
Abstract
COVID-19 has widely spread across the world, and much research is being conducted on the causative virus SARS-CoV-2. To help control the infection, we developed the Coronavirus GenBrowser (CGB) to monitor the pandemic. CGB allows visualization and analysis of the latest viral genomic data. Distributed genome alignments and an evolutionary tree built on the existing subtree are implemented for easy and frequent updates. The tree-based data are compressed at a ratio of 2,760:1, enabling fast access and analysis of SARS-CoV-2 variants. CGB can effectively detect adaptive evolution of specific alleles, such as D614G of the spike protein, in their early stage of spreading. By lineage tracing, the most recent common ancestor, dated in early March 2020, of nine strains collected from six different regions in three continents was found to cause the outbreak in Xinfadi, Beijing, China in June 2020. CGB also revealed that the first COVID-19 outbreak in Washington State was caused by multiple introductions of SARS-CoV-2. To encourage data sharing, CGB credits the person who first discovers any SARS-CoV-2 variant. As CGB is developed with eight different languages, it allows the general public in many regions of the world to easily access pre-analyzed results of more than 132,000 SARS-CoV-2 genomes. CGB is an efficient platform to monitor adaptive evolution and transmission of SARS-CoV-2.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) 1-3 has infected more than 75 million people, and at least 1.6 million people in more than 200 countries have died from COVID-19. Many factors have contributed to the COVID-19 pandemic 4-6, and it has been predicted that the COVID-19 pandemic may last until 2025 7,8. The pathogen genomics platform Nextstrain has allowed analysis of genomic sequences of approximately 4,000 strains of SARS-CoV-2 and investigation of its evolution 9. As more than 210,000 SARS-CoV-2 strains have been sequenced (Figure S1), analysis of all strains has far exceeded the capacity of Nextstrain. New approaches are needed to accomplish this task.
To allow timely analysis of a large number of viral genomes, we first solved the problem that all viral genomes have to be re-aligned when nucleotide sequences of new genomes become available. This is extremely time-consuming. With the distributed alignment system (Figure 1), we dramatically reduced the total time required for the alignment. We also built the evolutionary tree on the existing tree and new genomic data in order to reduce the complexity of tree construction. With these modifications, hundreds of thousands of SARS-CoV-2 genomes can be timely analyzed with data easily shared and visualized on personal computers and smart phones (Figure 1).

The pre-analyzed genomic variant data can be freely accessed via https://bigd.big.ac.cn/ncov/apis/.
For genomic sequence alignments, 132,443 high quality SARS-CoV-2 genomic sequences were obtained from the 2019nCoVR database 10, which is an integrated resource based on CNGBdb, GenBank, GISAID 11,12, GWH 13, and NMDC. The sequences were aligned 14 to that of the reference genome and presented as distributed alignments. Genomic sequences of the bat coronavirus RaTG13 15, the pangolin coronavirus PCoV-GX-P1E 16, and the early SARS-CoV-2 strains collected before Jan 31, 2020 were jointly used to identify ancestral alleles of SARS-CoV-2. The evolutionary tree was rebuilt based on new data and the existing tree, and mutations in strains of each branch were indicated according to the principle of parsimony 17.
The genome-wide mutation rate of coronaviruses has been determined to be 10−4 − 10−2 per nucleotide per year 18. As this range of mutation rate is too wide, we decided to estimate more precisely the genome-wide mutation rate (μ) of SARS-CoV-2 in a timely manner and determined that μ = 6.753 × 10−4 per nucleotide per year (95% confidence interval: 4.581 × 10−4 to 9.253 × 10−4). This calculation did not require information on demography and the time of appearance of the most recent common ancestor (MRCA) of SARS-CoV-2. The estimated μ was lower than that of other coronaviruses, such as SARS-CoV (0.80 to 2.38 × 10−3 per nucleotide per year) 18 and MERS-CoV (1.12 × 10−3 per nucleotide per year) 19. It was also lower than that determined by other investigators (1.19 to 1.31 × 10−3 per nucleotide per year) 20. Various mutation rates were found in different regions of SARS-CoV-2 genome. The mutation rate of each gene is presented in Table S1.
Similar to Nextstrain 9 and the WashU Virus Genome Browser 21, the pre-analyzed genomic variant data on CGB are shared with the general public. The size of distributed alignments is 3,894 Mb for the high-quality 132,443 SARS-CoV-2 genomic sequences collected globally. The tree-based data format allows the compression ratio to reach 2,760:1, meaning that the size of compressed data file is as small as 1.41 Mb. This approach ensures low-latency access to the data and enables fast sharing and re-analysis of a large number of SARS-CoV-2 genomic variants.
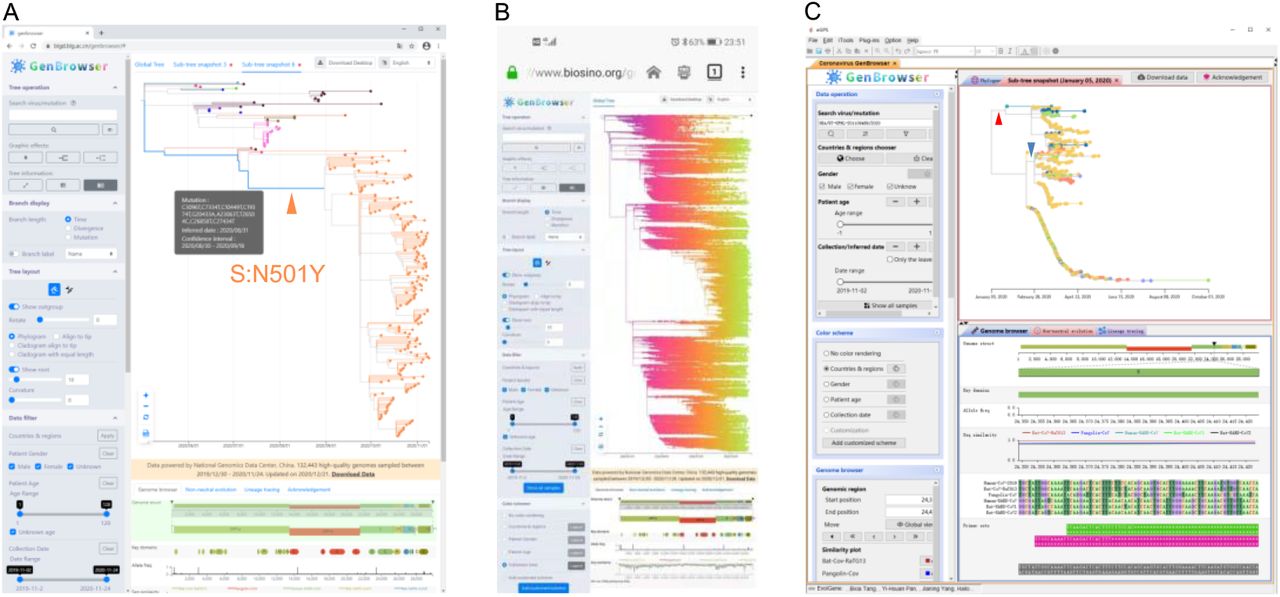
To visualize, search, and filter the results of genomic analysis, both desktop standalone and web-based user-interface of CGB were developed. Similar to the UCSC SARS-CoV-2 Genome Browser 22 and the WashU Virus Genome Browser 21, six genomic-coordinate annotated tracks were developed to show genome structure and key domains, allele frequencies, sequence similarity, multi-coronavirus genome alignment, and primer sets for detection of various SARS-CoV-2 strains. To efficiently visualize the results of genomic analysis, movie-making ability was implemented for painting the evolutionary tree, and only elements shown on the screen and visible to the user would be painted. This design makes the visualization process highly efficient, and the tree of more than 132,000 viral strains can be visualized even on a smart phone.
CGB detects on-going positive selection based on frequency trajectory of a selected allele. It has been shown that the spike protein G614 variant has a fitness advantage 23. Our analysis using CGB confirmed this finding even when the frequency of this mutation was very low (< 10%). Moreover, two previously identified variants (ORF1b:P314L, and N:A220V) 24 and five potentially advantageous variants were also identified even though their frequency was lower than 10% (Figure 2, Table S3). Thus, CGB is an efficient monitoring platform for detecting advantageous variants before they become widely spread (Figure S12).
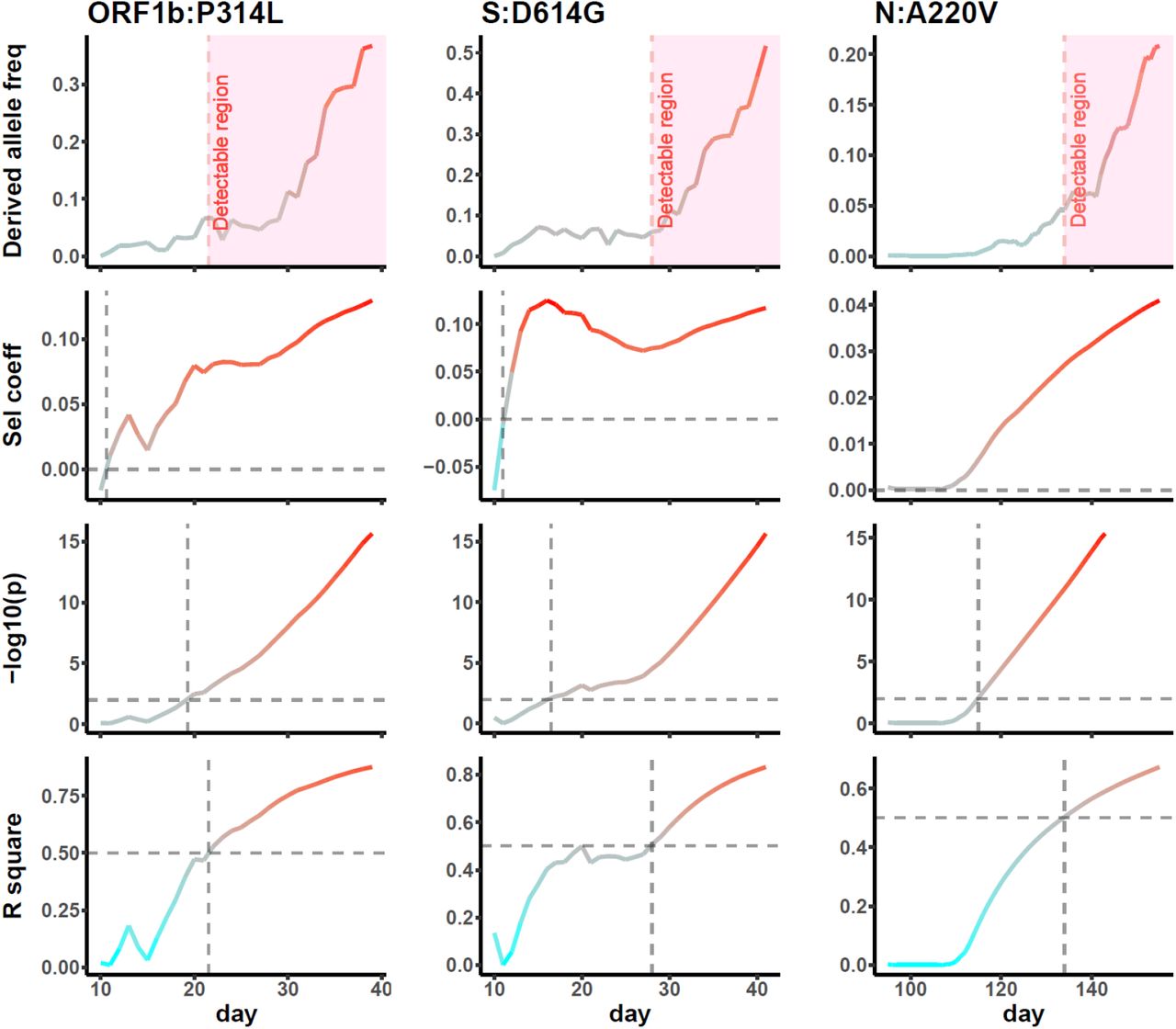
The x-axis displays number of days since the first appearance of derived allele in the global viral population. Predicted adaptation is marked in pink. Dashed gray crossings denote meaningful top right corners with a positive selection coefficient, p < 0.01, and R2 > 50%.
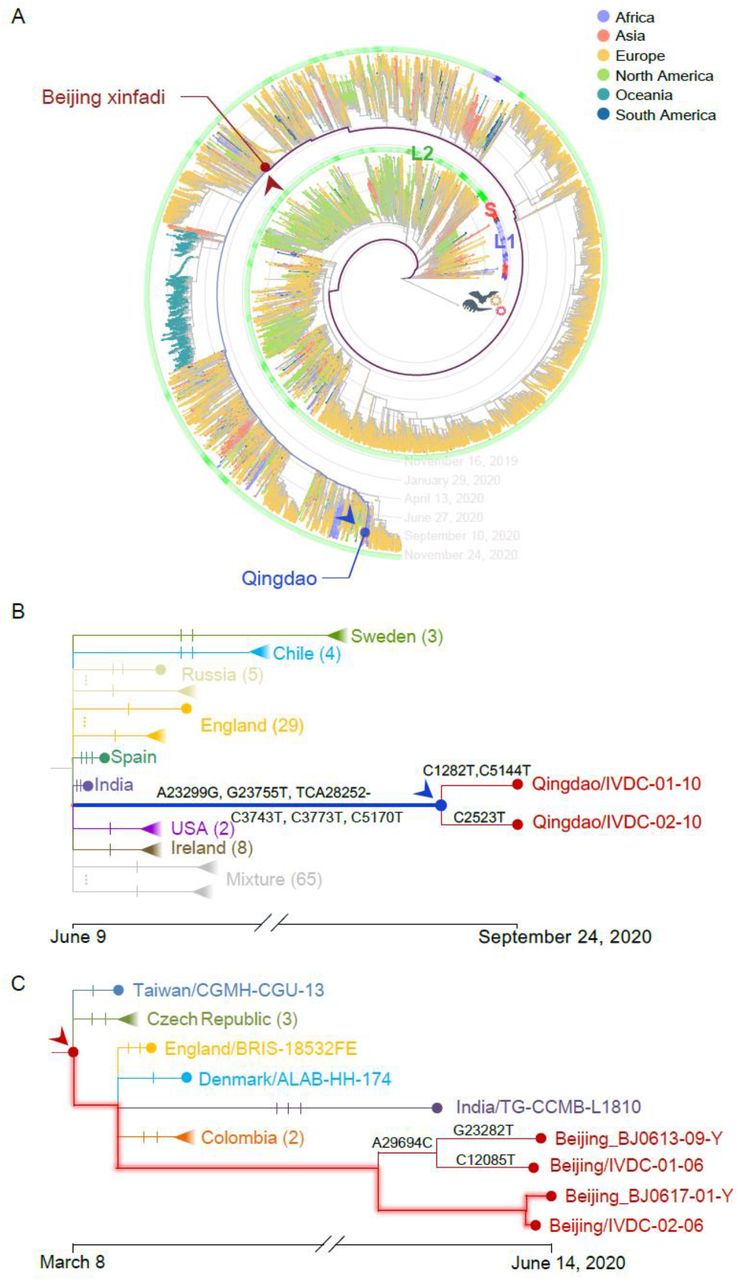
CGB is also an efficient platform to investigate local and global transmission of COVID-19 (Figure 3). There was a recent outbreak in Qingdao, China 25 after two dock workers were found to have asymptomatic infections on September 24, 2020. CGB lineage tracing revealed that the sequence of a sample collected from the outer packaging of cold-chain products is identical to that of the most recent common ancestor of the two viral strains isolated from the two dock workers (Figure 3B), suggesting that infection of these two individuals was cold-chain related. However, this possibility remains to be determined.

A) The lineages of traced targets are shown in blue and dark-red lines. The tree of 132,443 viral strains was used. L/S lineage types 30 are marked with an outside circle.
B) Qingdao/IVDC-01-10 and Qingdao/IVDC-02-10 were the two SARS-CoV-2 strains collected on September 24, 2020 from two dock workers in Qingdao, China. The query strain (env/Qingdao/IVDC-011-10) was found on an outer packaging of cold-chain products on October 7, 2020 The environmental strain, marked with a blue solid circle with an arrow head, was found to be identical to the most recent common ancestor of the two strains from the two dock workers. Each notch of the branches represents a mutation. Mutations of the Qingdao strains are indicated.
C) The ancestral viral strain found in early March 2020 is marked with a dark-red solid circle with an arrow head. This strain is identical to the two strains (Beijing/IVDC-02-06, Beijing/BJ0617-01-Y) collected from two Xinfadi cases on June 11 and 14, 2020. The branches with no mutations are highlighted.
CGB lineage tracing also revealed the difficulty in the control of COVID-19 pandemic. There was a recent outbreak in Xinfadi, Beijing, China 26. The sequences of two viral isolates (Beijing/IVDC-02-06, Beijing/BJ0617-01-Y), collected from two Xinfadi cases on June 11 and 14, 2020, were found to be identical to the sequence of an ancestral strain (Figure 3C) dated on March 6, 2020 (95% CI: February 28 – March 17, 2020). This ancestral strain was found to spread to Taiwan, India, Czech Republic, England, Denmark, and Colombia and caused the outbreak in Beijing three months later. These two Xinfadi strains were also found to evolve significantly slowly (P = 0.0043 and 0.0051, respectively) because no mutations were detected during the three months.
CGB is a powerful tool for the identification of global and regional routes of virus transmission as it is specially designed to determine whether the mutation rate of a specific strain is lower than the average mutation rate of the entire set of strains. This lineage-specific reduced mutation rate could be due to a long period of dormancy caused by the yet to be confirmed cold-chain preservation 27 or other reasons. Among the 132,443 SARS-CoV-2 strains, 4,597 strains were found to evolve significantly slowly (P = 2.18 × 10−8∼0.0041, Supplemental excel file) and did not mutate within at least 100 days. This data showed that CGB can narrow the time period for tracing the transmission of a specific strain.
A study on the sequences of 453 SARS-CoV-2 genomes collected before mid-March 2020 suggested that the first COVID-19 outbreak in Washington State was due to a single introduction 28. However, results of CGB analysis suggest that the first Washington State outbreak was actually caused by multiple introductions (Figure S14).
All the timely-updated data are freely available at https://bigd.big.ac.cn/ncov/apis/. The free desktop standalone version provides the full function of CGB and has a plug-in module for the eGPS software (http://www.egps-software.net/) 29. Although the web-based tool is a simplified version of CGB (Figure 4) (https://www.biosino.org/genbrowser/ and https://bigd.big.ac.cn/genbrowser/), it provides a convenient way to access the data via a web browser, such as Google Chrome, Firefox and Safari. The web-based CGB package can be downloaded and reinstalled on any websites. For educational purpose, eight language versions (Chinese, English, German, French, Italian, Portuguese, Russian, and Spanish) are available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Web-based CGB tree visualization of an accelerated lineage in the UK, out of 132,443 SARS-CoV-2 genomic sequences, with the desktop version of Google Chrome.
B) Web-based CGB tree visualization of 132,443 genomes with the Android version of Firefox.
C) Tree visualization of a lineage (USA/UT-UPHL-201109489/2020) with the mostly reduced evolutionary rate and its neighbors with desktop standalone CGB. There are only two mutations (A20268G, red arrow head; C15324T, blue arrow head) happened in 966 strains within nearly 9 months.
Data Availability
All the timely-updated data are freely available at https://bigd.big.ac.cn/ncov/apis/. The free desktop standalone version provides the full function of CGB and has a plug-in module for the eGPS software (http://www.egps-software.net/) 29. Although the web-based tool is a simplified version of CGB (Figure 4) (https://www.biosino.org/genbrowser/ and https://bigd.big.ac.cn/genbrowser/), it provides a convenient way to access the data via a web browser, such as Google Chrome, Firefox and Safari.
Members of the language translation team
German: Ning He7, Jing Lv7, Ting Peng7
Italian: Ting Zhou7, Nan Yang7, Siyi Hou7
Portuguese: Huang Li7, Jingxuan Yan7, Chenglin Zhu7, Wenjing Liu7
Russian: Yuhong Guan7, Huanxiao Song7
Spanish: Qin Zhou7, Han Gao7, Jinglan He7, Tiantian Li7, Ruiwen Fei7, Shumei Zhang7
French: Yuyuan Guo7
Acknowledgments
We thank Ya-Ping Zhang for providing valuable advices and encouragement, and the researchers who generated and deposited the sequencing data of SARS-CoV-2 in GISAID, GenBank, CNGBdb, GWH, and NMDC, making this study possible. This work was supported by a grant from the National Key Research and Development Project (No. 2020YFC0847000).
References




