
ABSTRACT
Background Both SARS-CoV-2 reinfection and persistent infection have been described, but a systematic assessment of mutations is needed. We assessed sequences from published cases of COVID-19 reinfection and persistence, characterizing the hallmarks of reinfecting sequences and the rate of viral evolution in persistent infection.
Methods A systematic review of PubMed was conducted to identify cases of SARS-CoV-2 reinfection and persistent infection with available sequences. Amino acid changes in the reinfecting sequence were compared to both the initial and contemporaneous community variants. Time-measured phylogenetic reconstruction was performed to compare intra-host viral evolution in persistent COVID-19 to community-driven evolution.
Results Fourteen reinfection and five persistent infection cases were identified. Reports of reinfection cases spanned a broad distribution of ages, baseline health status, reinfection severity, and occurred as early as 1.5 months or >8 months after the initial infection. The reinfecting viral sequences had a median of 9 amino acid changes with enrichment of changes in the S, ORF8 and N genes. The number of amino acid changes did not differ by the severity of reinfection and reinfecting variants were similar to the contemporaneous sequences circulating in the community. Patients with persistent COVID-19 demonstrated more rapid accumulation of mutations than seen with community-driven evolution with continued viral changes during convalescent plasma or monoclonal antibody treatment.
Conclusions SARS-CoV-2 reinfection does not require an unusual set of circumstances in the host or virus, while persistent COVID-19 is largely described in immunosuppressed individuals and is associated with accelerated viral evolution as measured by clock rates.
BACKGROUND
After resolution of coronavirus disease 2019 (COVID-19) following SARS-CoV-2 infection, antibodies against SARS-CoV-2 persist in the majority of patients for 6 months or more [1]. Despite this, there have now been a number of reports of COVID-19 reinfection that span a broad range of age groups, time frame between infections and disease severity [2–8]. There remains a great deal of uncertainty over the viral characteristics of reinfection cases, including the degree of sequence heterogeneity and the location of new mutations between the initial and reinfecting variants. In addition, the diagnosis of COVID-19 reinfection has been complicated by the increasing reports of persistent COVID-19 infection, especially in immunosuppressed individuals. Like reinfection cases, persistent COVID-19 can also span the range of disease severity, from asymptomatic to severe disease, and recurrent symptoms can last for months [9–12]. Differentiating between persistent and reinfection can be challenging, and little is known about differences in the location of SARS-CoV-2 mutations in these scenarios. We performed an analysis of SARS-CoV-2 sequences from published cases of COVID-19 reinfection and persistence, characterizing the hallmarks of reinfecting sequences and the rate of viral evolution in persistent infection.
METHODS
Data search and selection criteria
We conducted a systematic literature review in Pubmed through February 5, 2021 for cases of persistent COVID-19 using the search term “((covid or sars-CoV-2) AND (persistent or persistence or prolonged)) AND (sequence or evolution)”. A search for COVID-19 reinfection reports was made using the terms “(covid or sars-CoV-2) AND (reinfection)”. Both peer-reviewed and preprint results were evaluated. We used the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) for reviewing literature and for reporting search results. For cases of reinfection, papers were included if the authors described it as a case of reinfection diagnosed >30 days after the initial infection and if whole genome SARS-CoV-2 sequences or sites of mutations relative to a reference sequence (e.g., Wuhan-Hu-1) from both infection time-points were available. Of the 249 results from the search, 10 articles met the inclusion criteria and were included in the present report along with 2 additional preprints that were identified (Supplemental Figure 1A).
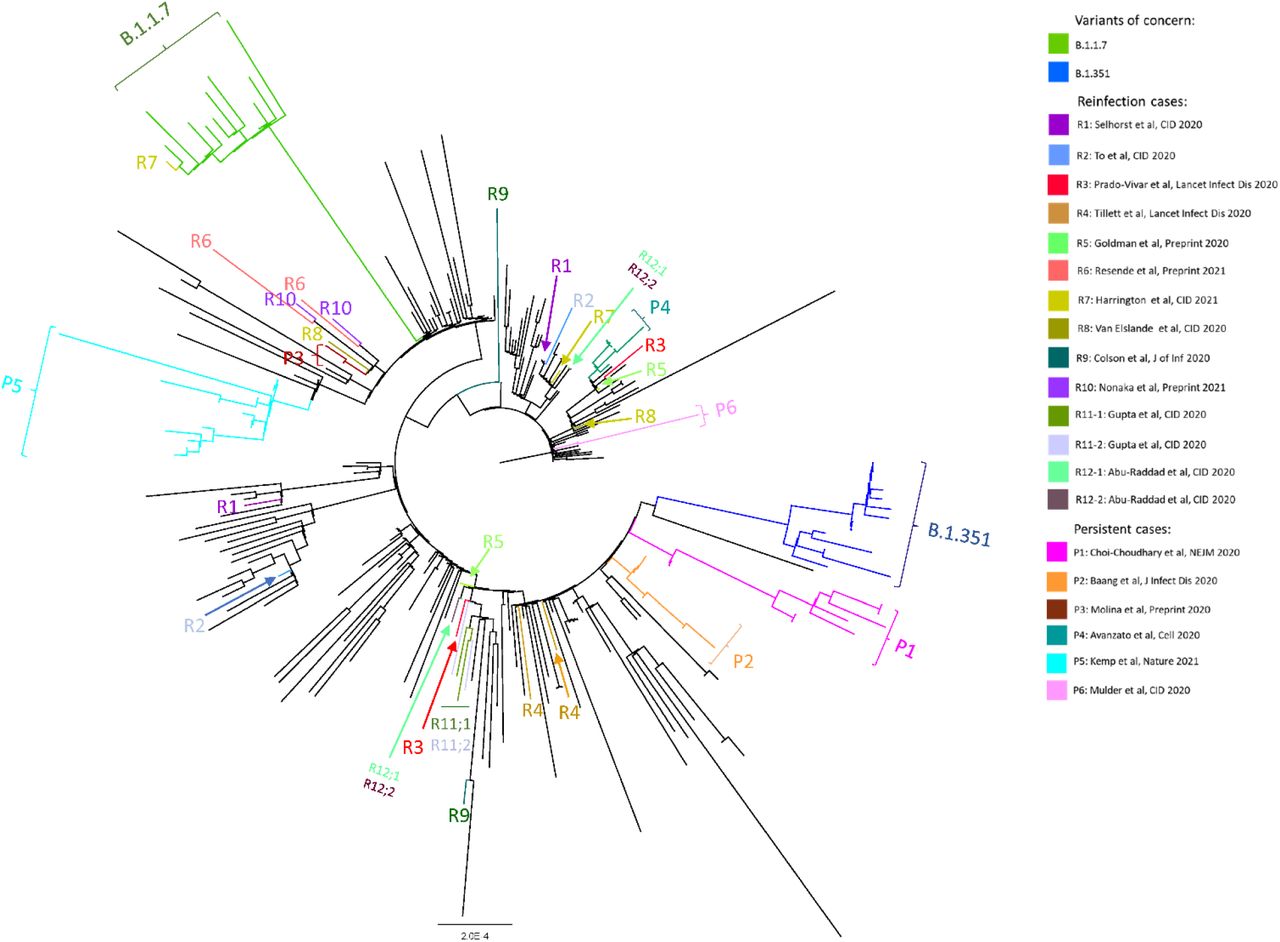
Maximum-likelihood phylogenetic tree of sequences from persistent COVID-19 cases (P1-P6), COVID-19 reinfection cases (R1-R12), the variants of concern B. 1.1.7 and B.1.351, and globally sampled sequences from GISAID.
Persistent cases were included if the authors described it as a case of persistent COVID-19 infection and if longitudinal whole genome SARS-CoV-2 sequences were available. The search returned 116 results, 4 of which met the inclusion criteria and were included in the present report along with one other preprint (Supplemental Figure 1B). Only sequences from direct patient nasopharyngeal or anterior nasal swabs were included in our analysis.
Sequences were downloaded and analyzed for mutations using NextClade (https://clades.nextstrain.org/) and snp-sites (https://github.com/sanger-pathogens/snp-sites). The degree of re-infection severity, either more or less severe compared to the first infection, was classified based on the reported symptoms and reported severity.
Sequencing dataset compilation and phylogenetic tree construction
The sequencing dataset contained a total of 266 globally representative SARS-CoV-2 genomes selected from GISAID and sequences from the reinfection and persistence cases (Supplemental Methods). The sampled sequences were chosen to be representative of global sequence diversity throughout the time course of the pandemic. Sequences of variants of concern B.1.1.7 and B.1.351 were also included. Nucleotide sequence alignment was performed using MAFFT (Multiple Alignment using Fast Fourier Transform) [13]. Best-fit nucleotide substitution was calculated using model selection followed by maximum likelihood (ML) phylogenetic tree construction using IQ-Tree with 1000-bootstrap replicates [13].
Mutation analysis
For reinfection cases, mutations were determined in two ways. First, amino acid changes were identified for the reinfection sequences relative to the first infection sequence. The frequency of amino acid changes within each gene was compared to the frequency of changes in the remainder of the genome by X2 test with Yates correction. The relationship between disease severity and number of amino acid changes in the genome was assessed using a Mann Whitney test. Second, to identify unique characteristics of reinfecting viruses, each of the first and reinfection sequences were compared to circulating sequences in the community as defined by the same NextStrain clade sampled within one month from the same geographic location uploaded to GISAID (https://www.gisaid.org/; Supplemental Table 1, Supplemental Methods). Rare mutations were determined as polymorphisms that were present only in the reinfecting sequence (not the initial variant) and found in less than 1% of contemporaneous community sequences. Mutation locations are graphically represented in Circos plots [14].
For persistent infections, sequence changes were assessed at two time intervals: sequences obtained before or after convalescent plasma or monoclonal antibody treatment. Sequences sampled before convalescent plasma or antibody treatment were compared to the first sequence sampled. For sequences sampled after convalescent plasma or antibody treatment, amino acid changes were determined relative to the last pre-treatment sequence. Linear regression was used to estimate the rate of viral changes within each of the two intervals. To compare the rate of intra-host viral evolution in persistent COVID-19 to the rate of community-driven evolution, we performed time-measured phylogenetic reconstruction as noted below.
Time-measured phylogenetic analysis
The temporal signal of the ML tree was examined in TempEst [15] regressing on root-to-tip divergence, and outliers were inspected in the distribution of residuals. A high degree of clock-like behavior in the whole dataset was observed (R2 = 0.726) in root-to-tip regression analysis with the slope rate as 7.33E-4 and the rough ancestral time of the sample was calculated as 2019.88. This suggests that the most recent common ancestor of the data set composed of only sequences from the persistent cases provides a realistic temporal signal and it is appropriate for an estimation of temporal parameters. No outliers were found in this sample. To compare the evolutionary rates between the reported persistent infections and the general population infections, time-measured phylogenetic reconstruction was conducted in Bayesian Evolutionary Analysis Sampling Trees (BEAST) v1.10.4 [16]. Five partitions, including four persistent patients and the global sequences, were used as separate groups of taxa, to estimate separate evolutionary rates. Due to large uncertainties with small samples, patients with only two viral sequences were excluded from this analysis. A general time reversible (GTR) model was applied with gamma-distributed rate variations among sites. A lognormal relaxed molecular clock was used with an initial mean of 0.0008 and a uniform prior ranging from 0.0 to 1.0. A logistic growth tree prior was applied. Three independent Bayesian Markov Chain Monte Carlo (MCMC) chains of 100 million generations were performed with a sampling step every 10,000 generations to yield 10,000 trees per run. To ensure a sufficient effective sample size ESS > 200, the convergence of three runs was diagnosed in Tracer v 1.7.1 (http://tree.bio.ed.ac.uk/software/tracer/) for all parameters. LogCombiner v1.10.4 as part of the BEAST software package was used to combine the multiple runs to generate log and tree files after appropriate removal of the burn-in from each MCMC chain. The comparison of the evolutionary rates from the combined log file is analyzed and visualized in R v4.0.2 (https://www.r-project.org/).
Statistical analysis
Nonparametric Wilcoxon rank sum or matched pairs signed rank tests were used to compare the number of amino acid changes between sequences. Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, San Diego, CA).
RESULTS
Sequence analysis of reinfection cases
A total of fourteen cases from twelve reports were included in this analysis (Table 1) [2–5, 7, 8, 17–22]. A broad range of age groups were represented and 79% were under the age of 65 years. Most (71%) of the cases had no reported comorbidities and none of the patients were immunocompromised. The interval between diagnosis of the first infection and the second infection ranged from 46 days to 250 days with a median of 110 days. Four patients had more severe illness during the second infection, while five had less severe symptoms on reinfection, including two who were asymptomatic on reinfection. Two cases were asymptomatic in both infections, one case reported the same severity for both infections and no information on infection severity was available for two cases (Table 1). Four cases reported reinfection with a virus from the same clade.
For the reinfection cases, phylogenetic analysis demonstrated distinct branches for the two sequences. We compared amino acid changes in the reinfecting viral sequence compared to the initial sequence and found a median of 9 amino-acid changes (range 6-20) compared to the original sequence (Figure 2A). The amino acid changes were distributed across the SARS-CoV-2 genome, with significantly lower frequencies of changes in ORF1a (P=0.008) and ORF3a (P=0.03), and higher frequencies of changes in S (P=0.02), ORF8 (P<0.001), and N (P=0.003) (Figure 2B). Each reinfection case had at least one substitution or deletion in the S gene (Supplemental Table 2). Next, we assessed whether reinfection with a more divergent second virus resulted in more severe disease. We found no significant differences in the number of amino acid changes in the reinfecting virus compared to the original viral variant when categorized by the severity of the reinfection (Figure 2C). Both the initial and reinfecting SARS-CoV-2 variants were similar to the sequences circulating in the community at the time of reinfection. The reinfecting viruses harbored fewer rare mutations compared to the initial infecting variant, with only a median of 1 rare amino acid compared to circulating variants in the community (Figure 2D-E).
Persistent cases

Comparison of viral sequences from reinfection cases. (A) Circos plot showing location of nucleotide changes in the re-infecting sequence for each of the 14 cases. Inner ring indicates nucleotide position. Synonymous changes are in green, nonsynonymous changes in orange, deletions in black. (B) Amino acid (AA) substitution frequency pooled across all reinfection cases for each SARS-CoV-2 gene. Dashed line indicates global substitution frequency across the whole genome. Substitution frequency for each gene was compared to the substitution frequency in the rest of the genome using a X2 test with Yates correction. * <0.05, ** <0.01 and ***<0.001 (C) Amino acid changes in the second infection relative to the first infection by clinical disease severity. Mutations shown for the whole genome, S gene, and receptor binding domain (RBD) P=0.8, Mann Whitney test. (D) Circos plot showing location of nucleotide mutations from the second infection relative to other viruses circulating at the same time in the same geographic region. Only rare mutations present in <1% of contemporaneous community sequences are shown. (E) Number of rare amino acids at each time point relative to circulating sequences in the community. P=0.01, Wilcoxon matched pairs signed rank test. ORF: open reading frame, S: Spike, E: Envelope, M: Membrane, N: Nucleocapsid.
Reclassification of one case
Mulder, et al. described as a case of reinfection in an 89-year-old female with Waldenström macroglobulinemia treated with B cell-depleting therapy [6]. The patient had two symptomatic episodes separated by 59 days with no RT-PCR testing between the two episodes and recrudescent symptoms shortly after receiving chemotherapy. The virus from the second timepoint clustered with the initial sequence (P6 in Figure 1) and both had the same two nucleotide substitutions and the same deletion in ORF1a relative to contemporaneous sequences. Neither substitution nor deletion was observed in other community sequences sampled at the time of the second episode. Given these features, we have classified this case as a persistent infection for this analysis.
Sequence analysis of persistent COVID-19 cases
A total of six reports describing persistent infection were retrieved from our literature search. Of these six cases, all but one had B cell immunodeficiency [6, 9–11, 23]. Four of these five were treated with B cell-depleting therapy for lymphoma or autoimmune disorders, while one had chronic lymphocytic lymphoma with acquired hypogammaglobulinemia (Table 2). The patient without immunodeficiency was an outlier: he was a young patient without a known immunosuppressing condition and with more than 180 days between symptomatic episodes [24]. Phylogenetic analysis showed the two sequences arising from the same root (P3, Figure 1), but uncertainty about whether this case represented reinfection or persistent infection led us to exclude it from this analysis. For the 5 participants included in this analysis as persistent infection, the median length of infection was 154 days and most cases (4/5) ended in death. One patient had asymptomatic disease throughout [10]. Three patients were treated with convalescent plasma at least once during their illness [10, 11, 23], and one patient was treated with the monoclonal antibodies casirivimab and imdevimab [9].
Phylogenetic analysis revealed that, for each of the five patients, sequences formed a distinct cluster (Figure 1). New mutations emerging over time were detected in all of the persistent COVID-19 patients with further changes identified after treatment with convalescent plasma or monoclonal antibodies (Figure 3). The rate of viral evolution was plotted for each patient both for the interval before and after convalescent plasma/antibody treatment. Overall, the rate of amino acid changes over time appeared faster before treatment (Figure 4a), but treatment with convalescent plasma or antibody cocktail treatment were insufficient to halt intra-host viral evolution (Figure 4b).
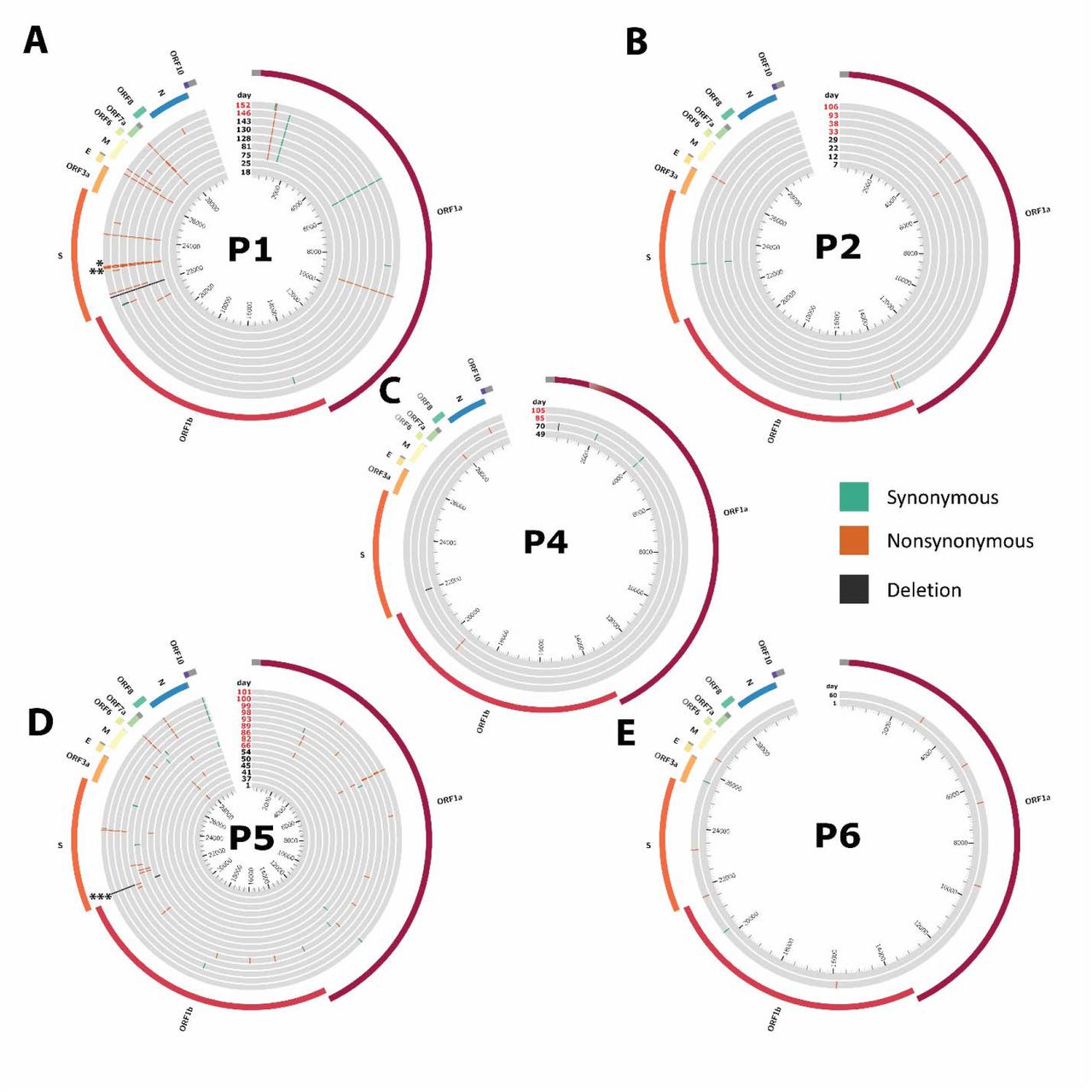
Circos plots mapping mutations for the persistent COVID-19 cases. Mutations are marked relative to the first timepoint. P1 * shows N501Y mutation (days 128-152). ** shows E484K mutation (days 75 and 81 only). P5 *** shows Δ69/Δ70 mutation. Synonymous mutations in green, nonsynonymous mutations in orange, deletions in black. Red text indicates timepoint was sampled after first convalescent plasma or antibody cocktail treatment. Inner ticks indicate nucleotide position. ORF: open reading frame, S: Spike, E: Envelope, M: Membrane, N: Nucleocapsid.

SARS-CoV-2 evolutionary rate in the persistent COVID-19 cases. (a) Amino acid changes in samples taken prior to convalescent plasma or monoclonal antibody treatment relative to first sampled sequence in each persistently infected patient. Regression line and 95% confidence interval of the slopes are shown. (b) Amino acid changes in samples taken after convalescent plasma or monoclonal antibody treatment relative to last sample taken prior to treatment in each persistently infected patient. Regression line and 95% confidence interval of the slope are shown. (c) Substitution rate (substitutions per site per year) of sampled global SARS-CoV-2 sequences relative to four persistent patients based on Markov chain Monte Carlo time-measured phylogenetic reconstruction.
We also performed time-measured phylogenetic reconstruction with the pre-treatment persistent sequences to compare the rate of intra-host viral evolution in persistent COVID-19 to the rate of community-driven evolution. SARS-CoV-2 evolution was faster in these persistent infection individuals compared to the rate in the general public population, though substantial uncertainties are shown in these estimates given the limited sequence sampling for each case (Figure 4C).
DISCUSSION
We conducted a systematic review and pooled analysis of sequences from reports of COVID-19 reinfection and persistent infection. Reports of reinfection cases demonstrate a wide range of situations: spanning a broad distribution of ages (from individuals in their 20s to >70 years), baseline health status, reinfection severity compared to the initial infection, and occurring as early as 1.5 months or >8 months after the initial infection. Common explanations for the presence of reinfection involves either waning SARS-CoV-2 antibodies or the presence of viral escape mutations [25, 26]. While most cases of SARS-CoV-2 reinfection did involve infection with a different clade (including the variant B.1.1.7), it is noteworthy that mutations were identified throughout the genomes and the frequency of mutations within the S gene was only modestly higher than the rate across the entire genome. In addition, individuals with more severe reinfections did not have significantly greater frequency of S gene mutations. Finally, the presence of rare mutations was uncommon in the reinfecting virus, which largely mirrored the contemporaneously circulating variants in the region of infection. The interpretation of this analysis is limited by the lack of immune profiling, but the results suggest that reinfection does not require an unusual set of circumstances with respect to the reinfecting virus.
While the number of immunosuppressed individuals with available sequences remains limited, the results suggest that the rate of viral evolution, meaning the rate at which non-synonymous mutations lead to changes in protein sequences, is accelerated within immunosuppressed individuals. In addition, treatment with convalescent plasma or monoclonal antibody cocktails was insufficient to fully halt of viral evolution and the emergence of viral escape has been documented [23, 27]. The results raise the possibility that novel variants, including those harboring escape mutations against current treatments, could arise from immunosuppressed individuals and suggest that immunosuppressed individuals should be a focus of public health efforts. Amongst the current reports of persistent COVID-19, B-cell dysfunction appears to be a common thread, including in reports that were not included in this analysis due to a lack of available full-length sequences [28–32]. It is important to note, though, that T cell function may also play a role in protection against SARS-CoV-2 [33] and a subset of these patients also included concurrent suppression of other aspects of the immune response. Additional studies are needed to fully define the type and intensity of immunosuppression that would place patients at greatest risk of persistent COVID-19.
Two factors generally differentiated between reinfection and persistent infection scenarios: first, reinfections have so far been largely described in immunocompetent individuals while the majority of persistent COVID cases have been in immunosuppressed patients. Secondly, phylogenetic analysis can generally differentiate between reinfection and persistent infection, especially in cases where persistent infection allowed the longitudinal collection of >2 sequences. However, given the slow rate of SARS-CoV-2 evolution and limited viral diversity [34], it can be challenging to differentiate between reinfection and persistent infection, especially in situations with limited sampling and/or duration between samples. Overall, our results demonstrate the need to further explore factors that increase the risk of breakthrough reinfections and persistent COVID-19. This line of investigation will have important implications for preventing the rise of novel variants and the durability of current available vaccines.
Data Availability
NA
Acknowledgements
We thank Jeremy Luban and Ronald Bosch for their feedback and discussion.
Footnotes
Funding: This study was funded in part by the NIH grant 106701.
Disclosures: Dr. Li has consulted for Abbvie.





{kind=link}
{kind=link}
{kind=link}
{kind=link}