
Abstract
Abstract Large-scale deployment of safe and durably effective vaccines can halt the COVID-19 pandemic.1–3 However, the high vaccine efficacy reported by ongoing phase 3 placebo-controlled clinical trials is based on a median follow-up time of only about two months4–5 and thus does not pertain to long-term efficacy. To evaluate the duration of protection while allowing trial participants timely access to efficacious vaccine, investigators can sequentially cross placebo recipients to the vaccine arm according to priority groups. Here, we show how to estimate potentially time-varying placebo-controlled vaccine efficacy in this type of staggered vaccination of placebo recipients. In addition, we compare the performance of blinded and unblinded crossover designs in estimating long-term vaccine efficacy.
Summary We show how to estimate potentially waning long-term efficacy of COVID-19 vaccines in randomized, placebo-controlled clinical trials with staggered enrollment of participants and sequential crossover of placebo recipients.
Introduction
A number of studies have been conducted around the world to evaluate the efficacy and safety of investigational vaccines against novel coronavirus disease-2019 (COVID-19).4–12 Interim results from several large-scale phase 3 randomized, observer-blinded, placebo-controlled clinical trials have demonstrated high vaccine efficacy,4–6 far exceeding the FDA and WHO thresholds of 50% reduction of symptomatic disease.13–14 However, those trials have been underway for only several months, and the data they have collected thus far can only speak to short-term vaccine efficacy. For example, the interim results for the Pfizer/BioNTech vaccine BNT162b2 and the Moderna vaccine mRNA-1273 that were presented for regulatory approval are based on a median follow-up time of approximately two months after the second dose;4–5 therefore, the reported 94-95% efficacy pertains only to an average of two months post vaccination.
Vaccine efficacy can wane over time because of declining immunologic memory or changing antigenicity of the pathogen. Thus, it is critically important to determine the duration of protection and the need for additional booster doses of vaccine. Certainly, after FDA issues Emergency Use Authorization (EUA), vaccine sponsors should continue to collect placebo-controlled data in any ongoing trials for as long as feasible.15
Although continuing blinded follow-up of the original treatment arms offers the ideal opportunity to evaluate long-term vaccine efficacy and safety, placebo recipients should be offered the vaccine at some point after an EUA. One strategy is the “rolling crossover”, which vaccinates placebo recipients around the same time as general population members in the same priority tier. Under this design, placebo participants are vaccinated at different times, with the timing of vaccination depending on enrollment characteristics that define their priority tier.
In this article, we show how to evaluate long-term vaccine efficacy under the rolling crossover design. Because of staggered enrollment, existing statistical methods do not produce valid estimates of time-varying vaccine efficacy even if the placebo group is maintained throughout the study. Our approach properly accounts for staggered enrollment and change of background incidence over time, as well as for the fact that higher-risk placebo volunteers may get vaccinated earlier than lower-risk ones during the crossover period. It provides unbiased estimation of the entire curve of placebo-controlled vaccine efficacy as a function of time elapsed since vaccination, up to the point where most of the placebo recipients have crossed over to the vaccine arm. We investigate the bias and precision in estimating long-term vaccine efficacy under various blinded and unblinded crossover designs and discuss how to perform sensitivity analysis when unblinded follow-up data are used.
Methods
Figure 1 provides a schematic illustration of the rolling crossover strategy in the context of the two mRNA vaccine trials. In this scenario, participants were screened and randomly assigned to vaccine or placebo over a 4-month period, and the vaccine was approved in the 5th month on the basis of interim results. Under the proposed design, crossover based on priority groups will occur over the next five months.

The endpoint of interest is time to symptomatic COVID-19 disease. We allow the background risk of disease to vary over the calendar time and to depend on baseline risk factors, such as age, gender, ethnicity, race, occupation, and underlying health conditions. In addition, we allow the relative risk of disease for vaccine versus placebo to depend on the duration elapsed since vaccination. We define the τ -day vaccine efficacy as the percentage reduction in the cumulative incidence of disease over the τ -day period for those who were vaccinated at the start of the τ -day period versus those who have not been vaccinated by day τ.
In Supplemental Appendix 1, we formulate the above framework through an adaptation of the popular Cox proportional hazards model16–17 and derive the maximum likelihood estimator for the curve of placebo-controlled vaccine efficacy as a function of time elapsed since vaccination. In addition, we show that the estimator is approximately unbiased and normally distributed, with a variance that can be estimated analytically, making it possible to construct valid confidence intervals for the vaccine-efficacy curve.
Results
We conducted a series of simulation studies mimicking the BNT162b2 vaccine trial. We considered 40,000 participants, who entered the trial at a constant rate over a 4-month period and were randomly assigned to vaccine or placebo in a 1:1 ratio. The vaccine received an EUA from FDA at the 5th month, by which time there were about 300 COVID-19 cases in the placebo group. To reflect the increase of COVID-19 cases since last summer and the expected downward trend in the spring due to herd immunity, we let the disease risk increase over the first 7 months and decrease afterward. We set the vaccine efficacy at 5 months to be 95% and the vaccine efficacy at 10 months to be 95%, 50%, or 0%.
We considered the statistically optimal design of keeping all participants on their original treatment assignments until the end of the trial, which was set to be slightly over 10 months. We refer to this design as Plan A, which serves as a benchmark. We also considered three blinded crossover designs:
B. Crossover occurs over months 6-10 according to priority tier groups.
C. 20% of participants follow Plan A, and the rest follow Plan B.
D. Crossover occurs for all participants in month 6.
Both B and C are priority-tier dependent rolling crossover designs. The difference is that under Plan B, all placebo recipients cross over to the vaccine arm, whereas under Plan C, 20% of participants choose for altruistic reasons to stay on their original treatment assignments. For Plan D, all placebo recipients are vaccinated over the course of a month without any priority tiering. With blinded crossover, placebo participants receive the vaccine and vaccine participants receive the placebo at the point of crossover, and all participants are followed until the end of the trial, which is 10.5 months since trial initiation. The designs of these simulation studies are detailed in Supplemental Appendix 2.
The results, based on 10,000 simulated datasets, are summarized in Table 1 and Figure 2. The proposed method yields virtually unbiased estimates of the vaccine-efficacy curves over the 10-month period for Plans A–C in all three scenarios (95%, 50%, 0%) of long-term vaccine efficacy; it also yields accurate variance estimates, such that the confidence intervals have proper coverage probabilities. When vaccine efficacy is constant over time, the standard errors for the estimates of vaccine efficacy under Plans B and C are slightly lower than those of Plan A. When vaccine efficacy wanes over time, the standard errors for the estimates of 5-month vaccine efficacy under Plans B and C are also slightly lower than those of Plan A; however, the standard errors for the estimates of 10-month vaccine efficacy under Plans B and C are higher than those of Plan A, with the standard errors being slightly lower under Plan C than under Plan B. Under Plan D, the estimates of 10-month vaccine efficacy are slightly biased, with high standard errors; these results are not surprising, because under this plan, the number of unvaccinated participants diminishes rapidly after month 6.
Estimation of Vaccine Efficacy (VE) by the Proposed and Standard Methods Under No Crossover (A) and Three Blinded Crossover Plans (B–D) in Three Scenarios of Long-Term Vaccine Efficacy

We also evaluated the performance of the standard Cox regression,16–17 with vaccine status as a potentially time-dependent covariate. Because it estimates an overall vaccine efficacy among individuals who have been vaccinated for different amounts of time, this method over-estimates long-term vaccine efficacy when vaccine efficacy decreases over time, although the estimation is unbiased when vaccine efficacy is constant over time.
We conducted a second series of simulation studies by considering unblinded crossover, under which participants are notified of their original treatment assignments and placebo recipients are vaccinated soon after. In this series, crossover occurs at the same time as in Plans B–D but is unblinded; we refer to these three plans as B’–D’ (Supplementary Appendix 2). Because vaccine recipients may engage in riskier behavior upon unblinding, we discarded the data collected after unblinding for both the vaccine and placebo groups by censoring each participant’s time to disease at their time of unblinding visit; the resulting analysis avoids any bias due to behavioral confounding.
The results for the second series of simulation studies, based on 10,000 simulated datasets, are summarized in Table 2 and Figure 3. The proposed method yields unbiased estimates of the vaccine-efficacy curves under Plans B’ and C’, but with much higher standard errors for estimates of long-term vaccine efficacy than under Plans B and C; the standard errors can be accurately estimated, although only up to month 10. Under Plan D’, the estimates of 5-month vaccine efficacy are unbiased; however, the estimates of 10-month vaccine efficacy are biased, especially when vaccine efficacy wanes over time. Naturally, long-term efficacy cannot be estimated as well under unblinded crossover as under blinded crossover because of limited blinded follow-up data, especially under Plan D’. Of note, the bias of the standard Cox regression in estimating waning vaccine efficacy is more severe under unblinded crossover than under blinded crossover.
Estimation of Vaccine Efficacy (VE) by the Proposed and Standard Methods Under Three Unblinded Crossover Plans (B’–D’) in Three Scenarios of Long-Term Vaccine Efficacy

Figure 4 displays the analysis results produced by the proposed methods for one of the trials simulated with 50% 10-month vaccine efficacy. The estimates of vaccine efficacy are close to the truth, and the 95% confidence intervals cover the truth mostly — up to the end of blinded follow-up. In terms of estimating long-term vaccine efficacy, Plan C is nearly as good as Plan A and is slightly better than Plan B, which is better than Plan D; Plans B, C, and D are substantially better than Plans B’, C’, and D’, respectively.

Discussion
For a preventive COVID-19 vaccine to be administered to millions of people, including healthy individuals, its safety and efficacy must be demonstrated in a clear and compelling manner. Although preliminary results from ongoing phase 3 clinical trials have revealed higher than expected efficacy of COVID-19 vaccines,4–6 additional follow-up is required to assess long-term efficacy and safety. Indeed, FDA does not consider issuance of an EUA, in and of itself, as grounds for stopping blinded follow-up in an ongoing clinical trial.15
We recommend the rolling crossover design, which allows placebo recipients to be vaccinated in a timely manner while still making it possible to assess long-term safety and efficacy. The standard Cox regression seriously over-estimates long-term vaccine efficacy when vaccine efficacy wanes over time, as shown in our simulation studies. We have developed a valid and efficient approach to evaluate the potential waning efficacy of a COVID-19 vaccine. The estimated curve of time-varying vaccine efficacy can be used to determine when a booster vaccination is needed to sustain protection; this information is also an important input parameter in mathematical modeling of the population impact of COVID-19 vaccines.
To ensure high-quality follow-up data, crossover should ideally be blinded, with placebo participants receiving vaccine and vaccine participants receiving placebo at the time of crossover. Unblinded crossover has practical benefits over blinded crossover: it reduces operational complexity and trial cost. However, unblinding can lead to differential exposure to SARS-CoV-2 between the original vaccinees and the placebo crossovers, which in turn can bias the estimation of vaccine efficacy.
This bias can be avoided by analyzing only the blinded follow-up data. However, discarding the unblinded follow-up data may substantially reduce the precision in estimating long-term vaccine efficacy. We suggest to estimate vaccine efficacy in two ways, one using all follow-up data and one using only blinded follow-up data, and compare the two results.
Another solution is to apply our methods to all follow-up data but perform a sensitivity analysis to assess the robustness of the results to potential unmeasured confounding caused by unblinding of trial participants. In Supplemental Appendix 3, we show how to apply a best-practice general methodology in epidemiological research18–19 to perform this sensitivity analysis. Specifically, we can assess how strong unmeasured confounding due to unblinding would need to be in order to fully explain away the observed vaccine efficacy. We can also provide a conservative estimate of vaccine efficacy that accounts for unmeasured confounding.
Recently, Follmann et al 20 advocated blinded crossover and continued follow-up of trial participants to assess vaccine durability and potential delayed enhancement of disease. They estimated an overall vaccine efficacy for the original vaccine arm in the post-crossover period (after all placebo recipients have been vaccinated) by imputing the case count for a counter-factual placebo group under several assumptions: that the vaccine and placebo groups remain balanced at the point of cross-over; that the crossover pattern and duration of follow-up in the second period are comparable to the enrollment pattern and duration of follow-up in the first period; and that the benefit of vaccination over time has the same profile between the original vaccinees and the placebo crossovers. We address the issue of long-term vaccine efficacy in a different way, using observed data to estimate the entire curve of vaccine efficacy as a function of time elapsed since vaccination, up to the point where most participants have been vaccinated. Our approach requires minimal assumptions and is applicable to both blinded and unblinded crossover plans, with any length of additional follow-up.
Data Availability
N/A
Authors’ Information
Dan-Yu Lin, Ph.D., is Dennis Gillings Distinguished Professor of Biostatistics, and Donglin Zeng, Ph.D., is Professor of Biostatistics, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, NC 27599-7420, USA. Peter B. Gilbert, Ph.D., is Member, Vaccine and Infectious Disease Division, Fred Hutch, Seattle, WA 98109-1024, USA.
Supplemental Appendix
1. Estimation of Vaccine Efficacy
Let T and S denote the time to symptomatic COVID-19 and the time to vaccination, respectively; both times are measured in days from the start of the clinical trial. In addition, let X denote baseline risk factors (e.g., age, occupation, comorbidities). We specify that the hazard function of T is related to S and X through a Cox proportional hazards model with a time-dependent regression coefficient
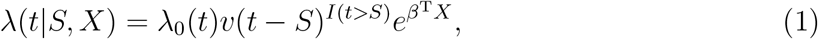 where λ0(·) is an arbitrary baseline hazard function, v(·) is a positive function characterizing the time-varying effect of vaccination, β is a set of regression parameters representing the effects of baseline risk factors, and I(·) is the indicator function. Under this formulation, the effect of vaccine on the risk of disease depends on the time elapsed since vaccination, but not on the specific date of vaccination or on the baseline risk factors.
where λ0(·) is an arbitrary baseline hazard function, v(·) is a positive function characterizing the time-varying effect of vaccination, β is a set of regression parameters representing the effects of baseline risk factors, and I(·) is the indicator function. Under this formulation, the effect of vaccine on the risk of disease depends on the time elapsed since vaccination, but not on the specific date of vaccination or on the baseline risk factors.
One may define the t-day vaccine efficacy (VE) as the proportionate reduction in the cumulative incidence of disease at time t for individuals who have been vaccinated for t days:
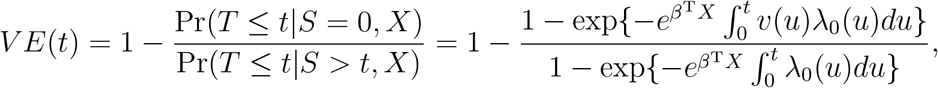 which is approximately
which is approximately
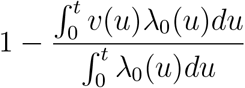 for rare disease. If λ0(·) is approximately constant, then
for rare disease. If λ0(·) is approximately constant, then
 where
where
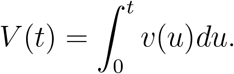
For simplicity, we define the time-varying vaccine efficacy by equation (2).
Suppose that the clinical trial enrolls a total of n participants. For i = 1, …, n, let Ri, Ti, Si, Ci, and Xi denote the entry time, the time to symptomatic COVID-19, the time to vaccination, the time to loss to follow-up, and the baseline risk factors for the ith participant. The data consist of (Ri, Yi, Δi, Di, DiSi, Xi) (i = 1, …, n), where Yi = min(Ti, Ci), Δi = I(Ti ≤ Ci), and Di = I(Si ≤ Yi).
We assume that (Ri, Si, Ci) are independent of Ti conditional on Xi. The likelihood takes the form
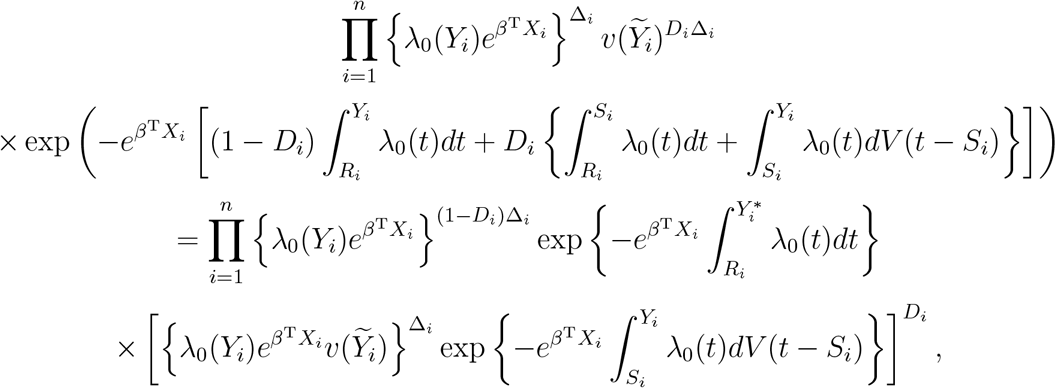 where
where 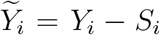 and
and 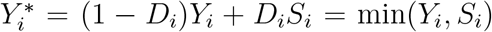 We approximate log λ0(t) through splines with m basis functions, B1(t), …, Bm(t), such that log
We approximate log λ0(t) through splines with m basis functions, B1(t), …, Bm(t), such that log 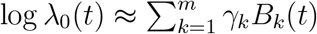
Let θ = (β, γ1, …, γm)T, and Zi(t) = [Xi, B1(t), …, Bm(t)]T. We perform the nonparametric maximum likelihood estimation,1 in which V (·) is treated as a step function jumping at the time points 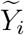 with Di = Δi = 1. Thus, we maximize the objective function
with Di = Δi = 1. Thus, we maximize the objective function
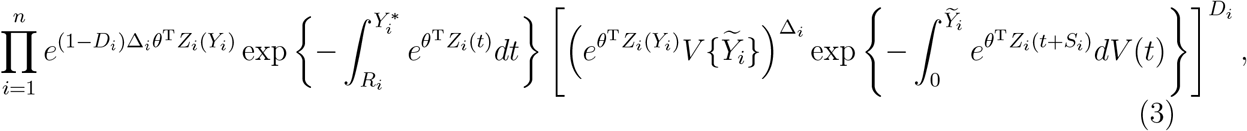 where V {t} is the jump size of V (·) at t.
where V {t} is the jump size of V (·) at t.
We first maximize the objective function in (3) for fixed θ to yield

Here and in the sequel, 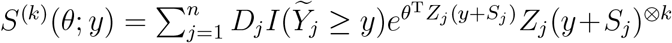 , where a⊗0 = 1, a ⊗1 = 1 and a ⊗2 = aaT for a column vector a. After plugging (4) into (3), we obtain the profile likelihood for θ. Differentiating the profile log-likelihood with respect to θ yields the estimating function
, where a⊗0 = 1, a ⊗1 = 1 and a ⊗2 = aaT for a column vector a. After plugging (4) into (3), we obtain the profile likelihood for θ. Differentiating the profile log-likelihood with respect to θ yields the estimating function
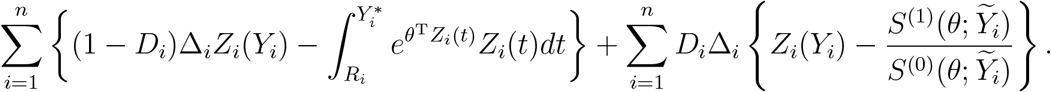
Denote the resulting estimator of 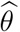 by θ. Replacing θ in (4) by
by θ. Replacing θ in (4) by 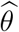 yields the estimator of V (t)
yields the estimator of V (t)
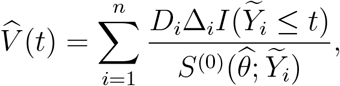 which is reminiscent of the Breslow estimator2 for the cumulative baseline hazard function under the standard Cox model. We then estimate V E(t) through equation (2).
which is reminiscent of the Breslow estimator2 for the cumulative baseline hazard function under the standard Cox model. We then estimate V E(t) through equation (2).
Using counting-process martingale theory and other mathematical arguments,3–4 we can show that 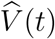 is consistent and asymptotically normal. In addition, the variance of
is consistent and asymptotically normal. In addition, the variance of 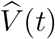 can be consistently estimated by n−1 n
can be consistently estimated by n−1 n 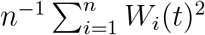 , where
, where
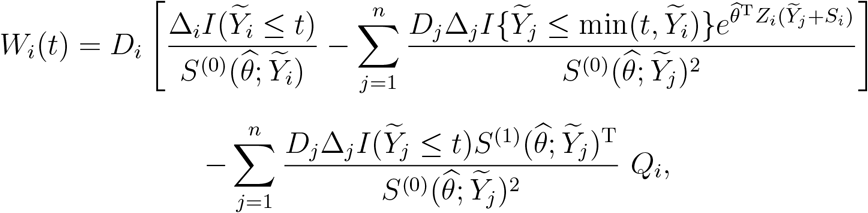 and
and
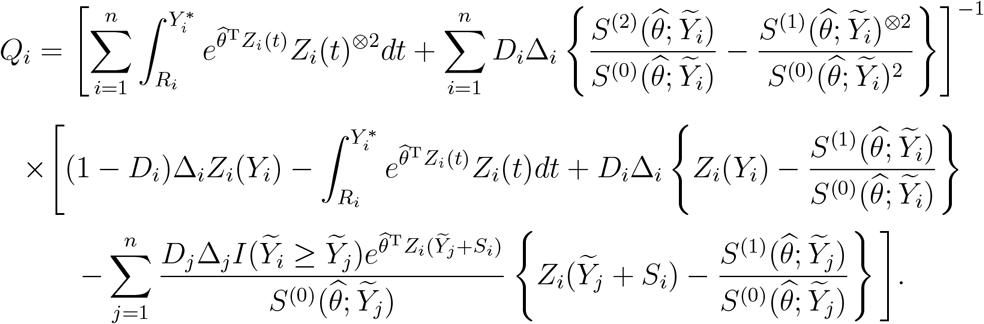
These results can be used to construct confidence intervals for V E(t). Specifically, we first construct the confidence intervals for log{V (t)} and then transform them to V E(t). We can also show the convergence of 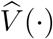 to a Gaussian process and construct simultaneous confidence bands for V E(·).4
to a Gaussian process and construct simultaneous confidence bands for V E(·).4
We have defined vaccine efficacy in terms of reduction in the cumulative incidence of disease. An alternative definition is reduction in the risk of disease.5 Specifically, we define the vaccine efficacy at time t by the proportionate reduction in the hazard rate at time t, i.e., ve(t) = 1 − v(t). We refer to V E(t) as t-day vaccine efficacy and to ve(t) as day-t vaccine efficacy, the former pertaining to cumulative vaccine effect and the latter to instantaneous vaccine effect. If the hazard ratio is constant over time, then the two definitions are equivalent. If the hazard ratio increases over time, then V E(t) is higher than ve(t). We can estimate v(t) and thus ve(t) through kernel smoothing of 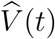
2. Designs of Simulation Studies
We assumed that 40,000 participants entered the study at a constant rate over four months, i.e., R ∼ Uniform(0, 4). We created a composite baseline risk score X, which takes values 1, 2, 3, 4, and 5 with equal probability. At study entry, half of the participants were assigned to vaccine and half to placebo. The statistically optimal design would be to maintain the original vaccine and placebo groups until the end of the study, which was set to be 10.5 months. We refer to this design as Plan A. We also considered three blinded crossover designs, under which placebo participants receive vaccine and vaccine participants receive placebo at the time of crossover and all participants are followed until the end of the study:
Plan B. Crossover occurs at month (11 − X + G), where G follows the exponential distribution with mean of 0.5.
Plan C. 20% of participants follow Plan A and 80% follow Plan B.
Plan D. Crossover occurs at month 6 + G, where G follows the exponential distribution with mean of 0.5.
Plan C mimics a scenario where all participants are offered the option of crossover but a small percentage (20%) choose to stay on the original assignments for the sake of science.
In addition, we considered three unblinded crossover designs:
Plan B’. Crossover occurs at month (11.5 − X).
Plan C’. 20% of participants follow Plan A and 80% follow Plan B’. Plan D’. Crossover occurs at month 6.5.
Under unblinded crossover, participants are notified of their original assignments at the time of crossover, and placebo participants receive the vaccine soon after. In practice, only placebo participants would cross over, since there is no need to give placebo to vaccine recipients after unblinding. In Plans B’–D’, the time of crossover is the time of unblinding rather than the time when placebo participants actually receive the vaccine. Because participants might change their behavior upon discovering their original treatment assignments, we discarded the follow-up data collected after unblinding by censoring each participant’s time to disease at their time of unblinding. This strategy avoids bias due to behavioral confounding, at the cost of reduced statistical efficiency.
We generated the event time T from model (1) with γ = 0.2,
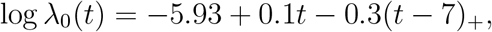 and
and
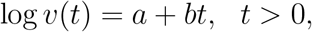 where a and b were chosen to achieve the desired VE values at 5 and 10 months. We censored T at the time of unblinding crossover under Plans B’–D’.
where a and b were chosen to achieve the desired VE values at 5 and 10 months. We censored T at the time of unblinding crossover under Plans B’–D’.
For each simulated dataset, we estimated log λ0(t) using a piece-wise constant function with 20 pieces placed at the equal quantiles of the observed event times. We then estimated V (t) using the proposed method and estimated V E(t) according to equation (2). For comparisons, we also fit a Cox model that includes X and time-dependent covariate I(S < t) to estimate VE by 1 minus the estimated hazard ratio of the time-dependent covariate.
3. Sensitivity Analysis
We suggest reporting the E-value7 as a summary measure of the evidence against the null hypothesis H0 : V E(t) ≤ 0. The E-value is the minimum strength of association on the risk ratio scale, i.e, RR(t) = 1 − V E(t), that an unmeasured confounder would need to have with both vaccination status and disease outcome in order to fully explain away a specific observed vaccine efficacy. Let 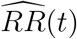 be the estimate of RR(t), and let UL(t) be the upper limit of the 95% confidence interval for RR(t). Then the E-value for
be the estimate of RR(t), and let UL(t) be the upper limit of the 95% confidence interval for RR(t). Then the E-value for 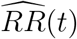 is given by
is given by
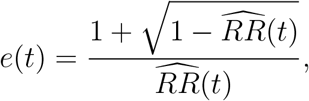 provided that
provided that 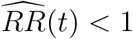 In addition, the E-value for UL(t) is computed as 1 if UL(t) ≥ 1 and as
In addition, the E-value for UL(t) is computed as 1 if UL(t) ≥ 1 and as
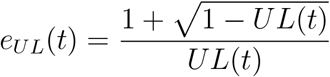 otherwise. E-values near one suggest weak support for a causal inference, and greater E-values provide increasing evidence for causality.
otherwise. E-values near one suggest weak support for a causal inference, and greater E-values provide increasing evidence for causality.
Suppose, for example, that 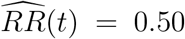 with 95% confidence interval (0.08, 0.75). Then e(t) = 3.4, meaning that the result of
with 95% confidence interval (0.08, 0.75). Then e(t) = 3.4, meaning that the result of 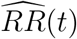 being less than one could be explained away by an unmeasured confounder associated with both vaccination status and disease by a risk ratio of 3.4-fold each after accounting for the vector X of measured confounders, but not by a weaker unmeasured confounder. In addition, eUL(t) = 2.5, which is the strength of unmeasured confounding at which statistical significance for V E(t) > 0 would be lost. These two E-values judge how confident we can be that V E(t) truly exceeds 0, accounting for potential unmeasured confounding due to unblinding as well as for sampling variability.
being less than one could be explained away by an unmeasured confounder associated with both vaccination status and disease by a risk ratio of 3.4-fold each after accounting for the vector X of measured confounders, but not by a weaker unmeasured confounder. In addition, eUL(t) = 2.5, which is the strength of unmeasured confounding at which statistical significance for V E(t) > 0 would be lost. These two E-values judge how confident we can be that V E(t) truly exceeds 0, accounting for potential unmeasured confounding due to unblinding as well as for sampling variability.
We can provide a conservative estimate of V E(t) that accounts for potential unmeasured confounding.8 Let RRUD(t) be the maximum risk ratio for disease when comparing any two categories of the unmeasured confounder U, within either the vaccinated group or the unvaccinated group, conditional on the vector X of observed covariates, and let RREU (t) be the maximum risk ratio for any specific level of the unmeasured confounder U when comparing the vaccinated and unvaccinated individuals. Define the bias or bounding factor
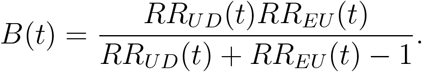
Then a conservative (lower bound) estimate of the vaccine efficacy is given by 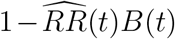 and a conservative confidence interval is obtained by multiplying the lower and upper limits of the confidence interval for RR(t) by B(t).
and a conservative confidence interval is obtained by multiplying the lower and upper limits of the confidence interval for RR(t) by B(t).
Acknowledgments
The authors thank Yu Gu and Bridget I. Lin for assistance. This work was supported by the National Institutes of Health grants R01 AI029168, R01 GM124104, P01 CA142538, and UM1 AI068635.
Footnotes
Added more details about estimating vaccine efficacy at a specific time point in Supplemental Appendix 1.





{kind=link}
{kind=link}
{kind=link}
{kind=link}