
Abstract
In the absence of reliable and effective prognostic biomarkers, endocrine therapy remains the standard of care for all advanced and metastatic estrogen receptor-positive (ER+) breast cancers. Attempts to develop biomarkers using the baseline tumor transcriptome or genome of advanced ER+ breast cancers have so far been unsuccessful due to predictive models with poor reproducibility in independent studies. Here we present an approach to develop a low-dimensional biomarker that estimates the risk of adverse events on endocrine therapy using the baseline tumor transcriptome of patients. Using a framework for supervised dimensionality reduction of the gene expression feature space, we constructed an endocrine response signature (ENDORSE) modeled on the survival outcomes of ER+ breast cancers from METABRIC. ENDORSE outperformed transcriptome-wide and knowledge-based signature models while significantly improving upon routine histopathological and genomic classifiers in cross-validation analyses. The ENDORSE risk estimate accurately predicted the outcomes for endocrine therapy in three independent clinical trials for ER+ breast cancers. Further, analysis of the phenotypes enriched in high-risk categories show endocrine resistance was not associated with rates of proliferation, but instead with a potential loss of DNA damage repair and cell-matrix interaction pathways.
Introduction
Estrogen receptor (ER)-positive tumors are the most common form of breast cancer 1. Endocrine therapy remains the backbone of initial therapy for metastatic ER+ breast cancers, which are now defined as tumors with 1-100% of cell nuclei stained positive for the ER 2,3. However, ER+ tumors are heterogeneous, both in terms of dependence on estrogen signaling for growth and survival and intrinsic or acquired resistance to endocrine therapy 4,5. Therefore, optimal clinical management of each ER+ breast cancer depends on accurate prediction of response to endocrine therapy and selection of companions for endocrine therapy. Several genomic tests are available for classifying breast cancers into molecular subtypes 6 or assessing the likelihood of benefit from chemotherapy in early-stage, node-negative ER+ breast cancers 7,8. However, these assays are not used in advanced or metastatic breast cancer. Therefore, the goal of our study is to develop a tumor expression-based prognostic biomarker for ER+ breast cancers on endocrine therapy, independent of the tumor stage or lymph node status.
Complex models, where the number of candidate features (genes) are much greater than the number of available samples (p >> n), can be difficult to interpret, are easy to overfit, and often overperform in training datasets with poor predictive ability in independent validations 9,10. Consequently, few biomarkers developed using the baseline tumor transcriptome are translated into clinical practice 11. To address these challenges, we developed a framework to model endocrine therapeutic response using a low-dimensional biomarker. We first systematically reduced the dimensionality of the feature space to derive a gene signature and model the survival outcomes of patients on endocrine therapy using a univariate gene set enrichment (GES) score calculated for each sample. The resulting endocrine response signature (ENDORSE) biomarker classified ER+ breast cancers into three risk categories. We demonstrate the robustness and reproducibility of ENDORSE through cross-validation analyses in the METABRIC dataset, in comparison with existing molecular classifiers PAM50 6 and IntClust 12, knowledge-based molecular signatures, and transcriptome-wide predictors. Further, we validated the performance of ENDORSE in three independent clinical datasets with advanced or metastatic ER+ breast cancers. As negative controls, we also tested ENDORSE in ER-METABRIC breast cancers and in an independent clinical trial with HER2+ breast cancers to demonstrate the specificity of the biomarker. Comparison of biological phenotypes showed that the biomarkers was not merely a proxy for proliferation while also outperforming proliferation-based signatures. Overall, our analyses demonstrate that the low dimensional ENDORSE biomarker is a reliable prognostic biomarker for advanced and metastatic ER+ breast cancers.
RESULTS
Developing a low-dimensional endocrine response signature
To develop the endocrine response biomarker, we used the baseline transcriptome from the METABRIC13,14 ER+ breast cancer cohort as the training dataset. The selection criteria for inclusion in the training data were hormone receptor status (ER+/HER2-), no treatment with chemotherapy, and cancer as the cause of death (Supplementary Table 1).
ER+ METABRIC patient characteristics
We used a LASSO-regularized Cox proportional hazards model to perform a cross-validation analysis with the survival outcomes of 833 cancers using baseline tumor expression profiles as the predictors. Genes with a positive coefficient in >50% of the cross-validations were retained to form an endocrine response gene set. Subsequently, we calculated a single sample gene set enrichment score (GES)15,16 for each sample using this gene set, and estimated the risk of death on endocrine therapy using a Cox proportional hazards model. The resulting ENDORSE risk estimates classified patients into three risk groups that predicted risk of adverse outcome (death) on endocrine therapy. Additionally, we modeled and compared the survival outcomes using the complete transcriptome and knowledge-based curated17 and hallmark molecular signatures18.
Stratifying METABRIC breast cancers based on ENDORSE scores
To demonstrate the practical utility of ENDORSE, we stratified the METABRIC ER+ breast cancers based on estimated risk of death on endocrine therapy. Cancers with an ENDORSE risk estimate or hazard ratio of ≥2 were classified into the high-risk category, cancers with an estimate ≤1 in to low-risk, and all cancers with intermediate hazard ratios in to the medium-risk category (Figure 1a). Analyses of the survival curves based on this classification shows significant differences in the survival rates (P=3.55×10−24) of cancers stratified by the ENDORSE risk estimates. In the METABRIC cohort, the majority of cancers were classified in the low or medium risk class, while only 16% of the patients were classified in high-risk class (Figure 1b).

A. Kaplan-Meier survival curves and accompanying risk table of METABRIC ER+ breast cancers stratified into low, medium and high-risk groups based on hazard ratios estimated using ENDORSE scores. B. Histogram and cumulative density function plots showing frequency distribution of samples based on estimated ENDORSE risk. C and D. A series of Kaplan-Meier survival curves showing stratification of METABRIC ER+ breast cancers based on reduced number of available genes (C) or samples (D) for calculating ENDORSE scores.
Next, to demonstrate the robustness of the ENDORSE risk estimates, we reanalyzed the Kaplan-Meier survival curves of METABRIC patients in scenarios where either genes or samples were missing from the training dataset (Figure 1c, d). We observed that even after reducing the number of genes down to 10%, the log-rank test P-values were within the same order of magnitude as the original stratification. Upon reduction of the sample size, the stratification yielded significant P-values even when sub-sampled down to 10% of the original dataset.
We further compared the performance of the ENDORSE risk estimates with clinical covariates, hormone receptor gene expression and other established breast cancer stratification models including PAM50 intrinsic subtypes6 and IntClust classes12. Univariate Cox models based on the ENDORSE scores and risk estimates outperform all other variables evaluated. Additionally, multivariate Cox-analysis with the ENDORSE signature along with hormone receptor expression, PAM50 subtypes and IntClust classes demonstrates that ENDORSE captures unique and significant proportion of the model variance over these predictors (Supplementary Tables 2-4). Thus, the ENDORSE scores are non-redundant from existing molecular classifications and augment risk stratification of ER+ breast cancers.
Multivariate ENDORSE Cox Models with PAM50 subtypes
Multivariate ENDORSE Cox Models with IntClust classes
Multivariate ENDORSE Cox Models with HR gene expression
Validating ENDORSE performance in independent clinical datasets
To demonstrate the reliability and reproducibility of the ENDORSE risk estimate guided stratification, we applied the Cox model coefficients obtained from the METABRIC models to ER+ breast cancers in three independent clinical trial datasets. The first independent clinical trial reported endocrine therapy treatment outcomes of 140 stage IV ER+ metastatic breast cancers, 78% of which had received prior endocrine therapy19. We calculated the ENDORSE gene set enrichment scores from the transcriptomic data obtained from the metastases and stratified the cancers based on METABRIC-derived ENDORSE coefficients (Figure 2a). Analysis of the survival curves indicates accurate stratification of the cancers based on the risk estimates (P = 3×10−4).

A. Kaplan-Meier survival curves of ER+ breast cancer metastases stratified based on ENDORSE risk estimates, along with survival risk table. B and C. Right panels show violin plots for ER+ breast cancers stratified based on ENDORSE risk estimates (X-axis) and Ki67 % (Y-axis). The dotted line indicates a Ki67 staining level of 10%, a threshold used in both studies to classify cancers as sensitive or resistant to therapy. The scatter plots in the left panel show correlation between continuous ENDORSE risk estimates (X-axis) and Ki67% staining (Y-axis). Liner fit along with 95% confidence intervals are also shown. D. Violin plots comparing ENDORSE risk scores in patients stratified based on trial-reported clinical response. The left panel represents ER+/HER2+ cancers while the right panel represents ER-/HER2+ cancers. E. Kaplan-Meier curves of ER-METABRIC breast cancers stratified based on ENDORSE risk estimates.
Next, we examined data from the NCT00265759 clinical trial which evaluated neoadjuvant aromatase inhibitor (AI) treatment in Stage II or III ER+ breast cancers 20. This study used 10% Ki67 staining at the end of treatment (2-4 weeks) as the threshold for sensitive or resistant cancers. We found that the ENDORSE risk estimates successfully stratified the cancers in agreement with reported Ki67 % at both baseline (P=2.7×10−8) and end of treatment (P=5.6×10−3) (Figure 2b). The third independent clinical trial evaluated fulvestrant response in advanced metastatic ER+ breast cancers previously treated with an antiestrogen (CONFIRM study)21. This study classified tumors as resistant or sensitive based on a 10% Ki67 staining threshold. Again, the stratification of these cancers based on ENDORSE risk estimates showed clear differences in Ki67% across the risk groups (P = 1.6×10−9), in addition to significant correlation with the continuous risk score (P = 2.5×10−11) (Figure 2c). Similar consistent patterns were observed with the continuous risk scores. In both the CONFIRM and NCT00265759 trial, patients stratified in low risk groups were consistently classified as sensitive based on the trial outcome data, while high risk tumors were classified as resistant (Figure 2b, c).
In addition to the endocrine therapy trials in ER+ breast cancer, we also applied the ENDORSE risk estimates to stratify ER-breast cancers or ER+ breast cancers on non-endocrine therapy as negative controls. First, we compared ENDORSE risk estimates in a trial for neoadjuvant trastuzumab and lapatinib in ER+/HER2+ or ER-/HER2+ breast cancers. In both molecular subtypes, there was no significant difference in ENDORSE risk estimates between the partial clinical response and residual disease groups (ER+ P = 0.465, ER-P = 0.824) (Figure 2d). Similarly, stratification of the ER-cancers based on the estimated ENDORSE risk resulted in no significant differences between the survival curves (P = 0.4) (Figure 2e). These results suggest the ENDORSE risk estimate is not a general biomarker for aggressive breast cancers, but specific to ER+ breast cancers on endocrine therapy.
Understanding the biology of high-risk cancers
To gain insights into the biology of high-risk cancers, we compared the gene set enrichment scores, gene-level mutation and copy number alteration frequencies across the METABRIC ENDORSE risk strata. We found that multiple pathways linked with functional loss of P53 and RB were elevated, and among the most significant pathways in the high-risk cancers (Figure 3a, Supplementary Figure 2). Concurrently, DNA-damage repair and cell cycle pathways that are closely associated with p53 and Rb loss were also enriched (Figure 3a). Among gene signatures that were enriched in the low/medium risk strata, we predominantly found pathways and complexes associated with extracellular matrix interaction, including integrin, laminin, hemidesmosome and basement membranes (Figure 3b, Supplementary Figure 1). To demonstrate that the ENDORSE risk strata were not solely driven by a proliferative signal22, we calculated the meta-PCNA proliferation index and analyzed the multivariate Cox models containing the proliferation index and the ENDORSE scores or risk groups as co-variates (Supplementary Tables 5-6). This analysis reinforces the notion that the ENDORSE scores capture information beyond mere proliferation.

{kind=link}
{kind=link}
{kind=link}
A-C. Violin plots comparing the single sample gene set enrichment scores of various pathways (Y-axis) in METABRIC tumors stratified by estimated ENDORSE risk. The low and medium risk tumors were combined in one category for comparison. A. Representative signatures for p53 loss, Rb loss, DNA damage repair and cell cycle. B. Tumor-extra cellular matrix interaction pathways. C. Bar plots representing gene-level mutation frequencies of various cancer-associated genes in low/medium vs. high-risk METABRIC tumors. The p-values from Chi-square test are shown above the bars, with bold letters indicating comparison significant at an FDR > 0.05 threshold.
Multivariate ENDORSE Cox Models with meta-PCNA
Multivariate ENDORSE Cox Models with meta-PCNA
We next analyzed the somatic mutation and copy number profiles of the METABRIC cancers. None of the copy number gain or losses were enriched in the high-risk tumors compared to low/medium risk tumors. However, the somatic mutation frequencies for a few cancer-associated genes were significant at the nominal threshold (Figure 3c, Supplementary Table 7). However, only two genes, TP53 (P = 6.6 x 10−5) and AKAP (P = 3.3 x 10−4), were significant at an FDR < 0.05 threshold. The functional impact of p53 mutation frequency in high-risk tumors was corroborated by the enrichment of p53 loss of function-linked gene signatures (Figure 3a, Supplementary Figure 2). Tumor sequencing efforts have shown high frequency of AKAP mutations in metastatic lesions compared to primary tumors 23, corroborating the observed enrichment in high-risk tumors.
Somatic mutation frequency
Discussion
Gene expression biomarkers have been successfully integrated in clinical practice for prognosis and guiding treatment decisions in early-stage breast cancers, as evident from the success of the MINDACT and TAILORx studies8,24. However, gene expression biomarkers to guide treatment decisions in metastatic ER-positive breast cancer, either in the first-line or later line settings, are lacking. Thus, biomarkers for predicting optimal treatment of metastatic ER-positive breast cancers, particularly after progression on first-line aromatase inhibitor/CDK4/6 inhibitor combinations, could have a large impact on people with ER-positive breast cancer.
The development of new biomarkers based on patient transcriptome data is challenging. Since the number of available genes to train the statistical models tend to be much larger than the number of available samples (p >> n), it is quite easy to create complex prediction models that contain a large number of predictor variables. Often, such models perform very well in the training datasets, but the performance cannot be replicated in independent test datasets due to overfitting. A number of approaches have been proposed to address this issue. Broadly, these can be classified into unsupervised and supervised approaches. The unsupervised approach typically relies on grouping or clustering the samples into based on similarity of gene expression profiles, followed by analysis of association with survival outcomes 25. Alternatively, a supervised approach is to perform dimensionality reduction prior to modelling the survival outcome or drug response using univariate or multivariate models 26.
In this study, we developed a biomarker for the prognosis of ER+ breast cancers using METABRIC data. We used a LASSO regularized Cox model for feature selection, effectively reducing the dimensionality of the gene expression data. In addition, we adapted a pathway signature approach in our framework, which further reduced the number of predictors down to a single integrated variable as the final biomarker. We evaluated the reliability and robustness of our biomarker through cross-validation analyses and simulations in the METABRIC ER+ cohort. Our analyses revealed that the univariate ENDORSE biomarker consistently outperformed multivariate models based on gene expression or literature-derived gene signatures (Figure 2). Furthermore, we found that reducing the number of available genes to calculate the ENDORSE risk estimates had a minimal impact on the ability of the biomarker to stratify METABRIC ER+ cancers. This distinct advantage of using the gene set enrichment scores (GES) over individual genes or pathway predictors can be explained by the algorithm for GES calculation. The ssGSEA method proposed by Barbie et al. replaces gene expression by their ranks, followed by calculating the differences in the empirical distribution functions of the signature genes vs. all other genes 16. The rank-based method helps mitigate issues encountered due to batch effects and differences in methods for transcriptome profiling, while the redundancy in the signature ensures the scores remain consistent even in the case some genes are missing in the data. Consequently, the ENDORSE biomarker was successful in stratifying ER+ breast cancers in multiple independent validation datasets from diverse gene expression profiling platforms.
The ENDORSE biomarker stratified cancers based on estimated risk of death due to the disease while on endocrine therapy. In addition to testing its potential to serve as a robust biomarker, we explored the biology of the high-risk tumors for possible hints into their mechanism. We found that high-risk tumors showed a consistent enrichment of pathways associated with loss of p53 and Rb, along with DNA damage repair and cell cycle progression. Mutations in the TP53 have long been associated with aggressiveness and chemotherapeutic resistance in hormone-receptor negative breast cancers 27,28. However, recent studies show that even though TP53 are infrequent in ER+ breast cancers, they have similar negative impact on patient outcome as hormone-receptor negative breast cancers 29. Similarly, inactivation of the tumor suppressor Rb has been associated with therapeutic resistance in ER+ breast cancers 30,31. A previous meta-analysis of gene signatures by Venet et al. associated with breast cancer outcomes showed that most signatures were redundant with proliferation 22. While proliferation is indeed an important feature of aggressive tumors, our analyses showed that proliferation signature did not encompass the entirety of the signal from the ENDORSE biomarker.
To determine the clinical utility and clinical validity of a biomarker, it must be tested in a clinical trial. According to the National Comprehensive Cancer Network (NCCN), options for metastatic ER-positive, HER2-negative breast cancer after progression on aromatase inhibitor plus CDK4/6 inhibitor include fulvestrant (which requires the breast cancer to still be estrogen dependent) with or withour alpelisib, exemestane (which also requires the breast cancer to still be estrogen dependent) plus everolimus, or chemotherapy 32. We will be testing the ENDORSE biomarker in a clinical trial to assign patients to the regimens targeting estrogen vs chemotherapy.
Funding
Funding for this research was provided by the National Cancer Institute of the National Institutes of Health through the U54 grant 1U54CA209978.
METHODS
Data retrieval, pre-processing and analysis
METABRIC gene expression, phenotypic and survival data were retrieved using cBioPortal for cancer genomics 33. Independent validation datasets used in this study were retrieved from the NCBI GEO portal under the following accession IDs: GSE12464719, GSE8741120, GSE7604021 and GSE130788. For each gene expression dataset, we removed genes with zero variance and summarized genes with multiple probes by mean expression. Each dataset was scaled, such that the mean of each gene across the samples was zero with standard deviation equal to 1. The analyses were performed in R 3.6.1, RStudio 1.2.1335.
Selecting samples for training models
The METABRIC cohort contained a total of 2509 samples. We constructed the training models for ER+ cancers and endocrine therapy response by filtering the samples on the following criteria: 1. The tumors were positive for estrogen receptor and negative for human epidermal growth factor receptor 2 (ER+ and HER2-) in their immunohistochemistry profile, 2. If the patient died, then disease was listed as cause of death, and 3. The patient did not receive chemotherapy alone or in combination with endocrine therapy. After filtering based on these criteria, we retained 833 samples with both gene expression and complete clinical data available.
Training features and model construction
We derived and compared four sets of features as potential predictors of long-term outcome of ER+ METABRIC tumors, including curated gene set and hallmark gene set enrichment scores, expression profiles of all genes and the ENDORSE gene signature (Figure 1). The curated gene sets and hallmark gene sets (v7.0) were retrieved from MSigDb17,18. For each gene signature, the gene set enrichment scores were calculated using the GSVA package for R15 using the ssGSEA method16. Next, the gene set enrichment scores (GES) or expression profiles of all genes, along with age at diagnosis, were used as input features in LASSO-regularized Cox regression models, with overall survival as the outcome variable34. The hazard function in the Cox model is defined as:
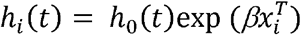 Where, X is a set of predictive features and h0 is an arbitrary baseline hazard function. We considered curated GES, hallmark GES and all genes each as individual sets of predictive features in separate analyses. The coefficient (β) for each predictor in the model can be estimated by maximizing the partial likelihood function L(β), defined as:
Where, X is a set of predictive features and h0 is an arbitrary baseline hazard function. We considered curated GES, hallmark GES and all genes each as individual sets of predictive features in separate analyses. The coefficient (β) for each predictor in the model can be estimated by maximizing the partial likelihood function L(β), defined as:
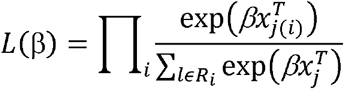 Where Ri is the set of indices of observations failing (events) at time ti. In the LASSO Cox model, the regularized coefficient is obtained by adding a penalty parameter λ to the log of the likelihood function.
Where Ri is the set of indices of observations failing (events) at time ti. In the LASSO Cox model, the regularized coefficient is obtained by adding a penalty parameter λ to the log of the likelihood function.
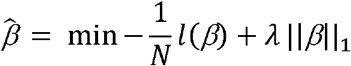 Where, l(β) = log L(β). The λ penalty parameter was determined using 10-fold cross-validation implemented in R package glmnet 36,37. The optimal λ for the curated or hallmark GES and gene expression feature sets were defined as the λ that minimized model deviance for each feature set. We defined the preliminary endocrine resistance gene set (ENDORSE) using the features with positive coefficients in the LASSO model for all genes, with optimal λ within one standard error from the minimum model deviance. We further expanded the signature by including genes that were positively correlated (Pearson’s correlation > 0.75) with the selected features in the ER+ METABRIC samples. Next, we calculate the GES for each sample using GSVA and use the GES as a predictive feature.
Where, l(β) = log L(β). The λ penalty parameter was determined using 10-fold cross-validation implemented in R package glmnet 36,37. The optimal λ for the curated or hallmark GES and gene expression feature sets were defined as the λ that minimized model deviance for each feature set. We defined the preliminary endocrine resistance gene set (ENDORSE) using the features with positive coefficients in the LASSO model for all genes, with optimal λ within one standard error from the minimum model deviance. We further expanded the signature by including genes that were positively correlated (Pearson’s correlation > 0.75) with the selected features in the ER+ METABRIC samples. Next, we calculate the GES for each sample using GSVA and use the GES as a predictive feature.
Repeated cross-validation, consensus ENDORSE signature and performance evaluation
To evaluate the performance of each set of predictive features and derive a consensus ENDORSE model, we performed 10-fold cross-validation analysis repeated 50 times. In this analysis, the features selected in the LASSO model from each set of predictors were evaluated in multivariate Cox model. In each of the 50 repeats, the samples were split into 10 equal parts, with one part serving as the test set and the remaining parts serving as the training set in each fold of cross-validation. In each fold, the feature selection was performed for curated, hallmark or ENDORSE GES and gene expression only using the training set. Then, the Cox model coefficients were derived for each set of selected features using only the training set. To evaluate performance, we applied the coefficients from the training model to the GES or gene expression data from the test data. We reported the concordance indices of the Cox model fit in the training and test data. In addition, we reported the correlation between the actual risk of event in the test data and the predict risk of event based on the coefficients derived from the training data and applied to the test data.
The consensus ENDORSE set was defined as the set of predictive features that were selected in more 50% of the repeated cross-validations. This resulted in the selection of 63 genes that comprised the consensus ENDORSE set for subsequent analysis involving risk or hazard ratio (HR) estimation and predictions in independent datasets. After the consensus ENDORSE set was defined, we obtained ENDORSE GES and Cox model coefficient for the complete ER+ METABRIC cohort.
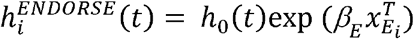 Where, XE is the set of ENDORSE GES and βE are the associated coefficients. We then stratified the tumors based on ENDORSE risk estimate by defining cancers with HR ≤ 1 as low-risk, HR ≥ 2 as high-risk, and those with HR between 1 and 2 as medium risk.
Where, XE is the set of ENDORSE GES and βE are the associated coefficients. We then stratified the tumors based on ENDORSE risk estimate by defining cancers with HR ≤ 1 as low-risk, HR ≥ 2 as high-risk, and those with HR between 1 and 2 as medium risk.
METABRIC survival models and simulations
The ENDORSE model was evaluated in the METABRIC cohort by stratifying the ER+ cancers based on estimated risk in the low, medium or high-risk categories. For comparisons with clinical variables and other breast cancer classification approaches, including PAM50 and IntClust, we reported the HR and p-values of the Cox model coefficients for these predictors in univariate analyses. In addition, we reported the results of multivariate analyses that included the PAM50 or IntClust classes along with the ENDORSE risk estimates.
We analyzed the survival curves of METABRIC ER+ tumors stratified based on ENDORSE risk estimates using the log-rank test. To simulate the effects of random dropouts or missing data, we sequentially reduced the total number of available genes to calculate the ENDORSE GES and recalculated the risk estimate. Here, we randomly sampled and removed 10% of the genes in each iteration until 90% of the genes were removed. In each iteration, we calculated the ability of the recalculated risk estimate to stratify the cancers based on the difference in survival curves using the log-rank test. Similarly, we simulated the impact of reduced sample set by sequentially reducing the number of samples available for recalculating the ENDORSE scores by 10% in each iteration and analyzed the difference in survival curves using the log-rank test.
Validation in independent clinical trial datasets
We evaluated the performance and ability of ENDORSE to predict treatment response in a set of three independent clinical trials. These trials were selected based on criteria that the trial cohort included any stage of ER+/HER2-breast cancers receiving endocrine therapy but did not receive chemotherapy. In addition, the trials should have reported a clinical assessment of treatment response or over survival outcomes, in addition to providing gene expression datasets. We found three datasets the met the above criteria (GSE12647, GSE87411 and GSE76040). The first trial (GSE12647) reported survival outcomes of 140 metastatic breast cancers on endocrine therapy. For this dataset, we predicted the estimated risk of event based on the ENDORSE coefficients derived from the METABRIC ER+ model. First, we calculated the ENDORSE GES for the test samples using the baseline expression of the tumors. Next, we predicted the estimate risk of event using the METABRIC-derived coefficients. We then evaluated the difference in survival curves for the different strata using the log-rank test.
For the two subsequent datasets (GSE87411 and GSE76040), we again predict the estimated risk based on ENDORSE GES from the baseline gene expression data. The NCT00265759 trial evaluated neoadjuvant aromatase inhibitor response in a cohort of 109 stage II-III ER+ breast cancers. This trial provided Ki67 data at both baseline and end of treatment and classified tumor with Ki67 > 10% at end of treatment as non-responders. The CONFIRM trial evaluated 113 metastatic ER+ cancers on fulvestrant, and also reported Ki6 staining data at end of treatment.
Since both trials reported Ki67 as a determinant for treatment response, we compared the Ki67 % across the ENDORSE risk strata or continuous risk scores as measure of performance evaluation.
In addition, we also evaluated the ENDORSE risk estimates in two negative control datasets. The first negative control dataset (GSE130788) evaluated trastuzumab and lapatinib response in 62 ER+/HER+ or 48 ER-/HER+ breast cancers. The trial reported clinical response (partial clinical response or residual disease) at the end of treatment. We predicted the estimated ENDORSE risk scores using baseline gene expression and compared across the clinical response classes. As an additional negative control, we also predicted and stratified the 429 ER-breast cancers in METABRIC based on ENDORSE estimates. Then, we compared the difference in survival curves using log-rank test.
Biological features associated with high-risk cancers
We assessed the features that may be important in explaining the biological difference between high-risk cancers compared to low/medium risk cancers by comparing the curated and hallmark enrichment scores, somatic mutation and copy number frequencies across the ER+ METABRIC. For the comparison of GES, we performed t-tests for each signature and adjusted the p-values using false discovery rate (FDR) or Benjamini-Hochberg method. In addition, we calculated the effect size of the difference in means using Cohen’s D method. An absolute Cohen’s D > 0.8 was considered as large effect.
The somatic mutations for the METABRIC ER+ cohort were summarized at the gene-level by first removing all synonymous variants as non-consequential, then binarizing the gene by mutation matrix based on presence of a mutation. Next, we filtered out genes with a sample mutation frequency < 0.05. We then constructed a 2 x 2 contingency matrix for each gene and the METABRIC ER+ tumor stratified based on low/medium risk or high risk. We calculated the p-values for enrichment using the Chi-square test, followed by FDR adjustment. Similar to the mutation analysis, we performed the Chi-square test on binary matrices of tumor gain or loss across all genes, followed by FDR adjustment.
To compare the information captured by the ENDORSE scores with the proliferative meta-PCNA signature 22, we calculated the proliferation index of the METABRIC ER+ tumors using the R-package ProliferationIndex 37. The package calculates the median expression of the meta-PCNA genes, as described by Venet et al. 22. Next, we performed multivariate Cox analysis of the ER+ tumors with the proliferation index and either ENDORSE risk groups or continuous risk estimates as covariates.
Data Availability
All data used in this manuscript are publicly available and listed under "Methods" section of the manuscript
Data availability and code
All training and validation datasets used in this study are publicly available and listed under “data retrieval, preprocessing and analysis”. The sample code for reproducing the analyses in this study are available at https://osf.io/bd3m7/?view_only=da4f860bd2474745880944fce1d433b1
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References




