with sarilumab did not achieve statistical significance, but benefit may be greater in patients receiving corticosteroids. A larger study is required to confirm this observed numerical benefit.
(ClinicalTrials.gov number, NCT04315298)
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel coronavirus first identified in December 2019,1 is the causative agent of coronavirus disease 2019 (Covid-19). While most patients with Covid-19 have mild disease, the leading cause of hospitalization and death is respiratory failure, including acute respiratory distress syndrome.2, 3 A key driver of this deterioration may be a dysregulated inflammatory response4 based on the observation of elevated circulating levels of inflammatory mediators such as C-reactive protein (CRP) and interleukin-6 (IL-6).5, 6 Early in the pandemic, small, uncontrolled studies reported that treating hospitalized patients with Covid-19 with two different IL-6 receptor (IL-6R) blocking antibodies, tocilizumab and sarilumab, resulted in potentially dramatic clinical improvement.7–12 More recently, two platform trials found that tocilizumab or sarilumab may improve clinical outcomes in patients with Covid-19, the majority of whom were also receiving corticosteroids.13, 14
On March 13, 2020, Covid-19 was declared a national emergency in the United States.15 On March 18, 2020, we initiated a clinical trial to evaluate the efficacy and safety of intravenous (IV) sarilumab, an anti–IL-6R monoclonal antibody, for the treatment of hospitalized patients with Covid-19 in the United States (ClinicalTrials.gov number, NCT04315298). Here we report the final results of this trial.
METHODS
TRIAL DESIGN, PATIENTS, AND TREATMENTS
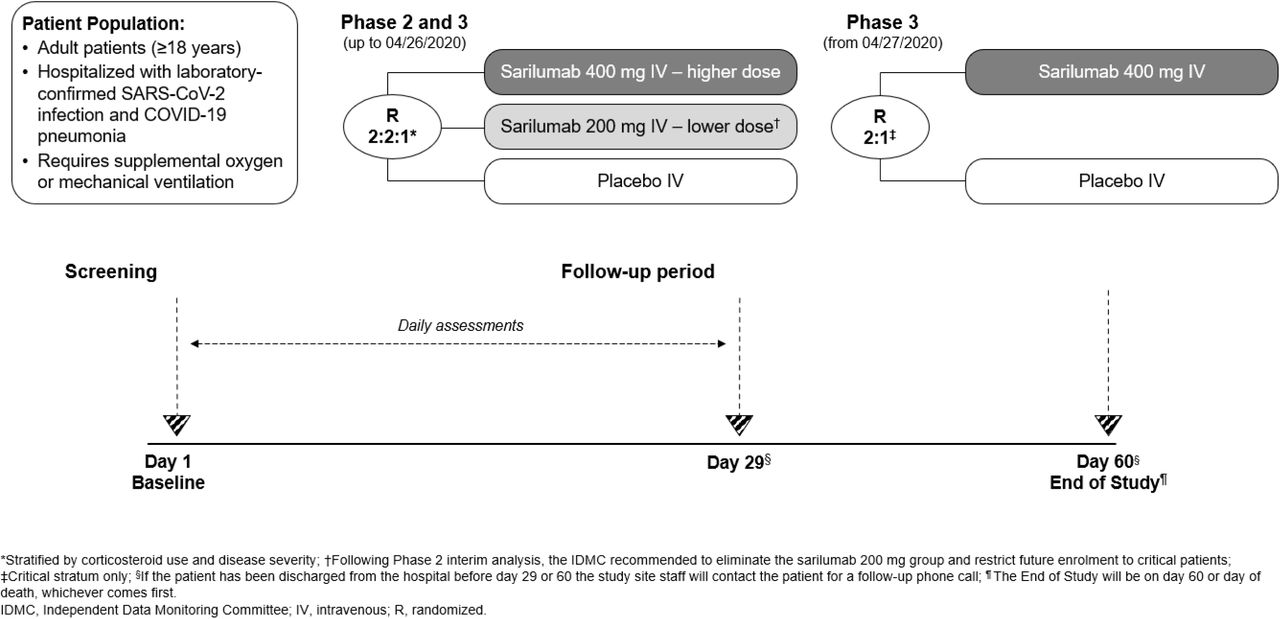
We conducted an adaptive, phase 2/3, randomized, double-blind, placebo-controlled, multicenter trial (65 sites in total, with 62 enrolling patients; see full list of sites in the Supplementary Appendix). Since the trial was being conducted during a pandemic with a novel coronavirus, the protocol allowed adaptations including closing treatment arms or randomization strata, modification of the provisional phase 3 end points, and sample size re-estimation before phase 3 database lock and study readout.
Eligible patients were ≥18 years of age and hospitalized with laboratory-confirmed SARS-CoV-2 infection requiring supplemental oxygen and/or assisted ventilation. All patients received local standard of care (SOC), including corticosteroids and open-label use of putative treatments for Covid-19 (see Supplementary Appendix).
In phase 2, patients were randomized in a 2:2:1 ratio to IV sarilumab 400 mg, sarilumab 200 mg, or placebo (instructions on dose preparation provided in the Supplementary Appendix). Randomization was stratified by corticosteroid use and disease severity (severe, critical, multi-system organ dysfunction [MSOD], and immunocompromised state). Further details provided in the Supplementary Appendix.
TRIAL ADAPTATIONS
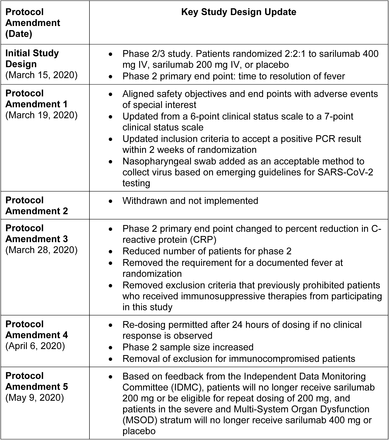
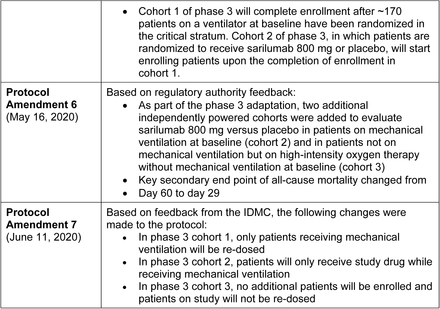
A summary of protocol amendments and study adaptations are found Supplementary Appendix. The phase 2 and phase 3 parts of the study used the same entry and stratification criteria (Fig. S1) until a pre-specified phase 2 interim analysis of 457 patients indicated potential benefit of sarilumab 400 mg in patients in the critical stratum (high-flow supplemental oxygen and/or mechanical ventilation) and potential harm of sarilumab 400 mg in patients in the severe (low-flow supplemental oxygen) and MSOD strata (see Supplementary Appendix for complete definitions of disease severity strata). Subsequently, the Independent Data Monitoring Committee recommended discontinuation of enrollment into the severe and MSOD strata and elimination of the 200-mg dose. Thereafter, the phase 3 protocol was amended to restrict enrollment to critical patients receiving mechanical ventilation with further randomization (2:1) to sarilumab 400 mg and placebo (cohort 1; Fig. S2) and to add a new cohort of critical patients receiving mechanical ventilation randomized to sarilumab 800 mg or placebo (cohort 2) and a new cohort of critical patients not receiving mechanical ventilation, but requiring high-flow oxygen or non-invasive ventilation, randomized to sarilumab 800 mg or placebo (cohort 3). In addition, the following adaptations were implemented prior to database lock: the phase 3 primary analysis population was changed to patients randomized to the critical stratum receiving MV (without extracorporeal membrane oxygenation [ECMO]) who were randomized to the sarilumab 400-mg group or placebo, the primary end point was changed from the time to ≥2-point improvement in clinical status using a 7-point ordinal scale to the proportion with ≥1-point improvement from baseline to day 22, and the sample size was re-calculated.

Results from the phase 3 immunocompromised stratum (n=35 patients randomized and treated) are not shown; results for cohorts 2 and 3 (sarilumab 800 mg) are reported in the Supplementary Appendix.
END POINTS
The pre-specified phase 2 primary end point was the percent change from baseline in CRP level at day 4. Following the interim analysis from the phase 2 part of the study, the pre-specified primary end point for phase 3 cohort 1 was the proportion of critical patients receiving MV at baseline with ≥1-point improvement in clinical status on a 7-point ordinal scale from baseline to day 22 (Clinical Status Scale in the Supplementary Appendix).16 In this population, a 1-point improvement indicates that the patient was alive and no longer receiving MV. Other pre-specified end points included the proportion of patients who died by day 60 and the proportion of patients who recovered (discharged or alive without supplemental oxygen use) by day 22. All outcomes were assessed by the site investigators, who were blinded to treatment assignment and serum IL-6 levels conducted at a central laboratory. Safety end points are described in the Supplementary Appendix.
STUDY OVERSIGHT
Details are provided in the Supplementary Appendix.
STATISTICAL ANALYSIS
The primary analysis population for phase 2 included all randomized patients who received study drug and had high baseline IL-6 levels (above the upper limit of normal) in all disease strata. The primary analysis population for phase 3 included all patients with critical Covid-19 receiving MV without ECMO at baseline who were randomized to the sarilumab 400-mg group or placebo group. Patients in phase 2 were not included in the phase 3 portion of the study.
Hypothesis tests of superiority of sarilumab 400 mg versus placebo were performed with the stratified Cochran–Mantel–Haenszel (CMH) test for two proportions. The stratification factor was the use of corticosteroids at baseline. P values and strata-adjusted confidence intervals (CIs) from the CMH test were reported with overall type 1 error controlled at 0.05 (two-sided). The primary and key secondary end points in the phase 3 study were tested sequentially in a hierarchical manner, while preserving the overall significance level at 0.05 (two-sided). Additional statistical details are provided in the Supplementary Appendix and the statistical analysis plan.
Post hoc analyses were conducted in the pooled data from phase 2 and phase 3 (cohort 1) to descriptively analyze the end points of the proportion of patients with ≥1-point improvement in clinical status to day 22, proportion alive and not receiving MV, and time to death. All post hoc analyses were unstratified.
RESULTS
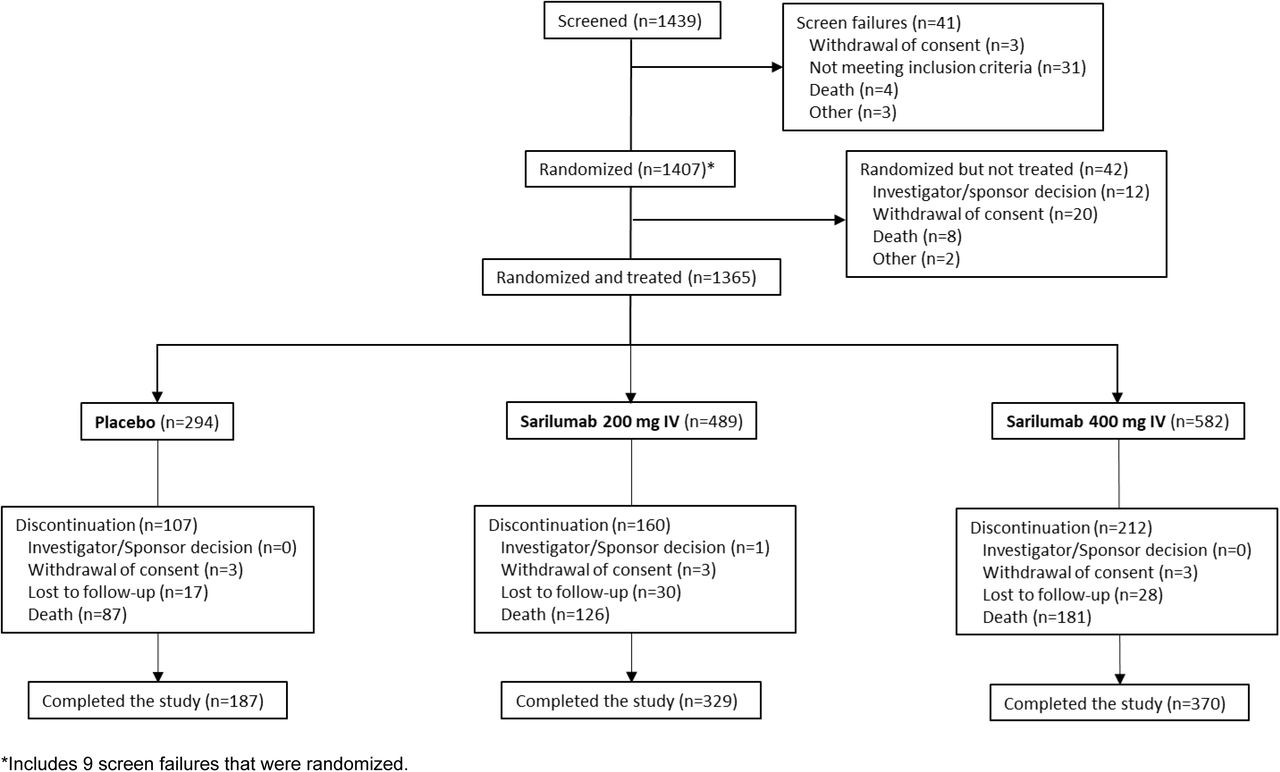
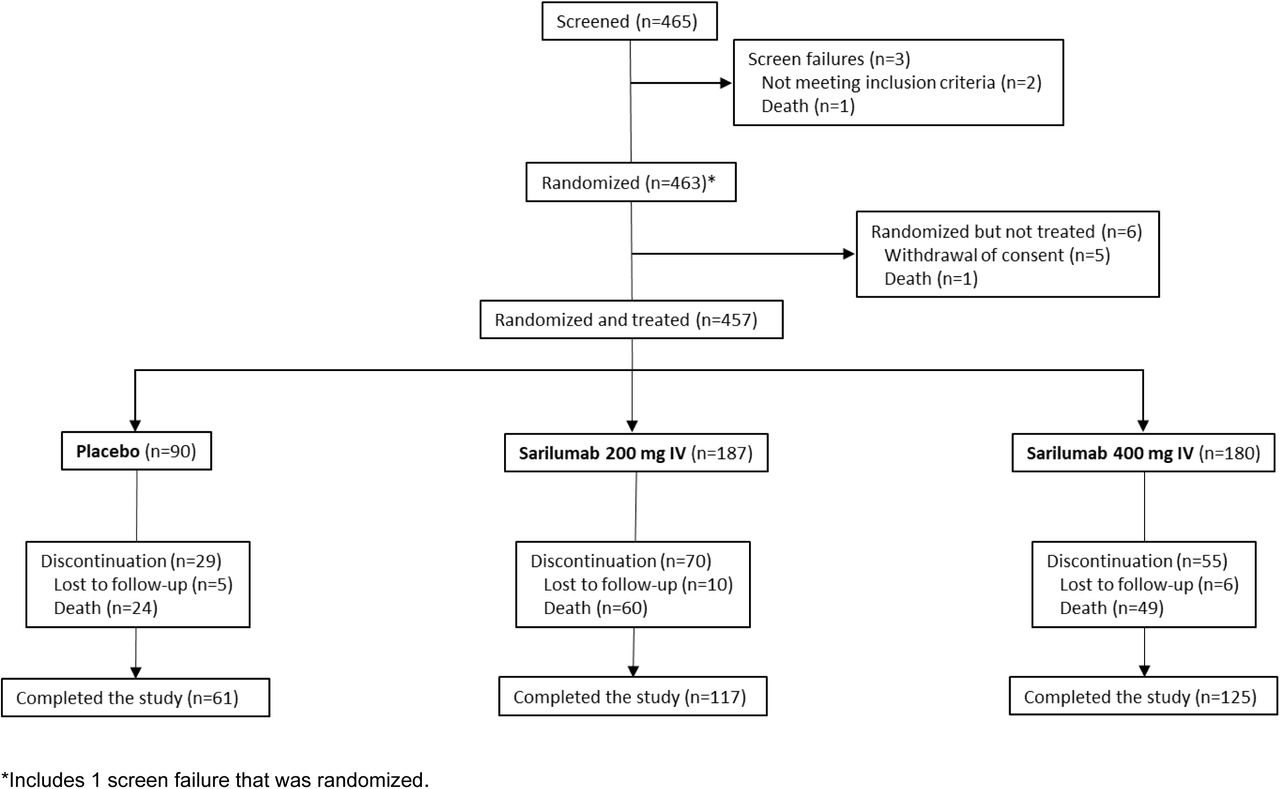
From the period of March 18, 2020, to July 2, 2020, 457 patients were randomized and treated in phase 2 (placebo, n=90; sarilumab 200 mg, n=187; sarilumab 400 mg, n=180), and 1365 patients were randomized and treated in phase 3 cohort 1 (placebo, n=294; sarilumab 200 mg, n=489; sarilumab 400 mg, n=582; Fig. S2). A summary of phase 2 and phase 3 analysis populations by disease severity strata is presented in Table S1. Results of the phase 2 portion of the study are presented in the Supplementary Appendix.
BASELINE DEMOGRAPHICS AND CHARACTERISTICS
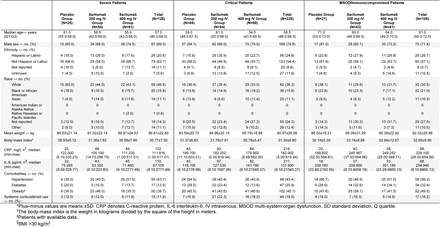
In phase 3, 750 (54.9%) patients were randomized to the critical stratum (298 [21.8%] receiving MV at baseline, 450 [33.0%] were not receiving MV, 2 [0.3%] receiving ECMO); 347 (25.4%) were randomized to the severe stratum, and 233 (17.1%) to the MSOD stratum. The median age of patients in the critical stratum was 61 years, 68% were male, and the median duration of illness was 9.0 days. The median serum CRP, serum IL-6 concentration, and viral load in nasopharyngeal swabs were 168.80 mg/l, 110.91 pg/ml, and 3.95 log10 copies/ml, respectively. At randomization, 34.3% of critical patients were receiving systemic corticosteroids, and this was balanced across treatment groups (Table 1).
Demographics and baseline characteristics for phase 3 severe and MSOD strata and for the pooled phase 2/3 dataset are presented in Tables S2 and S3, respectively.
PHASE 3 EFFICACY OUTCOMES
Primary Outcome
In the pre-specified primary analysis among critical patients receiving MV, 43.2% of patients in the 400-mg group and 35.5% patients in the placebo group had ≥1-point improvement in clinical status (alive not receiving MV) at day 22 (risk difference [RD] +7.5%; 95% CI, –7.4 to 21.3; P=0.3261; Table 2; Fig. 1A), representing a relative risk improvement of 21.7%.

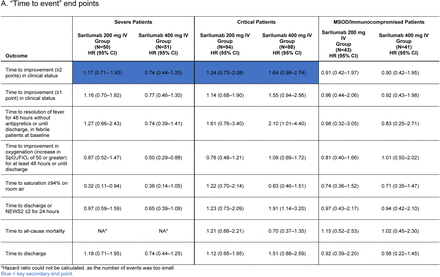
A) Risk difference for various population strata achieving ≥1 point improvement in clinical status from baseline to Day 22 using the 7-point ordinal scale; hazard ratios for the risk of death in various population strata; C) hazard ratios for the risk of death in various population strata by steroid use.
*Key secondary endpoint – phase 3 (cohort 1)
†Primary endpoint – phase 3 (cohort 1)
‡Hazard ratio could not be calculated, as the number of events was too small.
§Unstratified analysis.
CI, confidence interval; EITT, exploratory intention-to-treat; ITT, intention-to-treat; IV, intravenous; MSOD, multi-systemic organ dysfunction.
Secondary Outcomes
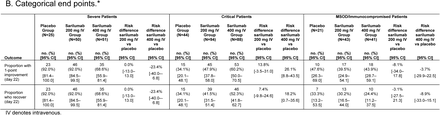
Among critical patients receiving MV at baseline, mortality by day 29 was 36.4% in the 400-mg group and 41.9% in the placebo group (RD –5.5%; 95% CI, –20.2 to 8.7; relative risk reduction 13.3%; key secondary end point) and mortality by day 60 was 39.4% in the sarilumab 400-mg group and 51.6% in the placebo group (RD –11.9%; 95% CI, –26.4 to 2.9; relative risk reduction 23.7%; Table S4). Time to death in critical patients receiving MV at baseline is shown in Figure 2. Recovery by day 22 occurred in 31.8% of critical patients receiving MV at baseline in the 400-mg group and 25.8% in the placebo group (RD +5.7%; 95% CI, –8.4 to 18.2; relative risk reduction 23.3%; key secondary end point; Table S4).

Kaplan Meier curve of time to death in the phase 3 (cohort 1) for A) severe patients (EITT Population); B) critical patients on mechanical ventilation at baseline (ITT Population); C) critical patients not on mechanical ventilation at baseline (ITT Population).
Among critical patients not receiving MV at baseline, 57.4% of patients in the 400-mg group and 64.8% of patients in the placebo group had ≥1-point improvement in clinical status at day 22 (RD –7.5%; 95% CI, –18.3 to 3.9; Table S4). Mortality by day 29 was 26.5% in the 400-mg group and 15.7% in the placebo group (RD +10.7%; 95% CI, 0.9 to 19.3; Table S4), and mortality by day 60 was 29.9% in the 400-mg group and 25% in the placebo group (RD +4.8%; 95% CI, 5.8 to 14.6; Table S4). Time to death in critical patients not receiving MV at baseline is shown in Figure 2. Recovery by day 22 occurred in 54.4% of patients in the 400-mg group and 63.9% in the placebo group (RD –9.5%; 95% CI, –20.4 to 2.0; Table S4).
POST HOC ANALYSES
In post hoc analyses of pooled phase 2 and phase 3 critical patients receiving MV, 46.4% of patients receiving 400 mg and 31.3% of patients receiving placebo had ≥1-point improvement in clinical status at day 22 (RD 15.1%; 95% CI, 2.6 to 27.6; Fig. 1A). Similar findings were observed for mortality (Fig. 1B). No benefit in clinical status was seen in critical patients not receiving MV at baseline (RD –2.6%; 95% CI, –12.9 to 7.7; Fig. 1A).
Among critical patients receiving MV and corticosteroids at baseline, 44.0% (22 of 50) of patients died in the 400 mg-group compared with 68.2% (15 of 22) in the placebo group (HR, 0.49; 95% CI, 0.25 to 0.94; Fig. 1C). Among critical patients receiving MV without corticosteroids at baseline, 37.2% (48 of 129) died in the 400 mg-group compared with 39.7% (23 of 58) in the placebo group (HR, 0.92, 95% CI, 0.56 to 1.52; Fig. 1C).
SAFETY
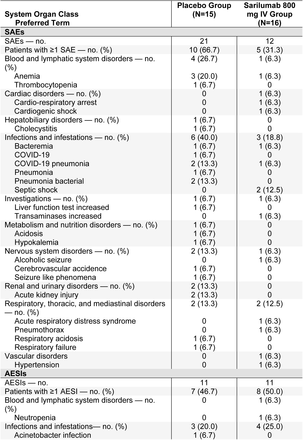
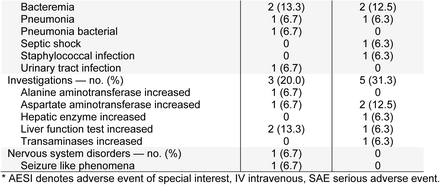
In the phase 3 portion of the study in critical patients, the safety profiles of sarilumab 200 mg and 400 mg were comparable and were similar to placebo (Table S6). Adverse events were consistent with advanced Covid-19 and its complications. Serious adverse events (SAEs) of cardiac arrest, Covid-19, respiratory failure, acute respiratory failure, septic shock, and acute kidney injury were reported in a similar proportion of patients in the sarilumab dose groups in comparison with placebo group, suggesting that these events were related to Covid-19 (Table 3).
Adverse events of special interest (AESIs) reported in more than 5% of the patients in any treatment group were elevations of liver function tests, reported numerically more often in patients receiving sarilumab than placebo (Table 3). None of the liver transaminase elevation cases met Hy’s law criteria.
In the phase 3 portion of the study, patients with severe disease or MSOD exhibited a similar safety profile to critical patients, and no new safety findings were observed (Table S7). Numerically, more patients reported elevation of liver function tests and SAEs in the sarilumab 200-mg and 400-mg dose groups in comparison with placebo (Table S8), which is expected, based on the known safety profile of sarilumab.17
HUMAN GENETIC STUDIES
To independently explore the association between IL-6 signaling and Covid-19 outcomes, we focused on a common partial loss of function allele in the IL6R gene (rs2228145:C, 39% allele frequency in Europeans) that is associated with 7% lower CRP levels,18 reduced surface IL-6R levels,19 50% higher IL-6R shedding,20 and 34% higher serum protein levels of soluble IL-6R (sIL-6R) (see Human Genetic Studies Methods in the Supplementary Appendix). This allele reduces risk of rheumatoid arthritis by 7%21 and coronary artery disease by 5%,22 while it increases the risk of asthma by 9%23 and atopic dermatitis by 15%.24
Using analysis from the COVID-19 Host Genetics Initiative,25 we first assessed the association between the partial loss of function variant (rs2228145:C) and risk of hospitalization, comparing individuals with inpatient hospitalization and polymerase chain reaction (PCR)–confirmed SARS-CoV-2 infection (n=12,888) against population-level controls with either a negative PCR test result or no PCR test (n=1,295,966). We observed a nominal association between the partial loss of function allele (rs2228145:C) and reduced odds of hospitalization (pan-ancestry meta-analysis per-allele odds ratio, 0.96; 95% CI, 0.93 to 0.99; P=0.007; Table S9).
We also considered two severity phenotypes, 1) hospitalization among PCR-confirmed SARS-CoV-2–infected individuals and 2) ventilation or death among SARS-CoV-2–infected individuals as compared with population controls, but these had less statistical power to detect associations. We found numerically lower odds of hospitalization among SARS-CoV-2–infected individuals (N=5773) compared with SARS-CoV-2–infected controls who were not hospitalized (N=15,497; TOR 0.96; 95% CI, 0.90 to 1.01; P=0.13) and lower odds of severe Covid-19 among SARS-CoV-2–infected individuals who required ventilation or died (N=5780) compared with population-level controls (OR 0.97; 95% CI, 0.92 to 1.02; P=0.19). These results were not statistically significant, suggesting larger sample sizes may be required. Altogether, these genetic findings are consistent with approximately 3% to 4% risk reduction for hospitalization and severe disease and provide support for IL-6 signaling blockade in Covid-19 management.
DISCUSSION
In this phase 2/3 clinical trial conducted early during the Covid-19 pandemic, sarilumab did not lead to significant improvement in clinical status or mortality in hospitalized patients with Covid-19, the majority of whom were not receiving corticosteroids as SOC. However, in phase 3 cohort 1, a numerically lower proportion of critical patients receiving mechanical ventilation in the sarilumab 400 mg had died by day 29 compared with placebo (36.4% vs 41.9%, respectively), a smaller effect than that observed with dexamethasone in the RECOVERY trial.26 There was also no benefit of sarilumab seen in patients receiving either low- or high-flow supplemental oxygen or non-invasive ventilation. Because only a minority of patients in our trial (∼30%) were receiving concomitant corticosteroids, these overall outcomes may not be generalizable to the hospitalized population receiving sarilumab in addition to corticosteroids as part of SOC.
In our post hoc analyses where we pooled phase 2 and 3 datasets, critical patients receiving MV at baseline had a mortality rate of 68% with steroids alone (placebo group) compared with 44% with steroids and sarilumab 400 mg. An additive benefit of anti–IL-6R therapy to corticosteroids in patients with Covid-19 has previously been reported in two platform trials. The REMAP-CAP trial, in which >90% of patients were receiving corticosteroids as part of SOC, reported a mortality benefit of tocilizumab and sarilumab compared with SOC alone in hospitalized patients requiring organ support.13 The RECOVERY trial in hospitalized patients with Covid-19 with hypoxemia and CRP >75 mg/l, in which 82% of patients were receiving corticosteroids as SOC, demonstrated that tocilizumab led to a statistically significant reduction in mortality (29% in the tocilizumab group versus 33% in SOC only). Tocilizumab treatment of patients not receiving mechanical ventilation reduced the risk of progressing to mechanical ventilation or death (33% vs. 38% SOC). Similar to the REMAP-CAP trial, the majority (82%) of patients were receiving corticosteroids at baseline, and the mortality benefit of tocilizumab was not seen in patients not receiving corticosteroids.14
Discussion of the results from a separate, large genetic association study of Covid-19 outcomes is presented in the Supplementary Appendix.
Strengths of this trial included the randomized, double-blind, placebo-controlled design and stratification based on disease severity and corticosteroid use. In addition, unlike the RECOVERY trial, we systematically collected all SAEs and AESIs providing important safety information of the use of sarilumab of doses up to 800 mg in the hospitalized Covid-19 population.
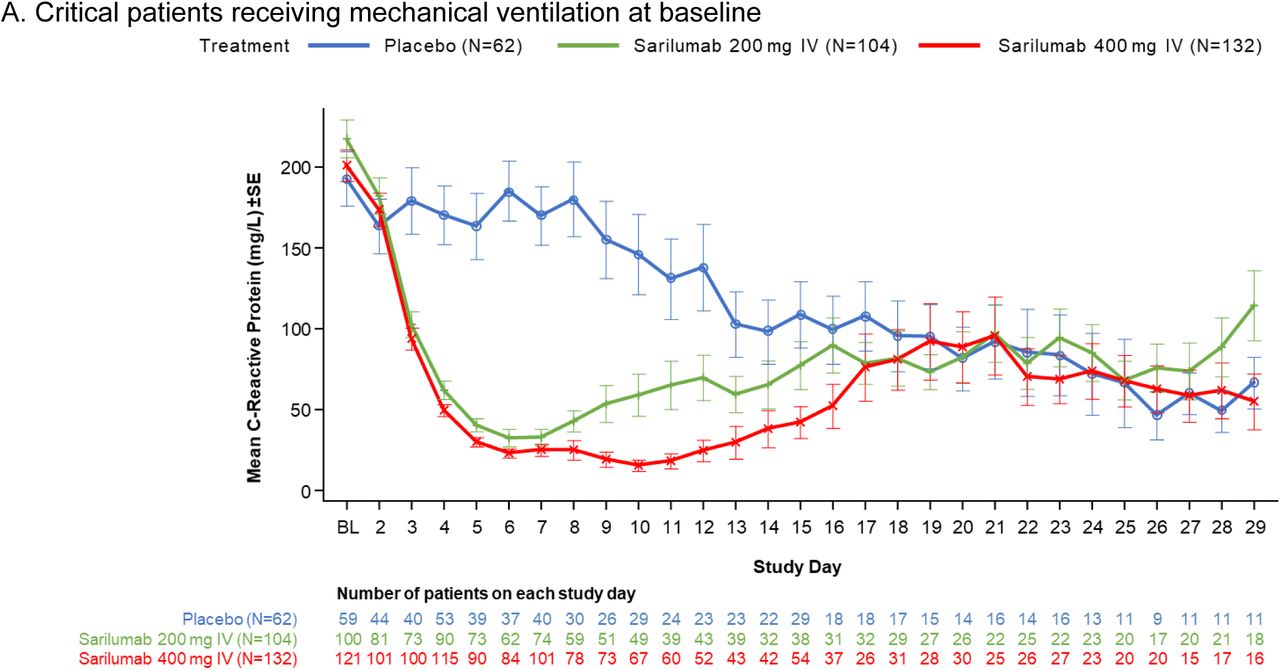
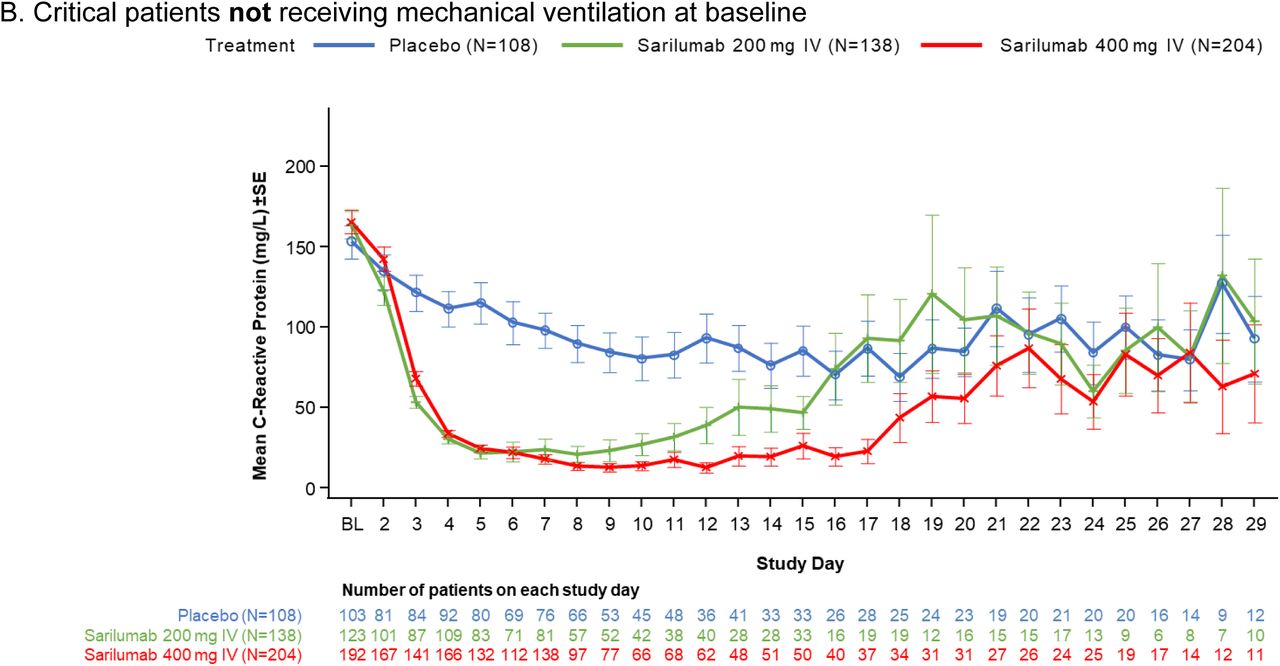
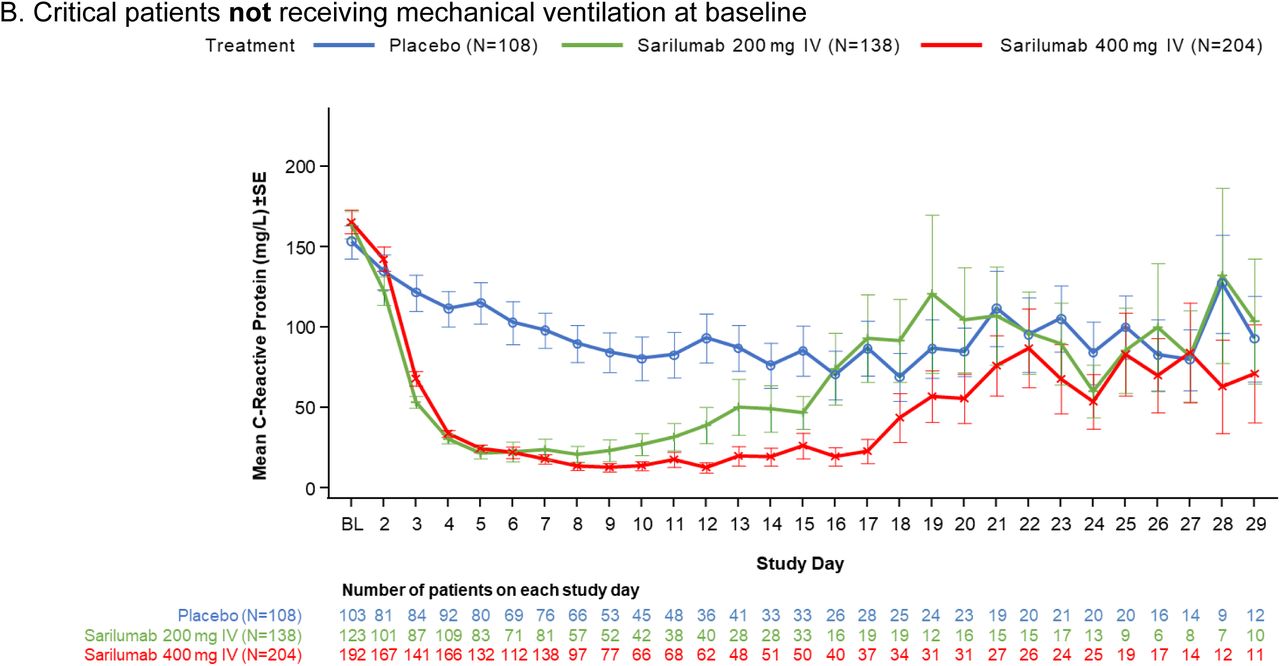
Our trial also has several limitations. The phase 3 sample size estimation based on the phase 2 interim data may have led to an underpowered study, which was further impacted by the 2:1 randomization ratio, leading to a small placebo treatment group. The heterogeneity in the critically ill population as seen in clinical trials in acute respiratory distress syndrome not related to Covid-19 highlights the need for large simple trials such as RECOVERY and SOLIDARITY to adequately assess efficacy of therapeutics in the hospitalized Covid-19 population.27, 28 Another limitation is that the 400-mg dose may be sub-therapeutic, as suggested by a reduction in CRP concentration in the first 7 days and a subsequent rebound (Fig. S3) thereafter, with similar findings reported in a separate trial using sarilumab 400 mg.29 Although an 800-mg treatment arm was included late in the trial, the small sample size makes interpretation challenging.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Based on results from the RECOVERY and REMAP-CAP trials, IL-6R inhibitors are currently recommended in combination with corticosteroids in certain hospitalized patients.30, 31 Within the REMAP-CAP study, although the point estimates for tocilizumab and sarilumab are similar, the smaller sample size in the sarilumab group and the lack of benefit of sarilumab in multiple small trials leave open the question about whether the benefit demonstrated with tocilizumab in RECOVERY is a class effect.32 The recent addition of tocilizumab and sarilumab into treatment guidelines will make conducting large, controlled trials with sarilumab challenging.30, 31, 33 Therefore, to further inform decisions on the use of sarilumab, a meta-analysis of multiple clinical trials with sarilumab to account for heterogeneous patient populations, SOC protocols, and treatment effects will be critical and is currently being conducted.34
Data Availability
Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Submit requests to https://vivli.org/.
FUNDING
Supported by Regeneron Pharmaceuticals, Inc. and Sanofi. Certain aspects of this project have been funded in whole or in part with federal funds from the Department of Health and Human Services, Office of the Assistant Secretary for Preparedness and Response, Biomedical Advanced Research and Development Authority, under OT number: HHSO100201700020C.
DATA SHARING
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
FINANCIAL DISCLOSURE
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Ethics declaration
Ethics approval was obtained from the following ethics review boards: WCG IRB, Puyallup, WA (IRB00000533); Oregon Health & Science University, Portland, OR (MOD00027616); Institutional Review Board University of Florida, Gainesville, FL (IRB202000779); Columbia University, Human Research Protection Office and IRBs, New York, NY (IRB-AAAS9615); Johns Hopkins Medicine, Office of Human Subjects Research, IRB, Baltimore, MD (IRB00246576/CIR00058074); NYU School Of Medicine, New York, NY (I20-00351_MOD03); Northwestern University IRB, Chicago, IL (STU00212239-MOD0012); Westchester Medical Center, NY Medical College, Office of Research Administration, Valhalla, NY (IRBReg#00000428); Portland Health and Services IRB Providence – St. Johns, Portland, OR (MOD2020001024); Cornell Weill Medicine IRB, New York, NY (IRB00009419); Providence St. Joseph IRB, Renton, WA (MOD2020001029); Providence Health and Services IRB, Portland, OR (MOD2020000519); Ascension St. John Hospital, Tulsa, OK (IRB#00001980); Geisinger IRB, Danville, PA (IRB#0008345).
SUPPLEMENTARY APPENDIX
Study Sites and Investigators

Regeneron Sarilumab-COVID-19 Study Team
Moetaz Albizem, Gayatri Anand, Dhanalakshmi Barron, Georgia Bellingham, Alison Brown, Colby Burk, Wilson Caldwell, Michael N. Cantor, S. Balachandra Dass, John Davis, Ajla Dupljak, Haitao Gao, Evelyn Gasparino, Lori Geisler, Ruchin Gorawala, Ingeborg Heirman, Olga Herrera, Matthew Houghton, Susan Irvin, Denise Kennedy, Yasmin Khan, Carol Lee, Kelly Lewis-Amezcua, Qin Li, Leah Lipsich, Adnan Mahmood, Marco Mancini, Jutta Miller, Michael Partridge, Christina Perry, Valancia Reddick, Arsalan Shabbir, Nirav Shah, Kenneth Turner, Dana Wolken, Joseph Wolken, Sonia Yanes, and Jeannie Yo.
Supplementary Methods
Inclusion and Exclusion Criteria
Inclusion Criteria
A patient must meet the following criteria to be eligible for inclusion in the study:
Male or female adult ≥18 years of age at time of enrollment
Hospitalized (or documentation of a plan to admit to the hospital if the patient is in an emergency department) with illness of any duration with evidence of pneumonia by chest radiograph, chest computed tomography or chest auscultation (rales, crackles), requires supplemental oxygen and/or assisted ventilation and meets 1 of the following criteria:
Phase 2 and Phase 3 Cohort 1: must meet at least 1 of the following at baseline (patients meeting more than one criterion will be categorized in the most severely affected category):
Severe disease
− Requires supplemental oxygen administration by nasal cannula, simple face mask, or other similar oxygen delivery device (i.e., above pre–COVID-19 baseline requirement, if any, by the patient)
Critical disease
− Requires supplemental oxygen delivered by non-rebreather mask or high-flow nasal cannula, OR
− Use of invasive or non-invasive ventilation, OR
− Requiring treatment in an intensive care unit
Multi-system organ dysfunction
− Multi-system organ dysfunction: use of vasopressors, extracorporeal life support, or renal replacement therapy
Note: patients receiving vasopressors for reasons other than circulatory shock (e.g., related to sedation or mechanical ventilation) may be randomized into the critical stratum
Immunocompromised
− Immunocompromised patients (or on immunosuppressant treatments)
Laboratory-confirmed SARS-CoV-2 infection as determined by a polymerase chain reaction (PCR) result from any specimen (or other commercial or public health assay) within 2 weeks prior to randomization and no alternative explanation for current clinical condition
Willing and/or able to comply with study-related procedures/assessments
Provide informed consent signed by study patient or legally acceptable representative
Exclusion Criteria
A patient who meets any of the following criteria will be excluded from the study:
In the opinion of the investigator, not expected to survive for more than 48 hours from screening
Presence of any of the following abnormal laboratory values at screening: absolute neutrophil count <2000 mm3, aspartate aminotransferase or alanine aminotransferase >5x upper limit of normal, platelets <50,000 per mm3
Treatment with anti–IL-6, anti–IL-6R antagonists, or with Janus kinase inhibitors (JAKi) in the past 30 days or plans to receive during the study period
Current treatment with the simultaneous combination of leflunomide and methotrexate
Exclusion criteria related to tuberculosis (TB)
Known active TB or a history of incompletely treated TB
Suspected or known extrapulmonary TB
Patients with suspected or known active systemic bacterial or fungal infections Note: Patients with a history of positive bacterial or fungal cultures but on enrollment do not have suspected or known active systemic bacterial or fungal infections may be enrolled
Participation in a double-blind clinical research study evaluating an investigational product or therapy within 3 months and less than 5 half-lives of investigational product prior to the screening visit
Exception: The use of remdesivir, hydroxychloroquine, or other treatments being used for COVID-19 treatments in the context of an open-label study, emergency use authorization, compassionate use protocol, or open-label use is permitted
Any physical examination findings, and/or history of any illness, concomitant medications, or recent live vaccines that, in the opinion of the study investigator, might confound the results of the study or pose an additional risk to the patient by their participation in the study
Known systemic hypersensitivity to sarilumab or the excipients of the drug product
Dose Preparation
Sarilumab (Kevzara®) was provided in a 200 mg pre-filled syringe containing 175 mg/mL of active drug and was administered intravenously (IV) over 1-hour via bags containing 100 mL of 0.9% sodium chloride solution, followed by flush with 0.9% sodium chloride for an additional 15 minutes.
The infusion was stored at room temperature prior to administration and was administered within 4 hours of preparation. Infusion through either central or peripheral lines was allowed. Direct mixing of sarilumab with other drug(s) was prohibited. When it was needed to administer sarilumab concomitantly with other drug(s) (i.e., if a common IV infusion set or central catheter [central line] was used), appropriate steps (e.g., flush before and after administration of sarilumab) was recommended to prevent sarilumab from mixing with other drug(s) while in the infusion set or central catheter.
Definitions of Disease Severity Strata
Severe stratum was defined as patients receiving low-flow supplemental oxygen. Critical stratum was defined as requiring supplemental oxygen by nonrebreather mask or high-flow nasal device, noninvasive ventilation, invasive mechanical ventilation (MV), or management in an intensive care unit (ICU). Multi-system organ dysfunction (MSOD) stratum was defined as use of vasopressors, extracorporeal life support, or renal replacement therapy.
Study Drug Administration and Data Collection
Sarilumab 200 mg, 400 mg, or normal saline (used as placebo for sarilumab) were administered intravenously over 1 hour. An unmasked pharmacist was responsible for preparation of all study interventions. Patients, care providers, outcome assessors, and investigators remained masked to assigned intervention throughout the study. Study drug was administered on day 1 (baseline). Following protocol amendment 4 (April 6, 2020), patients were eligible to be re-dosed for clinical worsening at 24 hours and weekly for up to four doses. Data were collected daily from baseline until day 29 or hospital discharge, and at the end of the study (day 60).
Summary of Key Protocol Amendments and Study Adaptations
Key amendments to the study in the course of adapting the phase 3 design based on phase 2 results.
Clinical Status Scale
The 7-point ordinal scale is an assessment of the clinical status. The scale is as follows:
1, Death
2, Hospitalized, requiring invasive mechanical ventilation or extracorporeal membrane oxygenation
3, Hospitalized, requiring non-invasive ventilation or high-flow oxygen devices
4, Hospitalized, requiring supplemental oxygen
5, Hospitalized, not requiring supplemental oxygen – requiring ongoing medical care (COVID-19–related or otherwise)
6, Hospitalized, not requiring supplemental oxygen – no longer requires ongoing medical care
7, Not hospitalized
Safety End Points
Safety outcomes included treatment emergent serious adverse events (SAEs) and the following adverse events of special interest (AESIs): grade 4 neutropenia (absolute neutrophil count <500/mm3); confirmed invasive bacterial or fungal infections; elevations in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) ≥3X ULN (for patients with normal baseline) or >3X ULN and at least 2-fold increase from baseline values (for patients with abnormal baseline); and grade 2 or higher hypersensitivity or infusion-related reactions.
Study Oversight
The protocol was developed by the sponsor (Regeneron Pharmaceuticals Inc.). Data were collected by the study investigators and analyzed by the sponsor. The local institutional review board or ethics committee at each study center oversaw trial conduct. All patients provided written informed consent before participating in the trial. The trial was overseen by an IDMC.
Ethics declaration
Ethics approval was obtained from the following ethics review boards: WCG IRB, Puyallup, WA (IRB00000533); Oregon Health & Science University, Portland, OR (MOD00027616); Institutional Review Board University of Florida, Gainesville, FL (IRB202000779); Columbia University, Human Research Protection Office and IRBs, New York, NY (IRB-AAAS9615); Johns Hopkins Medicine, Office of Human Subjects Research, IRB, Baltimore, MD (IRB00246576/CIR00058074); NYU School Of Medicine, New York, NY (I20-00351_MOD03); Northwestern University IRB, Chicago, IL (STU00212239-MOD0012); Westchester Medical Center, NY Medical College, Office of Research Administration, Valhalla, NY (IRBReg#00000428); Portland Health and Services IRB Providence – St. Johns, Portland, OR (MOD2020001024); Cornell Weill Medicine IRB, New York, NY (IRB00009419); Providence St. Joseph IRB, Renton, WA (MOD2020001029); Providence Health and Services IRB, Portland, OR (MOD2020000519); Ascension St. John Hospital, Tulsa, OK (IRB#00001980); Geisinger IRB, Danville, PA (IRB#0008345).
Statistical Methods
All analyses and tabulations were performed either using the Statistical Analysis Software (SAS) Version 9.4 or R language.
Adaptive Phase 2/3 Study Design
This randomized controlled clinical trial in hospitalized patients with COVID-19 across various disease severity stages followed a phase 2/3 adaptive design. Randomization was stratified by disease severity: 1) severe, 2) critical, 3) multi-system organ dysfunction (MSOD), and 4) immunocompromised, and by use of systemic corticosteroids. Due to the novel nature of COVID-19, efficacy end points at the time of study design were not well established. The phase 2/3 adaptive design of the study allowed for identification of early signs of clinical efficacy in the phase 2 data (e.g., reduction in C-reactive protein, and estimation of effects on clinical status measured on an ordinal scale). Interim phase 2 data analysis allowed adaptation of the phase 3 study design, including: 1) selection of clinical outcome end points in phase 3 (e.g., well-defined end points based on clinical status assessment of patients), 2) confirmation of the phase 3 population, 3) evaluation of further doses of sarilumab and 4) sample size adjustment for phase 3.
Although enrollment in the phase 2 and phase 3 portions of the study was seamless, the analyses of phase 2 and phase 3 portions of the study were kept separate. Each phase of the study was powered for separate objectives. No patients from phase 2 were included in the analysis of phase 3.
Sample Size Calculations in Phase 3 Portion of the Study
Based on the phase 2 interim data which included ∼460 patients randomized (2:2:1) and stratified by disease severity and use of systemic corticosteroids manner to sarilumab 400 mg IV, sarilumab 200 mg IV, or placebo. Based on IDMC recommendations, the phase 3 design was adapted to allow continued enrollment of patients in the critical disease severity stratum only with the sarilumab 400-mg dose and placebo groups. Enrollment in other disease severity categories and in the sarilumab 200-mg group was halted.
In the phase 2 portion of the study, in a subgroup analysis of the critical stratum based on being on a mechanical ventilator at baseline, in the mechanical ventilator group, approximately 17% of patients treated with placebo (i.e., standard of care) and 57% of patients treated with sarilumab 400 mg (on top of standard of care) achieved ≥1-point improvement by day 22. Therefore, the sample size for phase 3 cohort 1 was recalculated using the chosen phase 2 end point of the proportion of patients with ≥1-point improvement in clinical status on the ordinal scale at day 22 in patients in the critical stratum who were on mechanical ventilation at baseline. With a 2:1 randomization ratio (sarilumab 400 mg IV:placebo), an effect size the same as that observed in phase 2, and 170 patients in the critical stratum on mechanical ventilation, the comparison between sarilumab 400 mg IV (n∼113) and placebo (n∼57) would have >99% power. Assuming the rate on placebo is 17% and the rate on sarilumab 400 mg is 37% (i.e., a difference in proportions is one-half that observed in phase 2), then the sample size of 170 would provide approximately 80% power to detect this reduced difference. These calculations assumed α=0.045, allowing for an interim analysis at the 0.005 level. Therefore, this study planned to enroll approximately 450 critical patients to have approximately 170 patients within the critical stratum who are on mechanical ventilation and randomized to 400 mg IV or placebo.
Note that the total number of patients enrolled in phase 3 is larger than that to account for the patients who had been randomized to the severe stratum, MSOD stratum, or to the 200 mg IV dose group before the IDMC decision to discontinue further enrollment into those strata and dose groups. In addition, phase 3 adaptations also included two new cohorts of patients randomized 1:1 to sarilumab 800 mg IV (higher dose) or placebo, namely, cohort 2 consisting of patients who were on mechanical ventilation at baseline and cohort 3 consisting of patients not on mechanical ventilation but on high intensity oxygen therapy at baseline. These cohorts did not begin enrollment until after cohort 1 enrollment was completed and are not included in this analysis.
Analysis Populations
1) Intention-to-Treat (ITT) Population
The intention-to-treat (ITT) population included all randomized patients who received at least one dose of the study drug. Analysis of the ITT population was done according to the initial treatment assigned to the patient (as randomized).
The ITT population was the primary analysis population for phase 3. For phase 3, the specific primary population was the subset of patients in the critical stratum who were on mechanical ventilation (without extracorporeal membrane oxygenation [ECMO]) at baseline and randomized to sarilumab 400 mg or placebo. Demographics, baseline characteristics, and patient disposition was summarized in the ITT population.
2) Safety Population
The safety population (SAF) included all randomized patients who received at least one dose of the study drug. Analysis of the SAF population was done according to the treatment received (as treated). Determination of “as treated” will be based on the actual study drug received on day 1. The SAF population was used for analysis of all safety data, treatment exposure, medical history, and medication use.
Analysis of Primary Efficacy Variable and Secondary Efficacy Variables
Post adaptations to the phase 3 study design, the main focus of the efficacy analysis was pairwise comparison between sarilumab 400 mg and placebo in cohort 1 critical patients with mechanical ventilation without ECMO at baseline, in terms of the efficacy variable of proportion of patients with ≥1-point improvement in clinical status from baseline to day 22 using the 7-point ordinal scale. The primary analysis population was ITT.
Hypothesis test of superiority of sarilumab (400 mg) versus placebo was planned to be done using the stratified Cochran-Mantel-Haenszel (CMH) test for two proportions. Stratification factor was use of steroids at baseline (yes/no). Estimation of the treatment effect was provided as differences in proportions and confidence intervals calculated using the strata-adjusted confidence intervals from CMH method. P values and confidence intervals were reported with overall Type 1 error controlled at 0.05 (2-sided).
The statistical methods used for the key secondary efficacy variables were the same as the methods described for the primary efficacy variable.
These key secondary end points for the phase 3 cohort 1 portion of the study were to be tested sequentially in a hierarchical manner, while preserving the overall significance level at 0.05 (2-sided).
Proportion of patients with at least 1-point improvement in clinical status assessment from baseline to day 22 in patients with critical COVID-19
Proportion of patients who recover (discharged, or alive without supplemental oxygen use or at pre-COVID oxygen use) by day 22 in patients with critical COVID-19 receiving mechanical ventilation without ECMO at baseline
Proportion of patients who die through day 29 in patients with critical COVID-19 receiving mechanical ventilation without ECMO at baseline
Proportion of patients who die through day 29 in patients with critical COVID-19
Other secondary efficacy variables such as time-to-event end points were summarized with Kaplan-Meier estimates of medians and 95% confidence intervals on medians. Confidence intervals for hazard ratios comparing treatment groups were to be reported using Cox proportional hazards model with stratification factors mentioned earlier. Cumulative incidence rates were plotted with comparisons between groups to be descriptively tested using log-rank test.
Adjustment for Multiple Comparisons
Analysis of phase 2 data was strictly separately done from phase 3. No patients from phase 2 were used in the phase 3 analyses. Multiplicity control in phase 3 for testing primary and key secondary end points using a hierarchical testing order was pre-specified in the Statistical Analysis Plan. Overall type 1 error was controlled within each cohort at 0.05 (2-sided) level.
Human Genetic Studies Methods
Studies
We report association analyses from the COVID-19 Host Genetics Initiative (January 18, 2021, release, downloaded from https://www.covid19hg.org/results/).1
Definition of COVID-19 Outcomes
Risk of hospitalization was analyzed among RNA polymerase chain reaction (PCR)–confirmed SARS-CoV-2 positive cases (N=12,888) and controls who were either PCR-negative or not tested (N=1,295,966 population controls). Risk of severe COVID-19 was analyzed among individuals with a PCR-confirmed SARS-CoV-2 infection and record of mechanical ventilation or death due to COVID-19 (N=5,780) and population controls (N=1,115,203).
Association Analyses
The association between each phenotype and imputed variants was performed as described (https://www.covid19hg.org/results/r5/) with SAIGE v0.82 separately for each study and ancestry, with age, age2, sex, age-by-sex, and 10 ancestry-informative principal components included as covariates. Association results were combined across studies and ancestries with an inverse-variance meta-analysis using METAL.3
Phase 2 Study Results
Phase 2: Flow Diagram
Phase 2: Demographics and Baseline Characteristics (ITT Population)*
Phase 2: Primary Efficacy End Point (mITT population)*†

Phase 2: Summary of Selected Secondary End Points (ITT Population)
Phase 2: Overview of Adverse Events (Safety Population)*
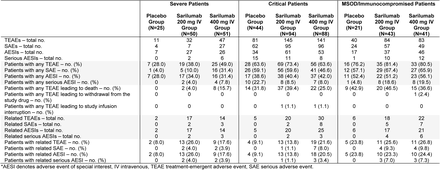
Phase 2: Treatment-Emergent Serious Adverse Events and Adverse Events of Special Interest Occurring in ≥5% of Patients in Any Group (Safety Population)*
Phase 3 (Cohort 2) Study Results
Phase 3 (Cohort 2): Flow Diagram

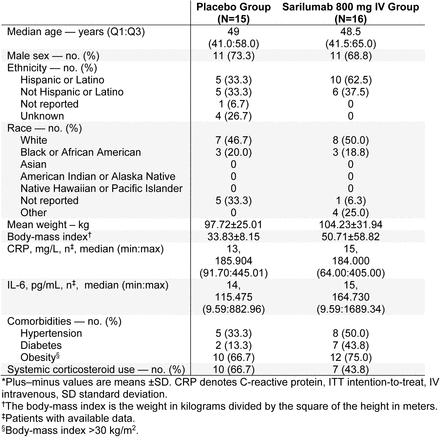
Phase 3 (Cohort 2): Demographics and Baseline Characteristics (ITT Population)*
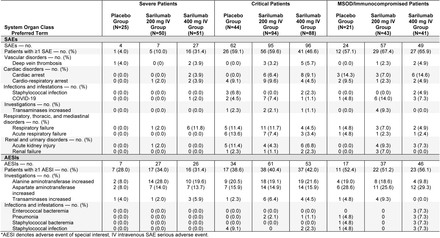
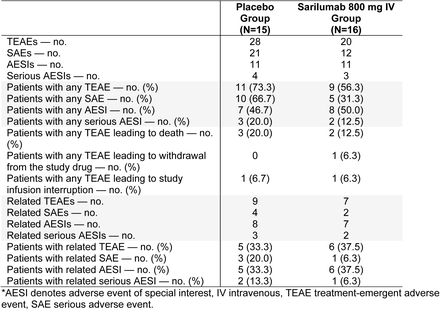
Phase 3 (Cohort 2): Overview of Adverse Events (Safety Population)*
Phase 3 (Cohort 2): Treatment-Emergent Serious Adverse Events and Adverse Events of Special Interest Occurring in ≥5% of Patients in Any Group (Safety Population)*
Phase 3 (Cohort 3) Study Results
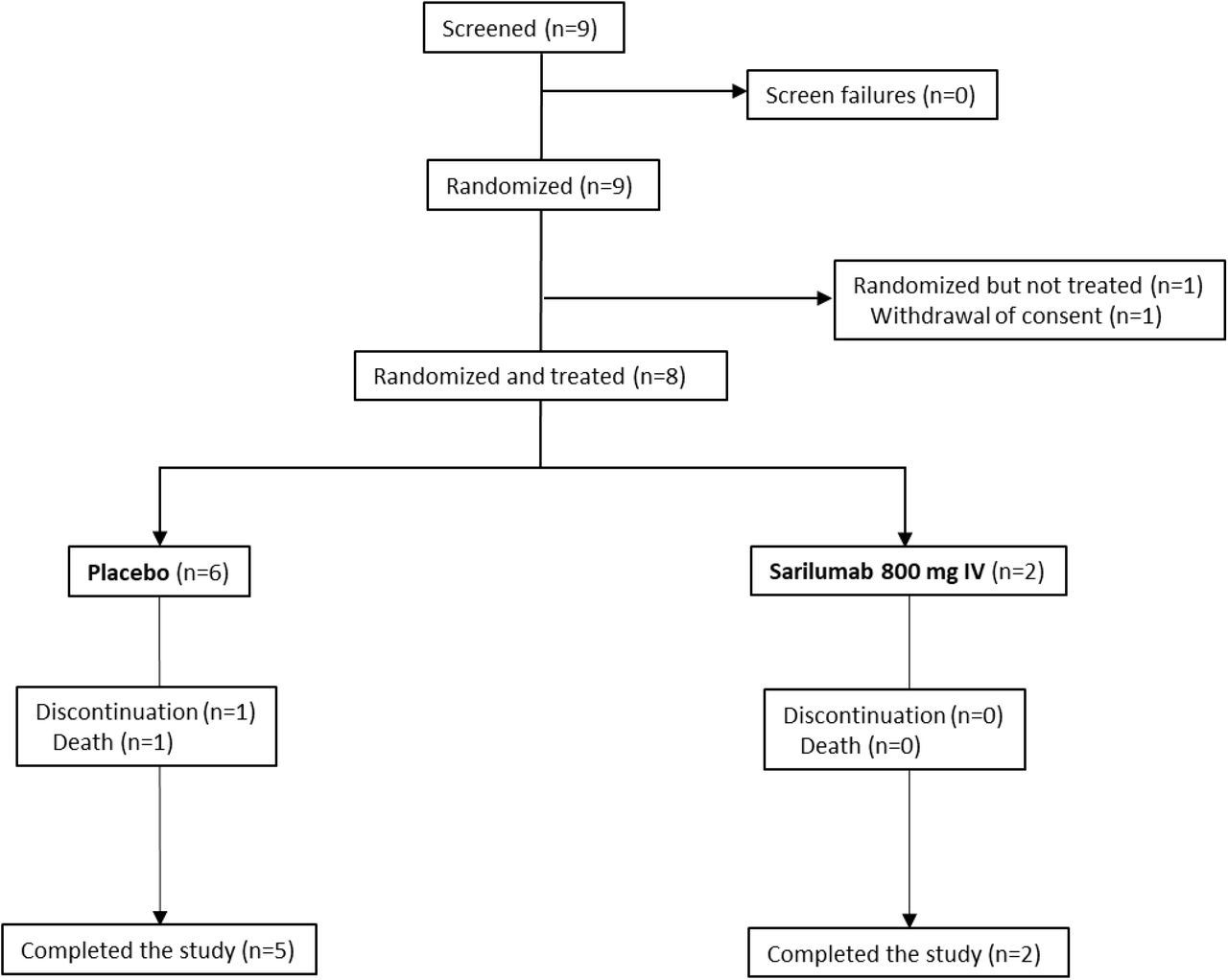
Phase 3 (Cohort 3): Flow Diagram
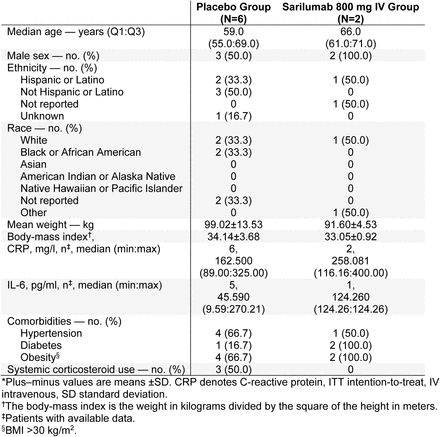
Phase 3 (Cohort 3): Demographics and Baseline Characteristics (ITT Population)*
Phase 3 (Cohort 3): Overview of Adverse Events (Safety Population)*
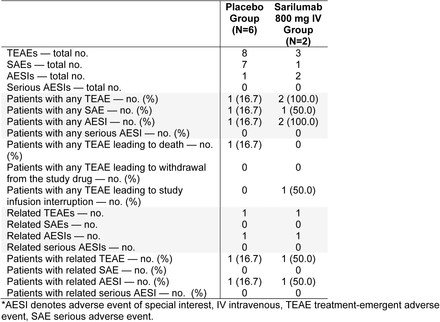
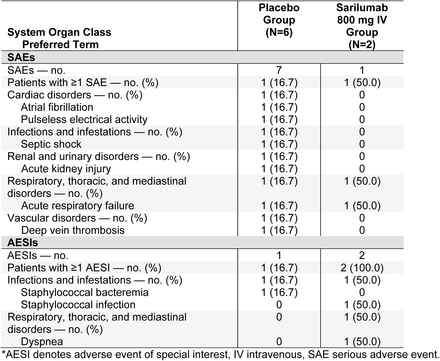
Phase 3 (Cohort 3): Treatment-Emergent Serious Adverse Events and Adverse Events of Special Interest Occurring in ≥5% of Patients in Any Group (Safety Population)*
ACKNOWLEDGMENT
The authors thank the patients, their families, and investigational site members involved in this study (principal investigators and subprincipal investigators listed in the Supplementary Appendix – Study Sites and Investigators); the members of the Independent Data Monitoring Committee (Steve Dahlberg, M.S., Mitchell Levy, M.D., Victor Ortega, M.D., Ph.D., Kevin Winthrop, M.D., Thomas Cook, Ph.D. [non-voting member], Caryn Trbovic, Ph.D., S. Balachandra Dass, Ph.D., and Brian Head, Ph.D., from Regeneron Pharmaceuticals) for assistance with development of the manuscript; and Prime, Knutsford, United Kingdom, for manuscript formatting and copy-editing suggestions.
We dedicate this article to the memory of Colby Burk, M.S., for his contributions and commitment to patients with Covid-19.
Footnotes
↵‡ Former employees of Regeneron Pharmaceuticals, Inc.
REFERENCES




