
Abstract
Background Genomic alterations in BRCA1/2 and genomic scar signatures are associated with homologous recombination DNA repair deficiency (HRD) and serve as therapeutic biomarkers for platinum and PARP inhibitors in breast and ovarian cancers. However, the clinical significance of these biomarkers in other homologous recombination repair-related genes or other cancer types is not fully understood.
Results We analyzed the datasets of all solid cancers from The Cancer Genome Atlas and Cancer Cell Line Encyclopedia, and found that the association between biallelic alterations in the homologous recombination pathway genes and genomic scar signatures differed greatly depending on gender and the presence of somatic TP53 mutation. Additionally, HRD cases identified by a combination of these indicators showed higher sensitivity to DNA-damaging drugs than non-HRD cases both in clinical samples and cell lines.
Conclusion Our work provides novel proof of the utility of HRD analysis for all cancer types and will improve the precision and efficacy of chemotherapy selection in clinical oncology.
Background
Homologous recombination repair (HRR) is one of the most accurate DNA repair mechanisms for DNA double-strand breaks (DSBs). Disruption of this mechanism (homologous recombination deficiency; HRD) leads to a high degree of genetic instability and accumulation of genetic mutations, thus playing an important role in the development and progression of cancer. Thus far, mutations in the BRCA1 and BRCA2 genes are considered principal drivers of HRD1; germline BRCA1/2 mutation carriers more frequently develop BRCA-associated cancers, i.e., those of the ovaries, breasts, prostate, and pancreas2. Both germline and somatic BRCA1/2 mutations are shown to be associated with high sensitivity to DNA-damaging drugs such as platinum, doxorubicin, and topoisomerase inhibitors1. After the discovery of synthetic lethality in BRCA1/2 mutated cancers by PARP inhibitors in 20053,4, several subsequent clinical trials validated this efficacy, leading to recent successive FDA approvals of PARP inhibitors for BRCA-associated cancers5,6.
In experimental studies, the suppression of HRR pathway signaling confers HRD properties in various cancers7, and thus gene mutations in the HRR pathway have been considered useful in predicting drug sensitivity associated with HRD. However, in a clinical setting, mutations other than BRCA1/2 have not been sufficiently proven to be useful8. Moreover, the efficacy of PARP inhibitors in non-BRCA-associated cancers remains low, even in patients with BRCA1/2 mutations9-11.
An alternative method for assessing HRD status is to detect characteristic patterns of genomic changes, referred to as genomic scar signatures. These indicators were developed in only the past decade, following remarkable improvements in sequencing techniques and the accumulation of large-scale multi-omics data12,13. As methods for scoring chromosome structural abnormalities due to HRD, the telomeric allelic imbalance (TAI) score14, the large-scale state transitions (LST) score15 and the loss of heterozygosity (LOH) score16 were developed, as well the sum of these scores which is titled the HRD score17. Furthermore, after the concept of mutational signatures was proposed18, a method for quantifying the characteristic mutational pattern in HRD tumors, generally referred to as mutational signature 3 (and referred to as Sig3 below), was developed19. In ovarian and breast cancers, platinum and PARP inhibitors have been found to be effective in tumors with high scores for these genomic scar signatures, even in the absence of BRCA1/2 mutations20.
For carcinogenesis, tumor suppressor genes typically require not only a loss-of-function mutation in a single allele but also a biallelic alteration to fully induce a loss of the wild-type suppressive allele21. Recently, several methods for analyzing allele-specific copy number alterations from SNP genotyping array data or whole-exome sequencing data have been developed22,23, and combining these methods with genomic scar analysis, some studies have reported that the BRCA1/2 mutation requires a loss of the non-mutated allele at the gene locus, termed locus-specific LOH, in order to possess its functional significance 24,25. However, these studies mainly focused on BRCA1/2 mutations in BRCA-associated cancers; other cancers have been insufficiently analyzed. Furthermore, the association between zygosity status and genomic scar signatures in HRR pathway gene mutations other than BRCA1/2 has yet to be investigated in detail. With the increasingly widespread use of gene panel and sequencing-based testing for personalized medicine, it is important to evaluate in full the significance of HRR pathway gene alterations and genomic scar signatures in a pan-cancer fashion.
Here, we comprehensively evaluate biallelic HRR pathway gene alterations and genomic scar signatures in all solid cancers from The Cancer Genome Atlas (TCGA) via an ensemble of analytical techniques. By including clinical information, we additionally examine the efficacy of DNA-damaging drugs in HRD cases. Not only is the clinical significance of HRD across cancer types clarified in a systematic way, but we also uncover a striking link between gender, TP53 mutations, and responses to DNA-damaging chemotherapeutic agents, which contributes to enhanced precision in personalized medicine based on HRD status.
Results
Correlations between HRR pathway gene alterations and genomic scar signatures
The HRD score17 was used as an indicator of chromosome structural abnormalities due to HRD. The HRD score’s underlying scores (TAI/LST/LOH) were strongly correlated (Additional file 1: Figure S1). Based on the literature25, we also calculated the Sig3 ratio (see Methods) as an additional indicator of somatic mutational patterns characteristic for HRD tumors. In what follows, the HRD score and Sig3 ratio are used as the measures of genomic scarring associated with HRD.
Using a combination of ASCAT22 and FACETS23 algorithms, allele-specific copy numbers at each locus of germline and somatic pathogenic variants in 29 selected HRR pathway genes were examined to determine whether they were accompanied by locus-specific LOH (see Methods for gene list). In short, the variants that were determined to have LOH by both algorithms, or to have LOH in one algorithm and to be unknown in the other, showed higher genomic scar scores than the other classification outcome groups (Additional file 1: Figure S2). Therefore, we defined these variants as those accompanying locus-specific LOH.
We examined the relationship between the genomic scar scores and the status of mutations with locus-specific LOH in HRR pathway genes; specifically, we considered the following six stratifications: germline(g)BRCA1, gBRCA2, gHRR (all HRR pathway genes excluding BRCA1/2, n=27, see Methods), somatic(s)BRCA1, sBRCA2, and sHRR (Fig. 1A,1B). When all of the cases were arranged in order of HRD score, mutations with LOH were significantly enriched in the high HRD score cases, while mutations without LOH were not; this trend was consistent in all six groups (Fig. 1A). The same trend was observed for the Sig3 ratio (Fig. 1B). Both the HRD score and Sig3 ratio were significantly higher in cases with LOH mutations than in cases with non-LOH mutations (Fig. 1A, 1B, box plot). For individual HRR pathway genes other than BRCA1/2, mutations with LOH in gATM, gBRIP1, gFANCM, gPALB2, gRAD51C, sATM, sCDK12, and sFANCD2 were enriched in cases with high HRD score or Sig3 ratio, whereas mutations without LOH were not (Additional file 1: Figure S3). Mutations in these LOH-detected genes have been previously reported to be found in tumors with molecular features similar to BRCA-mutant tumors, known as “BRCAness1.”

Association between HRR pathway gene alterations and genomic scar scores or tumor mutation burden (TMB). All samples were arranged in order of HRD score (A), Sig3 ratio (B), and TMB (C), respectively. The top and second panels show the distribution of mutations with and without locus-specific LOH, stratified by germline (prefix g) or somatic (prefix s), and BRCA1, BRCA2, or HRR pathway genes (except for BRCA1/2). The third panels show homozygous deletions in BRCA2, RAD51B, and HRR pathway genes (except for BRCA1/2 but including RAD51B). The fourth panels show BRCA1 methylation. The fifth panels contain the values used for ordering, and samples with MSI-high and somatic POLE mutation in C. The bottom box plots show comparisons of the scores between cases with and without locus-specific LOH in germline or somatic and BRCA1, BRCA2 or other HRR pathway gene mutations, respectively. *, **, and *** stand for P < 0.01, P < 1× 10−4, and P < 1× 10−6 in the Mann–Whitney U test, respectively. Asterisks in red mean a negative correlation with the corresponding value.
Alternatively, when all of the cases were arranged in order of tumor mutational burden, mutations without LOH in sBRCA1/2 and sHRR were strongly enriched in the hypermutated cases, including MSI-high and POLE mutated cases (Fig. 1C). Cases with somatic mutations without LOH had a remarkably high tumor mutational burden, suggesting that such mutations are neutral, known as “passenger,” mutations (Fig. 1C, box plot). In gBRCA1/2, cases with LOH mutations had relatively higher tumor mutational burden than cases with non-LOH mutations (gBRCA1 p=0.017, gBRCA2 p=0.014, Fig. 1C, box plot), which was consistent with previous reports that HRD tumors had moderately increased numbers of gene mutations26,27.
Homozygous deletions were annotated by combining two algorithms, ASCAT22 and ABSOLUTE28. Across all of the HRR pathway genes, cases determined to have homozygous deletion by both algorithms had higher genomic scar scores, whereas cases determined by only one algorithm had lower scores (Additional file 1: Figure S4A). Therefore, we defined homozygous deletion only when the results were matched in both algorithms. There were no homozygous deletions in BRCA1. Cases with BRCA2 homozygous deletion had higher genomic scar scores and lower gene expression (Fig. 1A, B, Additional file 1: Figure S4C). Also, RAD51B homozygous deletion was found in 20 patients with high genomic scar scores and reduced gene expression (Fig. 1A,1B, Additional file 1: Figure S4B, 4C). Although RAD51B mutations have been reported in a few cases in breast cancer, their pathogenic significance remains controversial29. To the best of our knowledge, there have been no reports on homozygous deletion in RAD51B.
Regarding the association between the genomic scar scores with promoter methylation of HRR pathway genes, only BRCA1 was significantly enriched in cases with a higher HRD score and Sig3 ratio (Fig. 1A,1B, Additional file 1: Figure S5). We, thus, used only BRCA1 methylation in subsequent analyses.
Differences in HRD status by cancer type
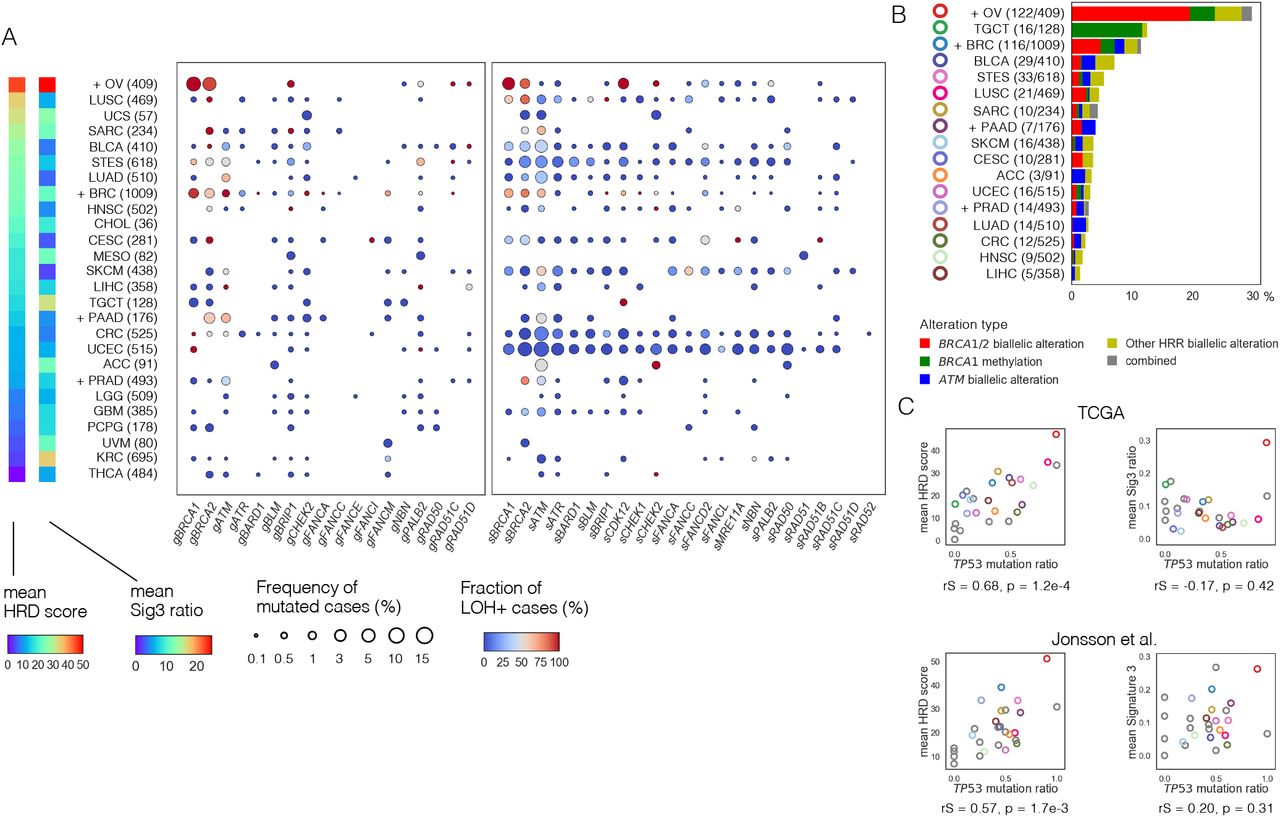
We next examined the mutation rate of HRR pathway genes and the frequency of locus-specific LOH for each cancer type. The TCGA ovarian cancer (OV) cohort contains only high-grade serous carcinoma12, which has the strongest HRD phenotype among the histopathological types of ovarian cancer. Consistent with previous reports24,25,30, BRCA1/2 mutations tended to show higher LOH ratios in BRCA-associated cancers, i.e., ovarian, breast, pancreatic, and prostate cancers. However, the mean HRD score and Sig3 ratio were not particularly higher in breast, pancreatic, and prostate cancers compared to other cancers (Fig. 2A). Similarly, the frequency of biallelic alterations in HRR pathway genes (i.e., germline and somatic pathogenic mutations with LOH, or homozygous deletion) and BRCA1 methylation by cancer type was not much higher in pancreatic and prostate cancers (Fig. 2B). After ovarian cancer, the next highest frequency of HRR alterations was observed in testicular germ cell tumors, in which most of the alterations were derived from BRCA1 methylation. As previously reported, all of these cases were of the non-seminoma type31 and considered to have HRD with platinum/PARP inhibitor sensitivity32,33. The biallelic alterations in ATM were observed the second most frequently after BRCA1/2 (Fig. 2B).

HRR pathway gene alterations per cancer type. A) Frequency of mutated cases and fraction of cases with locus-specific LOH for each HRR pathway gene per cancer type. Cancer types were arranged in order of the mean HRD score. The dot size indicates the frequency of mutated cases and the color indicates the fraction of involving locus-specific LOH in each gene. The prefix g in the gene name stands for germline and s for somatic. B) Types and proportions of biallelic HRR pathway gene alterations per cancer type. C) Correlation between TP53 mutation ratio with mean HRD score (left) and mean Sig3 ratio (right) by cancer type. The upper panels are from the TCGA dataset and the lower from the Jonsson et al. dataset. The dot color matches the color of the cancer type shown in B, otherwise gray. rS and p represent the Spearman correlation coefficient and its p-value, respectively. A),B) Plus signs indicate belonging to BRCA-associated cancers. Numbers in parentheses represent the number of cases. Cancer names are abbreviated with reference to Broad GDAC’s cohort names (https://gdac.broadinstitute.org/); Adrenocortical carcinoma (ACC), Bladder urothelial carcinoma (BLCA), Breast invasive carcinoma (BRC), Cervical and endocervical cancers (CESC), Cholangiocarcinoma (CHOL), Colorectal adenocarcinoma (CRC), Glioblastoma multiforme (GBM), Head and neck squamous cell carcinoma (HNSC), “Kidney chromophobe, Kidney renal clear cell carcinoma, Kidney renal papillary cell carcinoma” (KIPAN), Brain lower grade glioma (LGG), Liver hepatocellular carcinoma (LIHC), Lung adenocarcinoma (LUAD), Lung squamous cell carcinoma (LUSC), Mesothelioma (MESO), Ovarian serous cystadenocarcinoma (OV), Pancreatic adenocarcinoma (PAAD), Pheochromocytoma and paraganglioma (PCPG), Prostate adenocarcinoma (PRAD), Sarcoma (SARC), Skin cutaneous melanoma (SKCM), Stomach and esophageal carcinoma (STES), Testicular germ cell tumors (TGCT), Thyroid carcinoma (THCA), Uterine corpus endometrial carcinoma (UCEC), Uterine carcinosarcoma (UCS), and Uveal melanoma (UVM).
Previous reports showed that TP53 mutations were strongly associated with chromosomal instability across organs34 and elevated copy number change and HRD score35. Therefore, we hypothesized that differences in the genomic scar signatures between cancer types are related to the presence of TP53 mutations. We resultingly found a strong positive correlation between the mean HRD score and TP53 mutation ratio by cancer type (rS=0.68, p=1.2 × 10−4, Fig. 2C), whereas, there was no correlation between the mean Sig3 ratio and TP53 mutation ratio. These results were similarly observed in the external data set based on various cancers reported by Jonsson et al.25
Identification of pan-cancer HRD cases based on genomic scar signatures
The number of carriers of germline BRCA1/2 variants is equal in women and men36 though the lifetime incidence of cancer is much higher in women37. Therefore, we hypothesized that the effect of HRD on cancer development would be gender-dependent. In addition, a previous report showed that the genome-wide LOH scores of tumors with biallelic BRCA1/2 alterations differed greatly by cancer type and that prostate cancer, a male-specific tumor, was one of the lowest-scoring cancers30, although olaparib was shown to be effective in prostate cancer with BRCA1/2 mutations6.
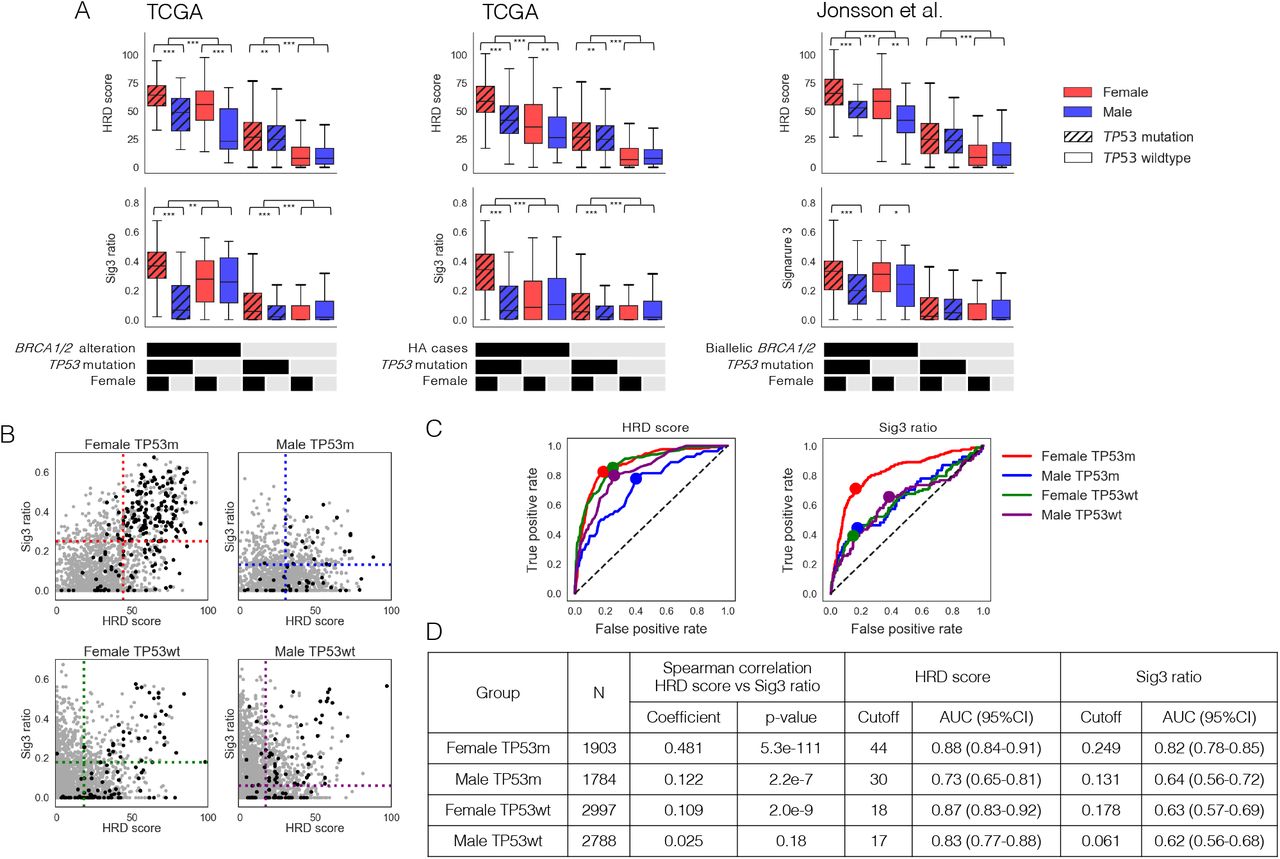
For the above reasons, we decided to evaluate the association between genomic scar scores and HRR pathway alterations separately by gender and TP53 mutation status. The results showed that cases with BRCA1/2 alteration, meaning germline and somatic BRCA1/2 biallelic alteration plus BRCA1 methylation, had the highest HRD score and Sig3 ratio in females withTP53 mutations (Fig. 3A, left). The results were also similar in cases with HRR pathway gene alteration (HA cases), meaning germline and somatic biallelic alteration in HRR pathway genes and BRCA1 methylation (Fig. 3A, middle). Also, in the Jonsson et al. dataset25, cases with biallelic BRCA1/2 alteration had the highest HRD score and Sig3 ratio in females with TP53 mutations (Fig. 3A, right). Furthermore, a similar trend was observed in cases with BRCA1/2 alteration even when excluding BRCA-associated cancers (Additional file 1: Figure S6A). On the other hand, even in the group without HRR alterations, the HRD score was elevated with the presence of TP53 mutation (Fig. 3A). These results indicate that stratification by gender and the presence of TP53 mutation is needed to identify HRD cases based on the genomic scar scores.

Identification of genomic scar high (GS) cases. A) Comparison of HRD score and Sig3 ratio in the eight groups divided by TP53 mutation, gender, and (left) biallelic BRCA1/2 alteration (including BRCA1 methylation), (middle) biallelic HRR pathway gene alteration, (right) biallelic BRCA1/2 alteration in Jonsson’s dataset. *, **, and *** stand for P < 0.05, P < 0.01, and P < 0.001 in the Mann–Whitney U test, respectively. B) Comparison of correlations between HRD score and Sig3 ratio and distributions of biallelic HRR pathway gene alterations among the four groups divided by gender and TP53 mutation. Black dots represent the HRR pathway gene-altered cases and gray dots the other cases. The dotted lines represent the optimal cutoff values determined by the ROC curves in C. C) Differences in ROC curves and optimal cutoff values for HRR pathway gene alterations in HRD score and Sig3 ratio among the four groups. D) Spearman correlation coefficients between HRD score and Sig3 ratio, optimal cutoff values, and AUCs on the ROC curves among the four groups. TP53m and TP53wt indicate with and without TP53 mutation, respectively.
The positive correlation between the HRD score and Sig3 ratio was strong in females with TP53 mutations but weak in males with TP53 mutations and females without TP53 mutations and was non-significant in males without TP53 mutations (Fig. 3B, rS=0.481, 0.122, and 0.109, 0.025, respectively). Similar results were still observed excluding BRCA-related cancers (Additional file 1: Figure S6B). After stratifying all cases into the four groups by gender and TP53 mutation status, we examined the optimal cutoff values for determining HA cases by ROC curves in the HRD score and Sig3 ratio, respectively. We found that both the optimal cutoffs and the AUCs significantly differed among the four groups, with the highest values for both in females with TP53 mutations (Fig. 3B, 3C, 3D). The cases that exceed both of the two cutoff values in each of the four groups were defined as genomic scar high (GS) cases.
Gene expression profile in GS and HA cases
The number of GS and/or HA cases was 341 in females with TP53 mutations, 187 in males with TP53 mutations, 233 in females without TP53 mutations, and 357 in males without TP53 mutations (Fig. 4A, Additional file 1: Figure S7). The rate of BRCA1/2 alterations was highest in females with TP53 mutations (Fig. 4A, 8.6%, 1.3%, 1.5%, and 1.2%, respectively, chi-square test p=6.5 × 10−67). The biallelic ATM alteration ratio was higher in the TP53 wild-type group than in the TP53 mutated group; ATM and TP53 mutation were significantly mutually exclusive (Fig. 4A, 0.19%, 1.2%, respectively, chi-square test p=1.4 × 10−7).
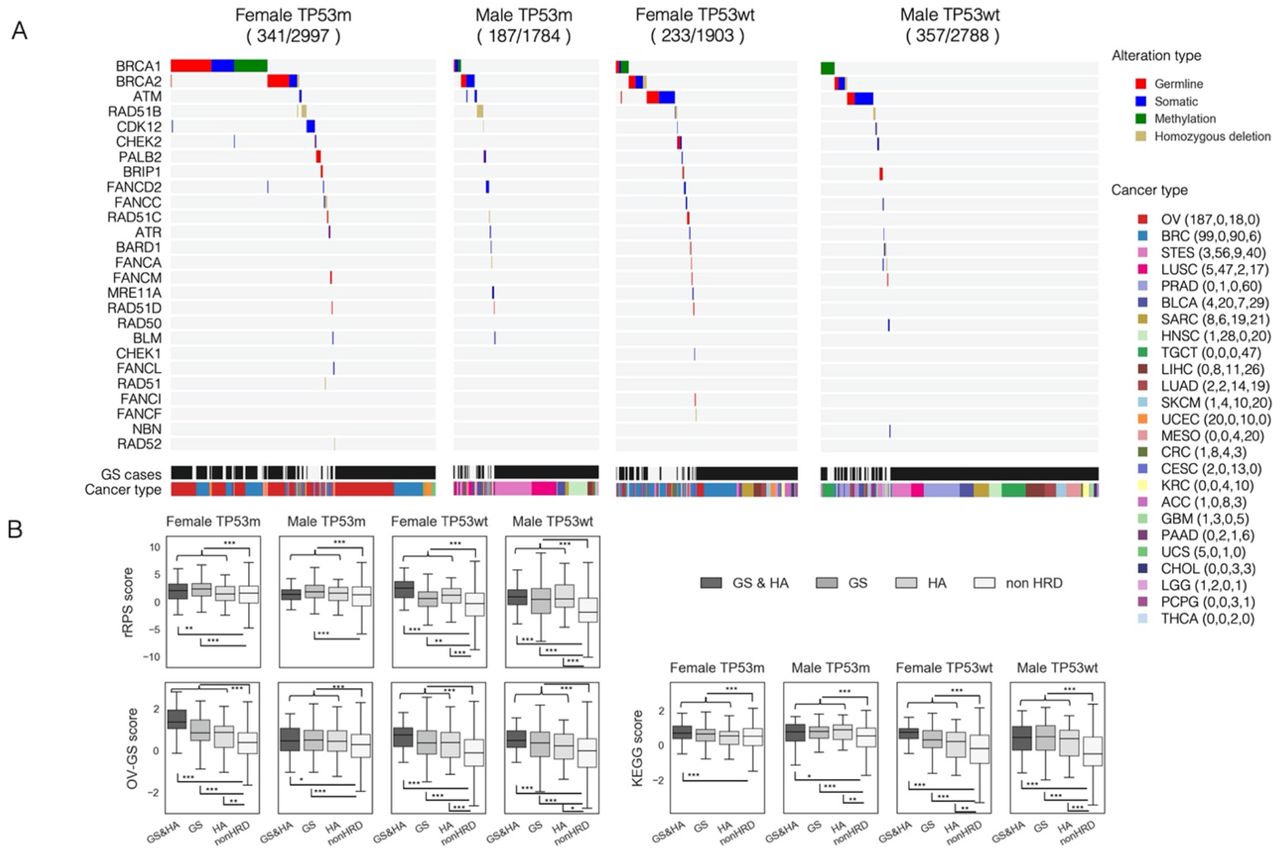
Characteristics of HRR pathway gene alteration (HA) cases and genomic scar high (GS) cases. A) Comparisons of the distribution of gene alterations and cancer types in HA and/or GS cases, after stratifying into the four groups by gender and TP53 mutation. HRR gene alteration types and cancer types are color-coded as shown on the right side. Numbers in parenthesis next to cancer types represent the number of cases in the four groups, respectively. B) Comparisons of the gene expression-based HRD assessment scores between GS and/or HA and non-HRD cases. The p-values by the Mann–Whitney U test comparing GS and/or HA to non-HRD in each of the four groups were 2.1 × 10−4, 3.3 × 10−4, 1.0 × 10−27, and 3.4 × 10−27 in the rRPS score, 3.9 × 10−23, 4.2 × 10−5, 7.8 × 10−15, and 4.0 × 10−15 in the OV-GS score, and 7.3 × 10−4, 3.3 × 10−7, 2.6 × 10−12, and 3.7 × 10−35 in the KEGG score, respectively. *, **, and *** stand for P < 0.05, P < 0.01, and P < 0.001 in the Mann–Whitney U test, respectively. TP53m and TP53wt indicate with and without TP53 mutation, respectively.
We next examined whether tumors defined as GS or HA cases had HRD properties in their gene expression profiles. By modifying the recombination proficiency score (RPS) in the previous report38, we calculated the reversed RPS (rRPS) score as a measure of HRD (see Methods). In addition, we identified differentially expressed genes (DEGs) from TCGA-OV cases based on both HRD score and Sig3 ratio (Additional file 1: Figure S8) and scored the enrichment of those DEGs in non-TCGA OV cases by the ssGSEA39 algorithm, referred to as the OV-GS score (see Methods). Furthermore, since the dysfunction of DNA repair mechanisms is reported to result in compensatory elevation of gene expression in that pathway40, using the KEGG homologous recombination pathway signature (hsa03440), we calculated the enrichment score of that signature by ssGSEA, referred to as the KEGG score (see Methods). All of the above three scores were significantly higher in GS and/or HA cases than in the other groups (Fig. 4B), and positively correlated with each other (Additional file 1: Figure S9). These results suggest that GS and/or HA cases, as defined by DNA alteration, have the characteristics of HRD in terms of gene expression profile. GS and/or HA cases are hereafter referred to as HRD cases.
In female cells, some of the molecules belonging to the DNA double-strand break repair pathway are reported to be involved in X-chromosome inactivation41-43. Therefore, we examined the association between HRD and X-chromosome inactivation. Expression of XIST, which is a non-coding RNA on the X chromosome with an essential function in X-chromosome inactivation, was significantly lower in female HRD tumors than non-HRD tumors (Additional file 1: Figure S10A). Additionally, the enrichment score of the whole X-chromosome gene set calculated by ssGSEA was significantly higher in female HRD tumors, whereas the score of a subset of genes reported to escape X-chromosome inactivation showed no significant elevation (Additional file 1: Figure S10A, see Methods). Also, we found that the global DNA methylation of the X chromosome was significantly lower in HRD tumors (Additional file 1: Figure S10A, see Methods). These differences were consistent even when BRCA-associated cancers were excluded (Additional file 1: Figure S10B). These results suggest that X-chromosome inactivation may underlie differences in HRD by gender.
The association between chemotherapeutic agents and survival outcomes in HRD cases
Recently, it has been theorized that cancers with defects in a specific DNA damage repair mechanism are sensitive to drugs that target the exact mechanism44. According to this theory, HRD tumors can be considered susceptible to drugs that increase DNA damage or replication stress45. To investigate the drug sensitivity of HRD cases, all of the cases were divided into two groups: those with (n=2979) and without (n=6460) a treatment history of DNA-damaging agents, including alkylating agents, antibiotics, antimetabolites, platinum, and topoisomerase. While HRD cases had a significantly better overall survival than non-HRD cases in the group with DNA-damaging agent use, HRD cases in the group without treatment had a significantly worse outcome than non-HRD cases (Fig. 5A, log-rank test p=5.1× 10−4, 1.1 × 10−10, respectively).

The prognosis of HRD patients differed with and without the administration of DNA-damaging agents. A) Comparison of overall survival between HRD and non-HRD cases by the Kaplan–Meier curve, in two groups divided by the history of DNA-damaging agent use. The number in the upper right corner represents the p-value in the log-rank test. B) Left: univariate and multivariate Cox proportional hazard model analyses with covariates of age, gender, TP53 mutation, clinical stage, and HRD cases. Right: forest plots showing adjusted hazard ratios with 95%CIs from the multivariate Cox analysis. C) Comparison of the two adjusted hazard ratios of HRD cases for overall survival among 13 cancer types that have five or more comparable cases. As in B, samples were divided into two groups by DNA-damaging agent use and analyzed by multivariate Cox proportional hazard model to calculate the adjusted hazard ratios with 95%CIs. D) Comparisons of MATH index and KEGG cell cycle scores between HRD cases and non-HRD cases, or in the four groups divided by gender and TP53 mutation. The p-values by the Mann–Whitney U test were 1.1 × 10−4) (upper left), 3.2 × 10−43 (upper right), and 1.5 × 10−13, 7.7 × 10−7, 9.3 × 10−9 and 4.4 × 10−14 (lower left), and 3.6 × 10−7, 1.4 × 10−9, 4.8 × 10−9, and 3.1 × 10−30 (lower right), respectively. *, **, and *** stand for P < 0.01, P < 1 × 10−5, and P < 1 × 10−7 in the Mann–Whitney U test.
Furthermore, in the Cox proportional hazard multivariate analysis with covariates of age, gender, stage, and TP53 mutation, HRD was a good independent prognostic factor in the DNA-damaging agent group (adjusted hazard ratio 0.78, p=6.6 × 10−3), whereas it was a poor independent prognostic factor in the non-DNA-damaging agent group (adjusted hazard ratio 1.58, p=2.8 × 10−9) (Fig. 5B). Similar analyses were performed for each of the 13 cancer types that contained five or more comparable cases in both groups on the use of DNA-damaging agents. As a result, in most cancer types, the adjusted hazard ratios of HRD were lower in the DNA-damaging agent group than in the non-DNA-damaging agent group (Fig. 5C, Additional file 2: Table S1). These results suggest that DNA-damaging agents would improve the prognosis of HRD cases, regardless of the cancer type.
Next, we examined why the survival outcome of HRD cases is worse in the absence of DNA-damaging agents than in non-HRD cases. The MATH index46, an indicator of intratumor heterogeneity, was higher in HRD cases than in non-HRD cases (Fig. 5D). This result suggests that HRD-induced genomic instability causes increased intratumor heterogeneity and may lead to a poor prognosis. Also, the gene expression-based ssGSEA score of the KEGG cell cycle signature (see Methods) was higher in HRD cases than in non-HRD cases (Fig. 5D). This result suggests that HRD tumors have a high proliferation capacity.
In addition, we performed a similar analysis without the stratification by gender and TP53 mutation status, using universal cutoff values for HRD score and Sig3 ratio (Additional file 1: Figure S11). Although the associations between gene expression profiles, drug administration and survival outcomes of HRD cases showed similar trends to those with the stratification, the characteristics of the determined HRD cases, such as differences in hazard ratios and p-values in the survival analysis, appeared to be weaker (adjusted hazard ratio: 1.37 vs 0.83, p=7.7× 10−4 vs 0.043, respectively, Additional file 1: Figure S11).
Validation of high sensitivity of DNA-damaging agents to HRD tumors in cell lines
Using datasets of human cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE), we performed exactly the same analysis as described above to reproduce the association between HRD status and chemotherapy sensitivity.
As a result, genomic scar scores were elevated only when HRR pathway gene mutations were involved in the locus-specific LOH (Additional file 1: Figure S12AB), mutations without LOH were enriched in hypermutators without elevation of these scores (Additional file 1: Figure S12C). Stratified analysis by gender origin and presence of TP53 mutation also showed similar trends to the clinical samples; the group of female origin with TP53 mutations tended to have the highest genomic scar scores, the groups with TP53 mutations tended to have higher HRD score in the absence of HRR pathway gene alterations (Additional file 1: Figure S13A), and only the group of male origin without TP53 mutations did not show a positive correlation between HRD score and Sig3 ratio (Additional file 1: Figure S13B, F). We determined the genomic scar high (GS) cell lines with the optimal cutoff values of HRD score and Sig3 ratio calculated from the AUC curve for each of the four groups divided by gender and TP53 mutation status (Additional file 1: Figure S13B, C, F). For samples with no information on gender or TP53 mutation, the cutoff values were calculated using data from all samples (Additional file 1: Figure S13D).
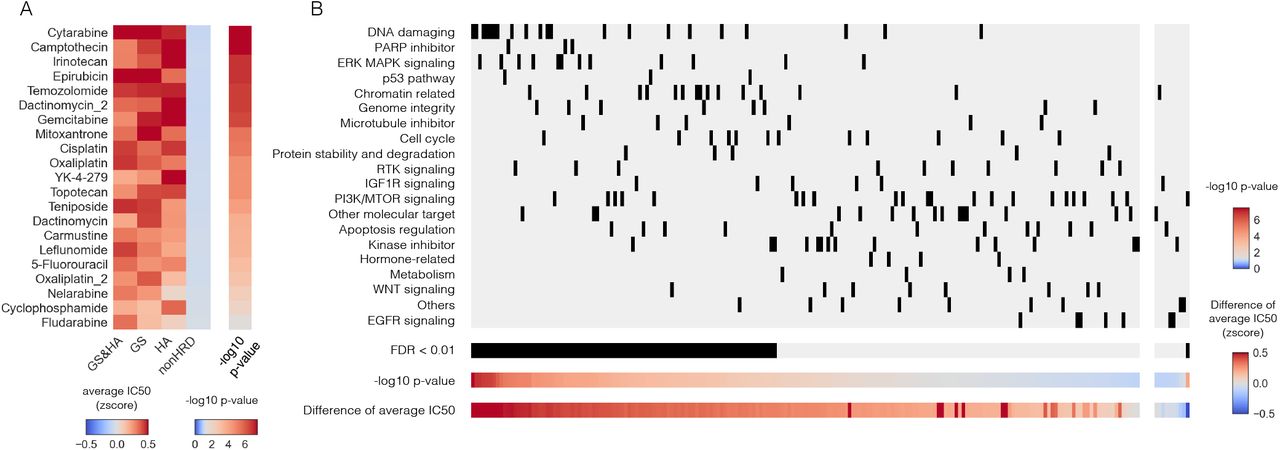
After identifying GS and HA samples (Additional file 1: Figure S13E), we examined the IC50 values of 198 compounds for these cell lines using the datasets of the Genomics of Drug Sensitivity in Cancer (GDSC). Consequently, almost all DNA-damaging drugs showed higher sensitivity to HRD tumors than to non-HRD tumors (Additional file 2: Table S5, Fig. 6A). Furthermore, this category of drugs showed higher sensitivity to HRD tumors compared to other types of drugs. (Fig. 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association between HRD status and chemotherapy sensitivity in CCLE cell lines. A) Most of the DNA-damaging agents showed higher sensitivity in HRD tumors than in non-HRD tumors. The left heatmap indicates the average IC50 for each group. The right heatmap shows p-values of the Mann–Whitney test comparing the IC50 of GS and/or HA samples to non-HRD samples. The center of the color bar is adjusted to P = 0.05. B) Association between drug sensitivity and drug type for cell lines with HRD status. The IC50 values were compared between HRD and non-HRD samples for 198 compounds examined in GDSC2. These compounds were divided into the left or right by positive or negative difference in average IC50, and then arranged in order of decreasing or increasing -log10 p-values on the x-axis. The drug categories were arranged on the Y-axis in order of decreasing mean of -log10 p-value. HRD samples showed significant sensitivity to several drugs (FDR<0.01), especially high sensitivity to DNA-damaging drugs.
Discussion
Many of the HRR pathway genes are considered to be tumor suppressor genes, which typically require biallelic alterations for actual loss of function21. In our study, most gene mutations in the HRR pathway showed increased genomic scar scores only when accompanied by locus-specific LOH (Figs. 1A,1B), whereas mutations without LOH were considered to be passengers (Fig. 1C). These results strongly suggest that to acquire the HRD phenotype, a genetic mutation in the HRR pathway must have the contralateral wild-type allele loss. Furthermore, the frequency of related locus-specific LOH in HRR pathway genes varied widely by gene and cancer type (Fig 2A). These observations are clinically important because they indicate that if a cancer has a genetic mutation in the HRR pathway, its pathogenic importance should be determined by assessing whether it is accompanied by locus-specific LOH, such as by gene panel testing. In addition, we used a combination of FACETS23 and ASCAT22 to examine the presence of locus-specific LOH and, similarly, used ABSOLUTE28 and ASCAT22 to test for homozygous deletions. These integrated analyses provided a clearer association between biallelic changes and genomic scar scores than when only a single method was used (Additional file 1: Figure S2, S4), showing that using ensembles of algorithms can improve robustness. While one study reported a benefit of PARP inhibitors in BRCA-associated cancers even with monoallelic BRCA1/2 alterations25, another study showed that tumors with germline BRCA1/2 mutations without locus-specific LOH have a low HRD score and low drug sensitivity24. Considering that all of these studies used only one algorithm, the differences in results may be due to difference in methods. Recently, several clinical trials of PARP inhibitors have been conducted in which HRR pathway gene mutations have been examined as biomarkers (e.g., NCT03209401, NCT03377556, NCT04123366). However, few have assessed whether these mutations are biallelic or not. Our data indicate that the clinical relevance of these variants is a very important consideration in future clinical trials and that a robust method for determining the zygosity of these variants needs to be established.
Biallelic ATM mutations were the second most common HRR alteration after BRCA1/2 (Fig. 3B). ATM is well known to have an important function in HRR from the results of many previous studies in Ataxia-telangiectasia. Recent studies have shown that ATM-deficient cells were sensitive to PARP inhibitors47,48, similarly that low ATM gene expression was associated with sensitivity to PARP inhibitors in some clinical trials49,50. We found that biallelic ATM and TP53 mutations were significantly mutually exclusive (Fig. 4A). The mutual exclusivity of these two has also been reported in hematologic51,52 and breast53 cancers. Since ATM has been reported to activate TP53 through the degradation of MDM2 after sensing DSBs and induce tumor suppressive effects including cell cycle delay, arrest, and apoptosis, this mutual exclusivity is plausibly due to the similarities between ATM and TP53 mutations in terms of disrupting the cell cycle checkpoint mechanism for cancer cells. Also, the relatively lower HRD scores in ATM-mutated cases (Additional file 1: Figure S3) may be explained by this mutual exclusivity.
In our analysis, HRD score was higher in tumors with TP53 mutations (Fig. 2C). From previous studies on TP53 mutations and HRD status, some reported that TP53 mutations activate the HRR pathway under certain circumstances54, while others suggest that they did not directly associate with HRD55,56. In our data, TP53 mutation did not correlate with the Sig3 ratio (Fig. 2C), while TP53 mutated cases, even without HRR alteration, had higher HRD score than TP53 wild-type cases (Fig. 3A). In a very recent study of prostate cancer, TP53 mutations were reported to be associated with increased HRD scores independently of HRR pathway gene alterations57. These observations suggest that the high HRD score in TP53 mutated cases is not necessarily due to true HRD but, rather, due to chromosomal instability caused by mechanisms other than HRD (e.g., loss of cell-cycle checkpoint mechanisms).
We observed that not only the two genomic scar scores themselves, but also their correlations, differed greatly depending on gender and TP53 mutation status (Fig. 3). Specifically, the correlation between HRD score and Sig3 ratio was the strongest in women with TP53 mutations, though weaker in the other groups (Fig. 3B). Also, the AUC values to determine HRR pathway gene-altered cases were lower in the groups other than women with TP53 mutations (Fig. 3C). Although the analysis using a pair of universal cutoffs without stratification by gender or TP53 mutation was still able to extract cases with HRD characteristics (Additional file 1: Figure S11), these cutoff values were impacted, resulting in over-estimation in women with TP53 mutations and under-estimation in the other groups, thus weakening the characteristics of the determined HRD cases (Additional file 1: Figure S11). The HRD score and Sig3 ratio were originally developed in a rather limited analysis to identify BRCA1/2 mutant tumors in breast and ovarian cancers14-16,18,19 and were reported to have low correlation19. Based on the findings after stratification, we propose that differences in gender and TP53 should be taken into account when using these genomic scores to determine HRD cases. For the groups other than women with TP53 mutations, a more appropriate measure other than the HRD/Sig3 scores could further strengthen analysis of HRD. Given that large-scale multi-omics databases are being constructed worldwide and that new methods for evaluating HRD using whole-genome sequencing are emerging58,59, there is opportunity to develop more robust methods for identifying HRD cases in pan-cancer contexts.
One of the major differences between the cells of men and women is the presence of X-chromosome inactivation. And the loss of X-chromosome inactivation in some female cancers has long been known as the loss of the Barr body60. In the present pan-tumor analysis, we found that the loss of X-chromosome inactivation was associated with female HRD tumor cells (Additional file 1: Figure S10). This suggests that gender differences in tumor HRD status and genomic scar scores are related to X-chromosome inactivation (Fig. 3A). There have been no reports on the association between the genomic scar-based HRD status and the X chromosome, and more detailed studies are needed in the future.
HRR pathway gene alterations were detected in only half of the genomic scar high (GS) cases (Fig. 4A). There are many possible causative factors for HRD other than those analyzed in this study. For instance, abnormalities in histone modifications and chromatin remodeling that operate in relation to DSB repair are reported to be strongly associated with HRD61. More recently, oncometabolites have been reported to result in HRD in cancer cells62,63. Thus, mechanisms that have not yet been fully elucidated may be involved in HRD, and further studies are warranted.
Recently, an emerging therapeutic strategy for cancer is the idea that cancers with defects in a certain DNA damage repair mechanism should be vulnerable to drugs that are targeting the same mechanism44. In this context, HRD tumors would then be susceptible to drugs that induce the DNA damage for which HRR is essential. Platinum compounds, alkylating agents, and some antibiotic-derived anticancer drugs (e.g., mitomycin C) have a function to generate inter- and intra-strand crosslinks between DNAs, resulting in the complex structures for which HRR is needed to restore sequence fidelity64. In practice, the efficacy of platinum agents for HRD tumors is well recognized in ovarian and breast cancers1. In addition, most antibiotic-derived anticancer drugs inhibit DNA polymerase, nucleic acid synthesis inhibitors deplete available deoxynucleoside triphosphate (dNTP) pools and reduce the speed of DNA synthesis at individual replication forks, and topoisomerase inhibitors bind to DNA and interfere with ongoing replication forks. In other words, all of these drugs hinder DNA replication and stall the replication fork, causing replication stress45. Since these stalled forks can only be repaired through HRR, replication stress will accumulate in HRR-dysfunctional cells up to a critical level after which replication forks collapse, leading to DNA double-strand breaks and cell death45. Therefore, in this study, we included drugs with these mechanisms as the DNA-damaging agents and found the high sensitivity of these drugs for HRD tumors in both clinical samples and cell lines.
In general, the standard of care for cancer treatment is selected on the basis of organ-specific clinicopathological factors derived from the results of the many randomized trials that have been conducted up to that time. However, our study demonstrated the potential benefit of DNA-damaging drug administration for HRD tumors as defined by pan-cancer genomic analysis (Fig. 5). The results indicate that treatment personalization beyond cancer type based on molecular phenotype including HRD status has substantial potential. Since the number of cases treated with PARP inhibitors in the TCGA cohort is highly limited, further investigations and publicly-accessible datasets are needed to determine if pan-cancer HRD analysis could predict sensitivity to PARP inhibitors.
Conclusion
This comprehensive pan-solid cancer HRD analysis revealed that stratified analysis by gender and TP53 mutation status can more accurately assess HRD status, and that DNA-damaging drugs could be beneficial for HRD cases across cancer types, suggesting that HRD is useful as a tumor-agnostic therapeutic biomarker. Based on the results, it is rational to expect improvements in the implementation of personalized cancer medicine based on HRD. In parallel, further diagnostic methods can be developed for the pinpoint identification of clinically significant HRD cases.
Methods
Patient selection and clinical data
Among the 10,967 cases integrated into the TCGA Pan-Cancer Atlas at cBioPortal, the cases with acute myeloid leukemia, diffuse large B-cell lymphoma, and thymoma, and the cases without the MC3 somatic mutation profiles65 were excluded. The remaining 9,847 cases were analyzed (Additional file 2: Table S2).
Clinical data were obtained from cBioPortal and Broad GDAC. Regarding the drugs administered in each case, we first reviewed and manually unified treatment annotations (e.g., for drug-spelling errors), then subsequently extracted individual drugs when they were part of combination therapies. Next, we separated the anti-tumor agents into the following categories: alkylating agents, antibiotics, antimetabolites, platinum, topoisomerase, microtubule inhibitors, molecular-target agents, immune checkpoint inhibitors, others, and those unresolvable from annotations. Finally, the drugs in the first five categories were defined as DNA-damaging drugs (Additional file 2: Table S3). Fewer than 10 cases were treated with PARP inhibitors, all priorly treated by DNA-damaging drugs.
Genomic scar scores
As an indicator of HRD based on chromosomal structural changes, LST, TAI, and LOH scores published in Pan-Cancer Atlas studies were obtained from the Genomic Data Commons (GDC) website. The sum of these three scores was used as the HRD score. In addition, as a score of HRD based on the pattern of single base substitutions, the mutational signature 3 value was calculated using the method previously described by Jonsson et al.25. To be more specific, we obtained somatic mutation profiles published in the MC3 project65 from the above-mentioned GDC website and, using 30 cancer mutational signatures from COSMIC version 2 (https://cancer.sanger.ac.uk/cosmic/signatures_v2) as a reference, calculated the contribution ratio to signature 3 as the “Sig3 ratio” by applying non-negative matrix factorization.
Definition of homologous recombination repair (HRR) pathway genes
Based on the literature66 we selected BRCA1, BRCA2, ATM, ATR, BARD1, BLM, BRIP1, CDK12, CHEK1, CHEK2, FANCA, FANCC, FANCD2, FANCE, FANCF, FANCI, FANCL, FANCM, MRE11, NBN, PALB2, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD52, RAD54L, and RPA1 as HRR pathway genes (n=29).
For germline gene mutations, we obtained the annotation information from the supplement data of the previous paper, in which 10,339 cases from TCGA were examined for pathological germline variants67 available on the GDC website. From the data, we extracted 412 cases who had germline mutations in the above HRR pathway genes with annotation of “likely pathogenic” or “pathogenic” in “Overall Classification” column.
For somatic gene mutations, we extracted the annotation information from cBioPortal except for both mutations and copy number alterations of unknown significance.
Locus-specific LOH status
The raw SNP genotyping array data of normal and tumor pairs were obtained from the GDC legacy archive, and the segmented genome-wide allele-specific copy number profiles were calculated using PennCNV68 and ASCAT22 The whole-exome sequencing data of normal and tumor pairs were obtained from the GDC data portal, and the allele-specific copy number was calculated using FACETS23 We checked the estimated copy numbers of the minor allele at the segment located in the locus of each of the above germline and somatic mutations; when the copy number of the minor allele was equal to zero, it was determined to be “LOH,” and when it was one or more, it was determined to be “non-LOH.” When it was unavailable, it was determined to be “unknown.” Then, we examined the concordance between the results from ASCAT and FACET for the presence of locus-specific LOH in each variant (Additional file 1: Figure S2). The variants that were determined to have LOH by both algorithms, or to have LOH in one algorithm and to be unknown in the other, showed higher genomic scar scores than the other classification outcome groups. From these observations, we determined these variants as those accompanying locus-specific LOH (Additional file 1: Figure S2).
Homozygous deletion status
From the allele-specific copy number profiles calculated by ASCAT22 as described above, segmented regions with a total copy number equal to zero were extracted as homozygously deleted regions in each sample. Additionally, we obtained segmented copy number data calculated by ABSOLUTE28 and published in the Pan-Cancer Atlas studies from the GDC website and extracted the regions annotated as “Homozygous_deletion” per sample. Using each annotation separately, for each HRR pathway gene in each case, a gene was determined to have homozygous deletion when part or all of the locus was contained in the above homozygously deleted regions. The results calculated from ASCAT and ABSOLUTE were combined to form the final annotation (Additional file 1: Figure S4A).
DNA promoter methylation
In a previous report, Knijnenburg et al. comprehensively investigated all of the TCGA samples for DNA promoter methylation status in 276 genes involved in DNA-damage repair and determined cases with methylation-driven gene silencing in stringent criteria35. From their published data, we found one or more methylated sample in BRCA1, RAD51C, and FANCC2 among the above HRR pathway genes (Additional file 1: Figure S5).
To quantify DNA methylation across the entire X chromosome, DNA promoter methylation data from the Illumina Infinium array published in The Pan-Cancer Atlas Studies were obtained from the GDC website. Among the 20601 probes, the average beta value of 856 probes on the X chromosome was calculated as the amount of X chromosome global methylation for each sample.
Tumor mutational burden, MSI-high annotation, somatic POLE, and TP53 mutation
From the somatic mutation profile published by the MC3 project65, the total number of non-synonymous mutations in each sample was counted and defined as tumor mutational burden (TMB).
Among the patients’ clinical data obtained from cBioPortal, annotations of tumor subtypes including MSI status exist for the TCGA-COAD, READ, UCEC, STAD, and ESCA cohorts. We recorded those cases as MSI-high for further analysis.
We also extracted somatic POLE and TP53 mutation annotation information from cBioPortal, discarding annotations marked as mutations and copy number alterations of unknown significance.
Gene expression analysis
From the GDC website, we retrieved the published batch effect-corrected mRNA gene expression data published in Pan-Cancer Atlas studies. Expression values of genes whose missing data exceeded 25% of all cases were excluded from the analysis. Otherwise, the missing data were completed with the median value in all other cases as the representative value.
In the original work38, the Recombination Proficiency Score (RPS) was calculated as the sum of the microarray-based gene expression values of the Rif1, PARI, Ku80, and RAD51 genes after log2-transformation and median normalization, and being multiplied by −1 for each gene. We modified this method to simply sum up the RNA-based gene expression values of the same four genes after log2-transforming and z-scaling in each gene and named the value reversed RPS (rRPS) score as an indicator of HRD.
The configuration of the TCGA-OV derived HRD signature was as follows. First, we selected 243 ovarian cancer cases for which the Sig3 ratio, HRD score, and mRNA gene expression data were all available. Next, we selected cases that were in the top 50% of both the Sig3 ratio and HRD score, and cases that were in the bottom 50% of both. Between those two groups, differentially expressed genes were analyzed by limma+voom69. Finally, we extracted the 76 genes highly expressed in the top group according to certain criteria (logFC > 0.5, AveExpr > −2, adj.P.Val < 0.01) and designated them as TCGA-OV derived HRD signatures (Additional file 2: Table S4). The enrichment score of the gene signatures for each sample was calculated using the ssGSEA algorithm39 and was designated as the OV-GS score. Because ovarian cancer was used as a training set, we assigned the OV-GS score values to cancer types other than ovarian cancer.
In addition, the gene sets of KEGG_HOMOLOGOUS_RECOMBINATION (hsa03440, 41 genes) and KEGG_CELL_CYCLE (hsa04110, 124 genes) were obtained from MsigDB (http://www.gsea-msigdb.org/gsea/msigdb/). The only gene found in common in the two KEGG gene sets was ATM. These signatures were also used for scoring by ssGSEA and designated as the KEGG-score and the KEGG cell cycle score, respectively.
To assess overall gene expression of the X chromosome, a list of 1666 gene names on the X chromosome was obtained from Ensemble BioMart Release 102 (GRCh38.p13). The enrichment score for each sample was calculated using ssGSEA and named as X score based on this gene set. We also obtained a list of 50 X-chromosome inactivation escaping genes from a previous report70 and calculated the X-chromosome inactivation score (XCI-esc score) per sample using ssGSEA.
MATH score
The MATH score for each sample was calculated from the somatic mutation profile published in the MC3 project65, by using the method described in the previous reports46.
Retrieving validation datasets
From the previous literature25 and the database of AACR Project GENIE (https://www.aacr.org/professionals/research/aacr-project-genie/), the clinical data, signature 3 values, HRD scores (sum of the LOH, LST, and TAI scores), and somatic mutation profiles from the 815 samples of 794 multiple cancer cases were obtained. Of these, two samples from two cases with hematological and lymphatic cancers were excluded from analysis.
Data analysis of CCLE cell lines
We obtained the gene mutation data from the DepMap portal (https://depmap.org/portal/) and determined that mutations marked as “damaging” in the “variant annotation” were pathogenic. The total number of nonsynonymous mutations in each cell line was defined as TMB. We obtained allele-specific copy number data analyzed by ABSOLUTE28 from the same data source and determined whether the above individual pathogenic mutations were accompanied by a locus-specific LOH: with LOH if the number of minor alleles at the locus was equal to 0, without LOH if the number was greater than one, and unknown if the number was unknown. For BRCA1 methylation, RNAseq gene expression and DNA methylation data were obtained, and samples with promoter methylation beta values of 0.3 or higher and gene expression below the median of all samples were determined to have methylation-associated silencing (Additional file 1: Figure S12D). HRD scores were calculated using scarHRD71 with the allele-specific copy number and average ploidy data available in the COSMIC cell line project (https://cancer.sanger.ac.uk/cell_lines). Based on copy number aberration analysis for each gene in the same project, genes with a total copy number of zero were determined to have homozygous deletions. The single-base-substitution pattern in the 96 trinucleotide contexts for each cell line was obtained from the published data of Petljak et al.72 and the Sig3 ratio was calculated using the same method as described above 25. The inferred MSI status was obtained from the published data of Ghandi et al.73. IC50 data of 198 compounds for each sample were obtained from Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene.org/) released as version 2. These compounds were classified into the following 20 categories based on the information of target molecules and target pathways (Additional file 2: Table S5): Apoptosis regulation, Cell cycle, Chromatin related, DNA damaging, EGFR signaling, ERK MAPK signaling, Genome integrity, Hormone-related, IGF1R signaling, Kinase inhibitor, Metabolism, Microtubule inhibitor, Other molecular target, Others, PARP inhibitor, PI3K/MTOR signaling, Protein stability and degradation, RTK signaling, WNT signaling, p53 pathway.
Statistical analyses
All statistical analyses in this study were performed in Python (3.8.6). The Mann–Whitney U test, chi-square test, and Spearman’s rank correlation coefficient test were performed using SciPy (1.6.0). Survival analyses including the Kaplan–Meier curve, log-rank test, and Cox proportional hazard regression model were performed using Lifelines (0.25.8). Unless otherwise noted, a p-value < 0.05 was considered statistically significant.
Data Availability
Open access data and processed data for TCGA samples are available from the cBioPortal (https://www.cbioportal.org/), the Broad GDAC (https://gdac.broadinstitute.org/), and the GDC Pan-Cancer Atlas studies (https://gdc.cancer.gov/about-data/publications/pancanatlas). Controlled access data including germline variant annotations, raw sequence data, and raw SNP array data are available through the dbGaP-authorized access system (study accession phs000178). CCLE cell line datasets and their drug sensitivity data are available in the DepMap portal (https://depmap.org/portal/), the COSMIC cell line project (https://cancer.sanger.ac.uk/cell_lines), and Genomics of Drug Sensitivity in Cancer (https://www.cancerrxgene.org/) . The processed data and codes to reproduce the main results of this work are available on the GitHub page (https://github.com/shirotak/pancancer_hrd_analysis). Other codes for preprocessing public or restricted-access data are available from the corresponding author upon reasonable request.
Availability of data and materials
Open access data and processed data for TCGA samples are available from the cBioPortal (https://www.cbioportal.org/), the Broad GDAC (https://gdac.broadinstitute.org/), and the GDC Pan-Cancer Atlas studies (https://gdc.cancer.gov/about-data/publications/pancanatlas). Controlled access data including germline variant annotations, raw sequence data, and raw SNP array data are available through the dbGaP-authorized access system (study accession phs000178). CCLE cell line datasets and their drug sensitivity data are available in the DepMap portal (https://depmap.org/portal/), the COSMIC cell line project (https://cancer.sanger.ac.uk/cell_lines), and Genomics of Drug Sensitivity in Cancer (https://www.cancerrxgene.org/). The processed data and codes to reproduce the main results of this work are available on the GitHub page (https://github.com/shirotak/pancancer_hrd_analysis). Other codes for preprocessing public or restricted-access data are available from the corresponding author upon reasonable request.
Additional files
Additional file 1
Figure S1. Correlations among LOH, TAI, LST, and HRD scores in all cases. Figure S2. Assessment of the locus-specific LOH. Figure S3. Distribution of germline and somatic mutations with or without locus-specific LOH in HRR pathway genes excluding BRCA1/2. Figure S4. Determination of homozygous deletion. Figure S5. Correlation between promoter methylation-driven gene silencing of HRR pathway genes and genomic scar signatures. Figure S6. Differences in genomic scar scores by gender and TP53 mutation in non-BRCA-associated cancers. Figure S7. Cancer types and the number of cases assigned to GS cases and/or HA cases in the four groups divided by gender and the presence of TP53 mutation. Figure S8. Identification of TCGA OV-derived HRD signature. Figure S9. Correlations among rRPS score, OV-GS score, and KEGG score. Figure S10. Differences in X chromosome inactivation by gender and HRD status. Figure S11. Reanalysis of HRD cases without patient stratification by gender and TP53 mutation. Figure S12. Association between HRR pathway gene alterations and genomic scar scores or tumor mutation burden in CCLE cell lines. Figure S13. Identification of genomic scar high (GS) cases in CCLE cell lines.
Additional file 2
Table S1. Multivariate Cox proportional hazard analysis in the two groups with or without DNA-damaging drug use. Table S2. Number of cases analysed in TCGA by cancer type. Table S3. Drug administration information for each case. Table S4. TCGA-OV derived HRD signature. Table S5. Categorized drug information of 198 compounds in GDSC2.
References




