
Abstract
Background Point-of-care (POC) tests for COVID-19 could relieve pressure on isolation resource, support infection prevention and control, and help commence more timely and appropriate treatment. We aimed to undertake a systematic review and pooled diagnostic test accuracy study of available individual patient data (IPD) to evaluate the diagnostic accuracy of a commercial POC test (FebriDx) in patients with suspected COVID-19.
Methods A literature search was performed on the 1st of October 2020 to identify studies reporting diagnostic accuracy statistics of the FebriDx POC test versus real time reverse transcriptase polymerase chain reaction (RT-PCR) testing for SARS-CoV-2. Studies were screened for risk of bias. IPD were sought from studies meeting the inclusion and exclusion criteria. Logistic regression was performed to investigate the study effect on the outcome of the RT-PCR test result in order to determine whether it was appropriate to pool results. Diagnostic accuracy statistics were calculated with 95% confidence intervals (CIs).
Results 15 studies were screened, and we included two published studies with 527 hospitalised patients. 523 patients had valid FebriDx results for Myxovirus resistance protein A (MxA), an antiviral host response protein. The FebriDx test produced a pooled sensitivity of 0.920 (95% CI: 0.875-0.950) and specificity of 0.862 (0.819-0.896) compared with RT-PCR, where there was an estimated true COVID-19 prevalence of 0.405 (0.364-0.448) and overall FebriDx test yield was 99.2%. Patients were tested at a median of 4 days [interquartile range: 2:9] after symptom onset. No differences were found in a sub-group analysis of time tested since the onset of symptoms.
Conclusions Based on a large sample of patients from two studies during the first wave of the SARS-CoV-2 pandemic, the FebriDx POC test had reasonable diagnostic accuracy in a hospital setting with high COVID-19 prevalence, out of influenza season. More research is required to determine how FebriDx would perform in other healthcare settings with higher or lower COVID-19 prevalence, different patient populations, or when other respiratory infections are in circulation.
Trial registration This work was based on a pooled analysis of anonymised data from two previous studies; the CoV-19POC study, described by Clark et al. (9), the “Southampton study” [ISRCTN:14966673, date registered: 18/03/2020]; and a study described by Karim et al. (13) the “Kettering study”.
Lay summary Tests to diagnose COVID-19 are crucial to help control the spread of the disease and to guide treatment. Over the last few months, tests have been developed that can detect the SARS-CoV-2 virus which causes COVID-19. These tests use complex machines in pathology laboratories accepting samples from large geographical areas. Sometimes it takes days for test results to come back. So, to reduce the wait for results, new portable tests are being developed. These point-of-care (POC) tests are designed to work close to where patients require assessment and care such as hospital emergency departments, GP surgeries or care homes. For these new POC tests to be useful, they should ideally be as good as standard laboratory tests so patients get their result quickly and can benefit from the best, safest care.
In this study we looked at published research into a new test, FebriDx, which can detect the presence of any viral infection, including infections due to the SARS-CoV-2 virus, as well as bacterial infections which can have similar symptoms. The FebriDx result was compared with that obtained on the same patient’s throat and nose swab and using the standard COVID-19 viral laboratory test. We were able to analyse data from two studies with a total of 523 adult patients who were receiving emergency hospital care with symptoms of COVID-19 during the early stage of the UK pandemic. Almost half of the patients were diagnosed as positive for SARS-CoV-2 virus using standard laboratory COVID-19 viral tests.
Our analysis demonstrated that the FebriDx POC test agreed 94 out of 100 times with the standard laboratory test results when FebriDx diagnosed the patient as free from COVID-19. However, FebriDx agreed only 82 out of 100 times with the standard laboratory test when FebriDx indicated that the patient had a COVID-19 infection. These differences have important implications for how these tests could be used. As there were far fewer FebriDx false results when the results of the FebriDx test were negative (6 out of 100) than when the results of the FebriDx test were positive (18 out of 100), we can have more confidence in a negative test result using FebriDx at the POC than a positive FebriDx result.
Overall, we have shown that the FebriDx POC test performed quite well during the first wave of the COVID-19 pandemic when compared with laboratory tests, especially when the POC test returned a negative test. For the future, this means that the FebriDx POC test might be helpful in making a rapid clinical decision whether to isolate a patient with COVID-19-like symptoms arriving in a busy emergency department. However, our results indicate it would not completely replace the need to conduct a confirmatory laboratory test in certain cases.
There are limitations to our findings. For example, we do not know if FebriDx will work in a similar way with patients in different settings such as in the community or care homes. Similarly, we do not know whether other viral and bacterial infections which cause similar COVID-19 symptoms, and are more common in the autumn and winter months, could influence the FebriDx test accuracy.
Background
The global SARS-CoV-2 pandemic (1) has put considerable pressure on healthcare services worldwide. Health and care providers require diagnostic strategies to rapidly identify patients infected with SARS-CoV-2 to implement the accurate segregation of positive and negative patients in health and care facilities, and to ensure early administration of evidence-based therapies to patients with coronavirus disease 2019 (COVID-19) (2). The risks of nosocomial infection are high (3) and mechanisms to ensure that SARS-CoV-2 has limited transmission within hospitals, care facilities, and the community are an urgent priority as many countries begin to face a second wave of infection.
There has been rapid development of novel clinical tests to support screening and diagnosis in both symptomatic and asymptomatic patients. In particular, there have been a number of molecular and antibody tests which manufacturers have developed for use at the point-of-care (POC) (4). POC tests for influenza have been adopted in many hospitals and other health and care facilities (5), where the rapid availability of results inform patient management, selection of the appropriate location for ongoing healthcare provision, and infection prevention and control (6, 7). In the emergency department setting for example, a rapid COVID-19 test result to aid triage of the patient into the appropriate COVID-19/non-COVID-19 sections of the hospital may contribute to a reduction in nosocomial infection, providing significant benefits to patient pathways, workflows, and outcomes (8).
There is limited published evidence on the accuracy, reliability, and usability of many of the available POC tests for COVID-19, in particular, when used in-context within clinical settings with varying disease prevalence. Some pre-existing POC tests may have a role in the management of patients with suspected COVID-19. The FebriDx lateral flow device (LFD) (Lumos Diagnostics, Sarasota, Florida, USA) is a CE marked POC test that detects two host response proteins, myxovirus resistance protein A (MxA) and C reactive protein (CRP), in finger prick blood samples. This combination of MxA and CRP is designed to distinguish between viral and bacterial respiratory infection (9-11). MxA is an intracellular protein that is exclusively induced by type I interferon (IFN) and not by other cytokines expressed during bacterial infection (12, 13). Type I IFNs are produced in response to a wide range of viral infections and are found to be elevated in the presence of most acute viral infections (10), therefore providing strong theoretical grounds to expect MxA to rise in response to SARS-Cov-2 infection. The manufacturer’s intended use includes recommendations to use in patients older than 2 years presenting within 3 days of an acute onset fever (exhibited or reported) and within 7 days of new onset respiratory symptoms consistent with a community-acquired upper respiratory infection (14).
We undertook a systematic review and pooled diagnostic test accuracy study of available individual patient data (IPD) to evaluate the diagnostic accuracy of the FebriDx LFD compared to contemporaneous reverse transcriptase polymerase chain reaction (RT-PCR) testing to understand the performance of FebriDx in the identification of patients with COVID-19. We did not limit this analysis to patients presenting within a certain number of days since the onset of symptoms in order to inform and identify all potential use cases within the pandemic.
Method
We performed a systematic review with a pooled diagnostic test accuracy study of available IPD, and followed the STAndards for Reporting Diagnostic accuracy studies (STARD) checklist (15) (supplementary material 1). Although not formally a systematic review and meta-analysis of individual patient data (IPD) (16), we also followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)-IPD checklist (PRISMA-IPD) (17) (supplementary material 2), where applicable. This study was conducted at pace as part of the CONDOR national test evaluation programme (18), and as a result, no protocol was developed, and the study was not registered.
Literature search
The inclusion criteria for studies were: published or un-published (preprint) diagnostic test accuracy studies; FebriDx used as the index test; RT-PCR used as a comparator test; and an adult population suspected of COVID-19 regardless of the time since symptom onset. The exclusion criteria were studies that were not a diagnostic test accuracy study.
OVID Medline, LOVE Platform (Epistemonikos, Santiago, Chile), and the references from the Living Systematic Review on SARS-CoV-2 (19) were electronically searched on the 30th of July 2020, and again on the 1st of October 2020, by one author (JS) to identify diagnostic test accuracy studies reporting diagnostic accuracy statistics of FebriDx versus RT-PCR for SARS-CoV-2. The following keywords: “COVID-19”, “2019-nCOV”, “SARS-COV-2”, “novel coronavirus disease” AND “FebriDx” were used. No date, location or language restrictions were applied to the search results, and all of the databases included pre-prints.
The abstracts of the search results were accessed and screened against the inclusion and exclusion criteria by two authors independently (SGU and KG). The two authors discussed, compared, and combined their findings, and if there was disagreement, adjudication was provided by a third author (AJA). If there was insufficient information in the abstract to exclude the study based on the inclusion and exclusion criteria, then a conservative approach of accessing the full text to perform the screening was taken to mitigate the risk of erroneously excluding relevant studies.
Risk of bias (RoB) assessments were performed on the manuscripts of the identified studies that passed eligibility screening against the inclusion and exclusion criteria using the QUADAS-2 tool (20) for the quality assessment of diagnostic accuracy studies by two authors independently (SGU and KG). The QUADAS-2 binary prompts were deemed insufficient to fully capture the potential bias of the studies. The descriptive sections of the tool were therefore used to capture any other potential sources of bias not considered by the binary prompts. These additional aspects contributed to the final assessment for RoB. The two authors held a discussion to compare and combine their findings, and if there was disagreement, this was adjudicated by a third author (BCL).
The Chief Investigators (CIs) of the identified studies that passed eligibility screening against the inclusion and exclusion criteria were approached via email to provide anonymised IPD. If IPD were not available, or not provided, then the study was excluded from further analysis. The minimum data set that was requested from the CIs is outlined in supplementary material 3. Any queries relating to the study or provided data were communicated and resolved via email. The RoB assessments were performed prior to making contact with the CIs, and any additional information provided by the CIs not included in the manuscripts did not impact upon the RoB assessments.
Data processing
Following receipt of the IPD, the FebriDx and RT-PCR results were summarised in 2×2 contingency tables for each study independently and also pooled across all studies. The following assumptions were applied. For FebriDx: a viral result (i.e. MxA positive [≥40 ng/ml], regardless of CRP positivity) was considered positive; a non-viral result (i.e. MxA negative [<40 ng/ml], CRP positive [≥20mg/L]) was considered negative; and a negative result (i.e. MxA negative [<40 ng/ml], CRP negative [<20mg/L]) was considered negative. For RT-PCR: if COVID-19 was detected the result was considered positive (COVID-19 + ‘other’ were also considered positive if studies used a respiratory panel that also tested for other viruses); and if COVID-19 was not detected the result was considered negative.
Data analysis
A complete case analysis approach was taken whereby cases with a completed and valid FebriDx and RT-PCR result pair were included, regardless of missing data within other fields. This was undertaken to maximise the available sample size for the pooled analysis. Cases that had missing FebriDx or RT-PCR results were therefore excluded, in addition to cases that had a final indeterminate FebriDx or RT-PCR result for the pooled analysis. However, to estimate overall test yield to allow interpretation of diagnostic accuracy estimates in spite of this, we included all cases with missing or invalid FebriDx test results.
To determine if the study populations were similar, and to quantify any baseline and outcome imbalance, the distributions of variables were compared statistically between the included studies. Non-parametric two-sample Wilcoxon rank sum tests (two-sided) were used to compare numerical data, whilst two-sample tests for equality of proportions with continuity corrections were used to compare categorical data. Statistical significance was set at an alpha level of <0.05. Numerical data were summarised using the median and interquartile range, whilst categorical data were summarised using counts and proportions.
To formally determine whether it was appropriate to pool the diagnostic accuracy results from the included studies, logistic regression was undertaken to investigate the study effect on the outcome of the RT-PCR test result, whilst controlling for the FebriDx test result. Thus a main effect for study, and an interaction effect between the FebriDx test result and study were included in the model. The following logistic regression model was constructed using a binomial error distribution and logit link function:
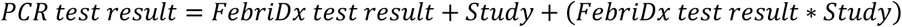 Diagnostic accuracy measures with 95% confidence intervals (CIs) were calculated from the 2×2 contingency tables for each study independently and also pooled across studies (supplementary material 4). A sub-group analysis was also performed, with the pooled data stratified by the time tested since the onset of symptoms into two groups: 0 to 7 days, and >7 days. This was undertaken to determine whether the time tested since the onset of symptoms had an impact on the diagnostic accuracy of FebriDx. The following diagnostic accuracy measures were calculated across all study groups:
Diagnostic accuracy measures with 95% confidence intervals (CIs) were calculated from the 2×2 contingency tables for each study independently and also pooled across studies (supplementary material 4). A sub-group analysis was also performed, with the pooled data stratified by the time tested since the onset of symptoms into two groups: 0 to 7 days, and >7 days. This was undertaken to determine whether the time tested since the onset of symptoms had an impact on the diagnostic accuracy of FebriDx. The following diagnostic accuracy measures were calculated across all study groups:
Test yield (the proportion of all patients tested with FebriDx that had a valid test result from FebriDx)
Apparent prevalence (the proportion of all patients with COVID-19 according to FebriDx)
Estimated true prevalence (proportion of all patients with COVID-19 according to RT-PCR)
Test sensitivity (proportion of patients with COVID-19, according to RT-PCR, that test positive by FebriDx)
Test specificity (proportion of patients without COVID-19, according to RT-PCR, that test negative by FebriDx)
Diagnostic accuracy (proportion of all FebriDx tests that give a correct result according to RT-PCR)
Positive predictive value (PPV) (probability that patients with a positive FebriDx test have COVID-19 according to RT-PCR)
Negative predictive value (NPV) (probability that patients with a negative FebriDx test do not have the disease according to RT-PCR)
Positive likelihood ratio (probability of a patient who has COVID-19 according to PCR testing positive by FebriDx, divided by the probability of a patient who does not have the disease according to RT-PCR testing positive by FebriDx)
Negative likelihood ratio (probability of a patient who has COVID-19 according to RT-PCR testing negative by FebriDx, divided by the probability of a patient who does not have the disease according to RT-PCR testing negative by FebriDx)
Results
Fifteen studies were identified from the literature search, with seven studies excluded following deduplication. Six studies were excluded following screening against the inclusion and exclusion criteria, leaving two studies for potential inclusion (supplementary material 5). The CIs of the two studies were able to provide IPD which were included in analysis. The two studies were from the UK: a study from Southampton (CI = TWC) (9) and a study from Kettering (CI = RVR) (13). In addition to receiving the requested data underpinning the publications, an additional, unpublished dataset was made available to us from the Southampton study using the same methods as their initial publication (9).
Risk of bias
The results of the RoB assessment are provided in supplementary material 6. In the ‘Patient selection’ domain, the Southampton study was assessed to have an unclear RoB due to uncertainty around which patients were re-tested and which patients were excluded, and whether those with no CRP line [<20mg/L] on the FebriDx LFD but with concurrent laboratory CRP results of ≥20mg/L were excluded. In the ‘Index test’ domain, the Kettering study was assessed to have an unclear RoB due to patients with FebriDx positive and RT-PCR negative tests considered to be positive for COVID-19 if clinical suspicion was high. Due to this, there was concern regarding the applicability of the index test to the focus of this study. In the ‘Flow and timing domain’, the Kettering study was assessed to have a high risk of RoB due to the exclusion of one third of the patients for having symptoms longer than 7 days, however this did follow FebriDx’s instructions for use (14).
Eligibility criteria, index test, and reference standard in the included studies
Table 1 presents a summary of the eligibility criteria, index test, and reference standard used within the Southampton and Kettering studies.
The eligibility criteria, index test, and reference standard of the included studies
Eligibility criteria
In the Southampton study, consecutive patients were approached for participation at Southampton General Hospital, University Hospital Southampton NHS Foundation Trust, between the 20th March 2020 and 29th April 2020. In the Kettering study, consecutive patients were approached for participation at Kettering General Hospital, Kettering General Hospital NHS Foundation Trust, between 16th of March and 7th of April 2020.
When combined, both studies recruited hospitalised patients over 16 years of age with suspected COVID-19 between the 16th of March and 29th April 2020, however the Southampton study also allowed patients to be recruited who did not have an acute respiratory illness (ARI) or did not meet the PHE definition of a suspected case, but where testing was considered necessary by the clinical team.
Index test
In both studies, the results of the FebriDx test were not shared with the clinical teams and the readers of the FebriDx test lines were blinded to the RT-PCR results, and vice versa (9). In both studies, the finger prick blood samples for FebriDx were taken at the same time as the nose and throat swabs for RT-PCR (9, 13). In the Southampton study, the FebriDx result was read independently by two investigators and disagreements were further adjudicated by a third investigator. In the Kettering study, the FebriDx result was read by one investigator, however if the result was inconclusive or negative, this was further adjudicated by two investigators.
Reference standard
The Southampton study used the QIAstat-Dx RT-PCR system (Qiagen, Hilden, Germany) for analysing combined nose and throat swabs, which gave a binary readout of positive or negative for the detection of targets including SARS-CoV-2 (9), in addition to Public Health England (PHE) laboratory RNA-dependent RNA polymerase (RdRp) and envelope protein (E) RT-PCR testing for SARS-CoV-2. However, only results from the QIAstat-Dx RT-PCR system were available for the additional unpublished data from the Southampton study following their initial publication (9), so the results from the QIAstat-Dx RT-PCR system were used for all patients from the Southampton study to maintain within-study consistency for the purposes of this analysis. The Kettering study used Public Health England laboratory RdRp and envelope protein (E) RT-PCR testing for SARS-CoV-2 to analyse nose and throat swabs (13).
Patient flow
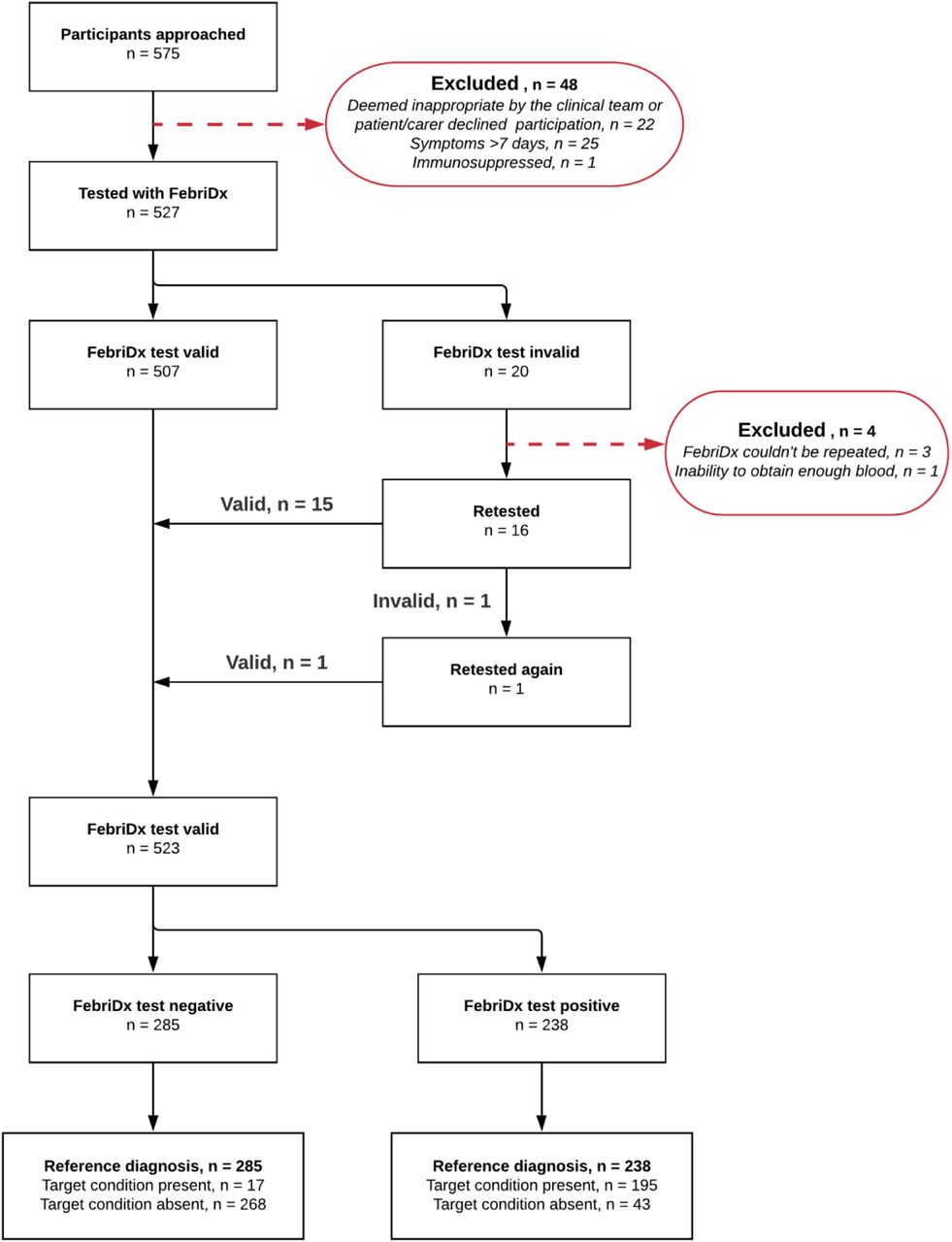
The flow of patients in the Southampton and Kettering studies are presented in supplementary material 7 and 8, respectively, and pooled data from both studies is presented in Figure 1.

In the Southampton study, 500 patients were considered for testing with FebriDx, with 22 excluded (4.4%) as it was deemed inappropriate by the clinical team, or where the patient/carer declined participation in the study. Out of the 478 patients tested with FebriDx, 19 tests were initially invalid (4%). FebriDx could not be repeated in 3 of the 19 initially invalid tested patients (15.8%). Out of the 16 initially invalid tested patients that were retested, 1 was invalid (6.3%), and was subsequently retested again where the patient then received a valid test result upon a second retest. Considering all 20 invalid tests, 16 were due to blood clotting in the collection tube (80%), whilst 4 were due to there being no CRP line [<20mg/L] on the FebriDx device but with concurrent laboratory CRP results of ≥20mg/L (20%). 475 patients remained for analysis, resulting in a test yield of 99.4% (supplementary material 7). The Kettering study approached 75 patients for testing with FebriDx, where 26 were excluded (34.7%), with 25 due to symptoms being longer than 7 days, and 1 due to being immunosuppressed. Out of the 49 patients tested with FebriDx, 1 test was initially invalid (2%) due to the inability to obtain enough blood. FebriDx could not be repeated in this patient as they were elderly, frail, and clinically unstable at the time of testing, and 48 patients remained for analysis, resulting in a test yield of 98% (supplementary material 8).
This resulted in 575 patients being approached for testing with FebriDx when pooled, with 48 excluded due to the aforementioned reasons (8.4%). Out of the 527 patients tested with FebriDx, 20 tests were initially invalid (3.8%). FebriDx could not be repeated in 4 of the 20 initially invalid tested patients (20%). Out of the 16 initially invalid tested patients that were retested, 1 was invalid (6.3%), and was subsequently retested again where the patient then received a valid test result upon a second retest. 523 patients remained for the pooled analysis, resulting in an overall test yield of 99.2%.
Patient population and outcome
The patient population and outcome in the Southampton study, Kettering study, and pooled data from both studies is summarised in Table 2.
Patient population and outcome in the included studies and pooled data from both studies
Symptom and outcome data were provided as requested for both studies, but due to methodological differences between the studies in how these data were defined and captured, they were excluded from statistical analyses. The Southampton study recorded symptoms in the prospectively stipulated categories presented in Table 2, whereas the Kettering study recorded free text symptoms. For presentational purposes only, two authors (SGU, AJA) reviewed each of the free text fields in the Kettering study independently and applied the same categories as the Southampton study, with no disagreements requiring adjudication by a third author. The Southampton study reported death at 30 days following admission, whereas the Kettering study reported death at the end of the index admission.
No statistically significant differences were found between the populations of the Kettering and Southampton studies in terms of age (p = 0.59), sex (p = 0.06), and symptom duration (p = 0.15), although the sex distributions almost reached statistical significance, with the Kettering study having a slightly higher proportion of males (66.7% vs. 51.4%).
Diagnostic test accuracy
Pooling data
In the logistic regression analysis to investigate the study effect on the outcome of the RT-PCR test result, none of the included terms were statistically significant: FebriDx test result (b = 18.6; p = 0.98); the study (b = 13.9; p = 0.98); and an interaction term between FebriDx test result and the study (b = −14.5; p = 0.98). The interaction term was therefore removed from the hierarchical model, and in the reduced model, as expected, the FebriDx test result was highly statistically significant (b = 4.24; p<0.001), and the study effect was still not statistically significant (b = −0.32; p=0.49). This result suggests little study effect on the outcome of the PCR test result, whilst controlling for the FebriDx test result, and supports pooling of data across the studies.
Results
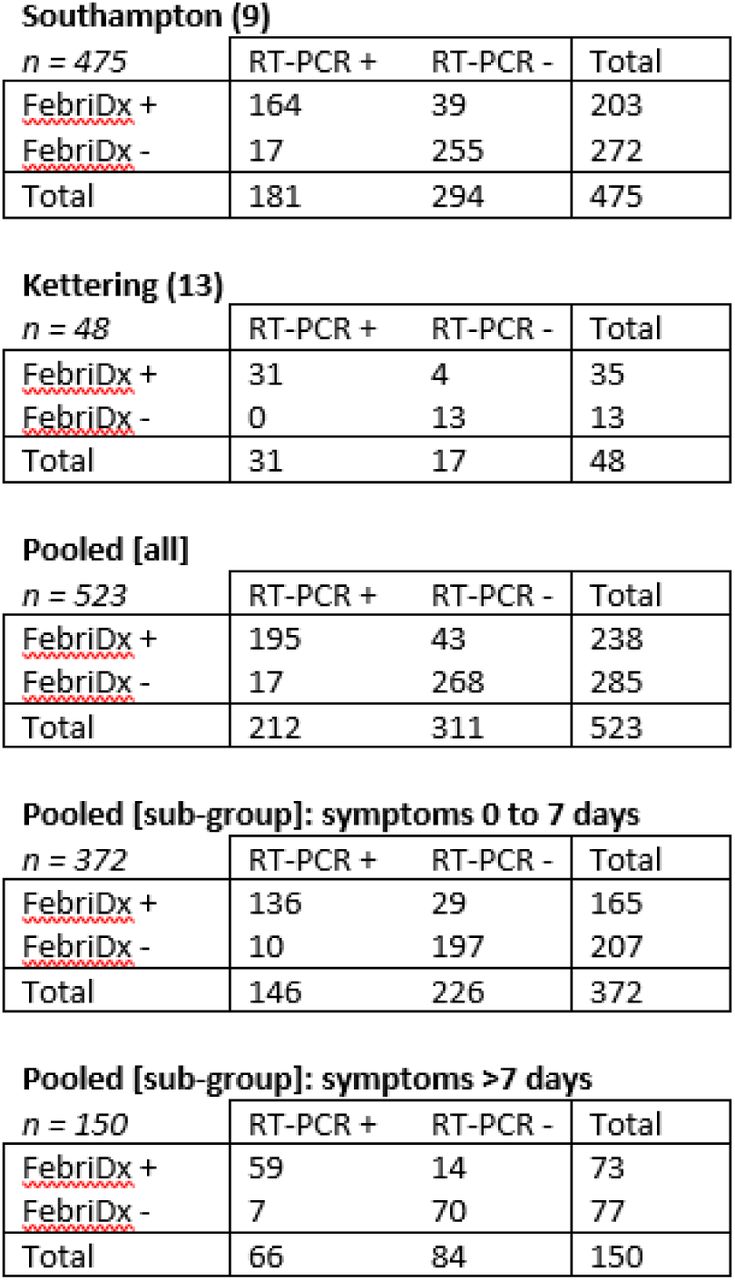
The Southampton and Kettering studies had a test yield of 99.4% and 98%, respectively, and when combined, a test yield of 99.2% was found. These valid tests were presented in 2×2 contingency tables for the Southampton study, Kettering study, and pooled data (including sub-group analyses) from both studies in Figure 2, with the diagnostic accuracy results calculated for the Southampton study, Kettering study, and pooled data (including sub-group analyses) from both studies presented in Table 3.
Diagnostic accuracy results for the included studies and pooled data from both studies
{kind=link}
{kind=link}
2×2 contingency tables from the Southampton study (Clark et al. (9) + additional unpublished data), Kettering study (Karim et al. (13)), and pooled [all and sub-group] data from both studies. One patient was excluded from the ‘Pooled [sub-group]’ analysis due to missing symptom duration data. Please note that data from the ‘Pooled [sub-group]: symptoms >7 days’ were from the Southampton study only
The estimated true and apparent COVID-19 prevalence were the only statistically significant differences between the studies in the diagnostic accuracy measures (both p<0.001), with the Kettering study experiencing a higher COVID-19 prevalence (estimated true prevalence = 0.646 (95% CI: 0.504-0.766)) than the Southampton study (estimated true prevalence = 0.381 (0.338-0.426)).
The pooled results from 523 patients gave an estimated sensitivity of 0.920 (0.875-0.950) and specificity of 0.862 (0.819-0.896) for the FebriDx test, with an overall diagnostic accuracy of 0.885 (0.855-0.910) at an estimated true prevalence of 0.405 (0.364-0.448) (Table 3). The PPV was 0.819 (0.765-0.863) and NPV was 0.940 (0.906-0.963).
One patient was excluded from the sub-group analyses of the pooled results due to missing symptom duration data. In these analyses, no statistically significant differences were apparent between the 372 patients that were tested between 0 and 7 days after symptom onset and the 150 patients that were tested more than 7 days after symptom onset in any of the diagnostic accuracy measures calculated (Table 3).
Discussion
In this systematic review and pooled analysis of IPD, we found that the FebriDx LFD had a pooled sensitivity of 0.920 (95% CI: 0.875-0.950) and specificity of 0.862 (0.819-0.896) for COVID-19 across two studies performed within acute hospitals in the UK when compared to RT-PCR on nose and throat swabs during the first wave of the SARS-CoV-2 pandemic. There were no other published data on the diagnostic accuracy of FebriDx, with the overall evidence base in terms of studies performed remaining small. The two studies did, however, include a total of 523 patients, achieving a large sample size for the pooled analysis.
Testing with FebriDx produced an initial test failure rate of 3.8% (20 out of 527 patients tested). In those patients where re-testing was performed, most were valid upon the first re-test (15 out of 16 patients), and only one patient required a second re-test to produce a valid result. Most test failures were described as being the result of blood clotting within the collection tube, and therefore not being released into the device for analysis.
The duration between onset of symptoms and the patient being tested did not seem to have an impact on the diagnostic accuracy of FebriDx, with no statistically significant differences evident between the two sub-groups of 0 to 7 days and greater than 7 days after symptom onset. However, only one of the studies included patients with symptom onset after 7 days, so this finding is not from a pooled analysis.
All patients included in the pooled analysis were from the acute hospital setting, and such findings must be extrapolated with caution to other patient groups and settings. Further context-specific evaluation would be required in order for FebriDx to be used in other patient groups where performance has not yet been demonstrated; such as children, immunocompromised and cancer patients, and those who are asymptomatic or pauci-symptomatic. Further, taking care homes as an example setting, the mean age of residents is 85 years (21), and all care home residents are significantly affected by frailty. The high prevalence of immunosenescence in this group is such that CRP and MxA results might be significantly attenuated (22). It is clear that hospital data cannot be extrapolated to such a group and further context-specific evaluation would be required in other settings.
In the context of older, more frail, community dwelling populations where delirium is a common, sensitive, but non-specific presentation of COVID-19 (23), the ability to rule out viral and bacterial infections as the cause of delirium during an outbreak may be even more important than the ability to detect them. Further work is therefore required to look at the NPV of FebriDx in populations where such information may be of use.
It should also be noted that specific treatments (neuraminidase inhibitors) are available and recommended for use during influenza outbreaks, and an increasing body of evidence supports different interventions (remdesivir and dexamethasone) for use in cases of COVID-19 (2, 24). The ability to distinguish between different types of viral infection is important and the value added by FebriDx in the context of already wide availability of influenza POC testing is unclear.
Although not a direct test for the presence of SARS-CoV-2 infection, the performance measures reported in this analysis are comparable to results from other studies of FebriDx in detecting the presence of respiratory viral infections (10). As a raised MxA level is not diagnostic of SARS-CoV-2 due to its non-specific response to a number of respiratory infections, the optimal use case of the FebriDx test is unlikely to be for ‘ruling in’ COVID-19. In a hospital setting, if FebriDx was used to cohort patients to wards incorrectly, then the unspecific nature of the result may lead to the exposure of patients to potentially serious co-infection. Recent evidence has suggested that the risk of death from co-infection of SARS-CoV-2 and influenza was nearly double that of SARS-CoV-2 alone, 43.1% vs 26.9% (25). However, the simplicity of a fingerprick blood test with a 10-minute turnaround time could enable rapid ‘rule out’ of COVID-19 in patients who have low concentrations of MxA. In a hospital setting, those patients could be potentially sent to non-COVID areas of a hospital.
In the pooled analysis presented in this study, the NPV was 0.940 at a prevalence of 0.405. The performance of FebriDx as a rule out test at a lower prevalence is likely to be better. In the community, the test could potentially be used to allow relatives to visit care homes residents; to facilitate air travel; or to support schools or universities. However, it is imperative to evaluate this test within the appropriate patient populations in-context for each of these use cases before making recommendations. In this study, we have only identified evidence relating to use in symptomatic patients presenting to hospital with suspected COVID-19.
Implications for practice
Whilst diagnostic performance measures were reasonably high, our findings are limited by the uncertain generalisability to longer durations of symptoms (more than a week), to different healthcare settings, and to different phases of the pandemic as prevalence rates will vary.
The utility of FebriDx may be limited to its ability to rule out acute COVID-19 infection because a positive result does not specify which respiratory viral pathogen is present. However, a sensitivity of 0.920 will lead to a false negative result in almost one in ten positive patients. This is an important consideration for settings with a high prevalence of disease in tested patients. Testing protocols would need to be developed carefully to ensure the correct use of the test, likely as a triage test in conjunction with RT-PCR.
If further evidence confirms the diagnostic characteristics of FebriDx in a hospital setting then it may be of greatest utility when deployed in a triaging capacity. For example, enabling the allocation of patients to wards based on the likely risk of SARS-CoV-2 whilst confirmatory RT-PCR testing is sought. This should, however, be used with caution due to the potential increased risk that co-infection poses to patients with SARS-CoV-2. The use of the FebriDx LFD should be carefully considered within the context of both clinical pathway needs and the patient pathways that it may influence.
Limitations
The reference standard of RT-PCR on nose and throat swab samples is imperfect, and while commonly used as a reference test, it is not a gold standard (26). RT-PCR has shown limited diagnostic performance characteristics, particularly with the production of false negative results in patients presenting in an emergency with suspected COVID-19 (27-29). An imperfect reference standard in this case, which most likely produced false negative results, would be likely to produce an underestimate of both sensitivity and specificity. If additional clinical and diagnostic data were available for both studies, this analysis would have benefitted from the use of a composite reference standard (30) or latent class analyses with instrumental variables to minimise the probability of such error or bias (31-33). Although both studies used RT-PCR as the reference standard, different RT-PCR tests were used across the studies. The accuracy of the different RT-PCR tests used will vary between methods, and as these methods were further developed during the pandemic, this leads to an inconsistent reference standard. This suggests that the pooled diagnostic accuracy estimates should be treated with caution.
Both studies were conducted during the first peak of the SARS-CoV-2 pandemic in the UK (March/April 2020), where the prevalence of COVID-19 was high. The pooled diagnostic accuracy results (particularly PPV and NPV) should only be interpreted within this specific phase of the pandemic, and not extrapolated to other phases where a lower prevalence of COVID-19 was evident. A key additional issue will occur when there are increased rates of several other viruses and bacteria circulating, most notably during the winter season. The possibility of co-colonisation/infection at such times will be a challenge for diagnostic accuracy evaluations. In particular, it is likely that the capacity to rule out COVID-19 will be compromised when other respiratory viruses are prevalent.
Conclusion
Based on a large sample of patients from two studies during the first wave of the SARS-CoV-2 pandemic, FebriDx had reasonable diagnostic accuracy in a hospital setting with high COVID-19 prevalence, out of influenza season. We cannot be certain how FebriDx would perform in other healthcare settings with higher or lower COVID-19 prevalence or at times year when other respiratory infections may affect diagnostic performance. Further evidence is needed on FebriDx’s diagnostic performance and utility in different populations, clinical and non-clinical settings.
Data Availability
An anonymised minimum dataset containing enough information to reproduce the diagnostic accuracy statistics is available from the corresponding author on reasonable request. The complete anonymised dataset is not currently available as it is still being used by the Southampton study team to produce further publications.
Declarations
Ethics approval and consent to participate
This work was based on a pooled analysis of anonymised data from two previous studies; the CoV-19POC study, described by Clark et al. (9), the “Southampton study” [ISRCTN:14966673, date registered: 18/03/2020]; and a study described by Karim et al. (13), the “Kettering study”. The Southampton study was approved by the South Central - Hampshire A Research Ethics Committee: REC reference 20/SC/0138, on the 16th March 2020. The protocol is available at: https://eprints.soton.ac.uk/439309/1/CoV _ 19POC _ Protocol _ v1.1 _eprints.pdf. The Kettering study was approved by the Kettering General Hospital Ethics Committee. Informed consent was obtained from all participants and failure to consent was considered an exclusion criterion in both studies.
Consent for publication
Informed consent was obtained from all participants.
Availability of data and materials
An anonymised minimum dataset containing enough information to reproduce the diagnostic accuracy statistics is available from the corresponding author on reasonable request. The complete anonymised dataset is not currently available as it is still being used by the Southampton study team to produce further publications.
Competing interests
MHW co-led an (unpublished) pilot study of FebriDx in 2019 for which free kits were provided by the manufacturer (Lumos). Charitable funding has been obtained to carry out an (as yet unstarted) follow on study: Clinical utility of FebriDx in determining whether or not patients presenting to a UK Accident and Emergency Department with symptoms of acute respiratory infection require antibiotic treatment (Jon Moulton Foundation (2020) - £151K (Co-Led by MHW)). The other authors declare no relevant conflicts of interest.
Funding
This study is part of the CONDOR platform (18) which is funded by the UKRI, Asthma UK and the British Lung Foundation. SGU, BCL, KG, JS, SG, DAP and AJA are supported by the National Institute for Health Research (NIHR) Newcastle In Vitro Diagnostics Co-operative. MHW is supported by the NIHR Leeds In Vitro Diagnostics Co-operative. DSL receives funding from the NIHR Community Healthcare MedTech and In Vitro Diagnostics Co-operative at Oxford Health NHS Foundation Trust. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Authors’ contributions
RB and AJA devised the project. SGU, BCL and AJA designed the analysis plan. JS designed and executed the literature search. SGU and KG conducted the RoB assessment. SGU conducted the analysis with supervision from BCL and AJA. SGU drafted the initial manuscript. All authors contributed to drafts and revisions of the manuscript.
Supplementary material
Supplementary material 1: ‘Supplementary material 1.pdf’ presents the STARD checklist.
Supplementary material 2: ‘Supplementary material 2.pdf’ presents the PRISMA-IPD checklist.
Supplementary material 3: ‘Supplementary material 3.pdf’ presents the minimum data set that was requested from the Chief investigators (CIs) of the included studies.
Supplementary material 4: ‘Supplementary material 4.pdf’ presents the methods for calculating the diagnostic accuracy measures.
Supplementary material 5: ‘Supplementary material 5.pdf’ presents the PRISMA-IPD flow diagram.
Supplementary material 6: ‘Supplementary material 6.pdf’ presents risk of bias (RoB) assessments for the included studies.
Supplementary material 7: ‘Supplementary material 7.pdf’ presents the flow of patients in the Southampton study.
Supplementary material 8: ‘Supplementary material 8.pdf’ presents the flow of patients in the Kettering study.
Acknowledgements
The authors would like to acknowledge Lumos Diagnostics for sharing information vital to the design of the study, however they had no part in the study design, analysis or of the development of the manuscript.
List of abbreviations
- AMU
- Acute Medical Unit
- ARI
- Acute respiratory illness
- b
- Regression coefficient (estimate)
- CI
- Chief Investigator
- CI
- Confidence intervals
- COVID-19
- Coronavirus disease 2019
- CRP
- C-reactive protein
- ED
- Emergency Department
- GICU
- General Intensive Care Unit
- HDU
- High Dependency Unit
- IPD
- Individual patient data
- IQR
- Interquartile range
- LFD
- Lateral flow device
- MxA
- Myxovirus resistance protein A
- NLR
- Negative likelihood ratio
- NPV
- Negative predictive value
- p
- P value
- PHE
- Public Health England
- PLR
- Positive likelihood ration
- POC
- Point-of-care
- PPV
- Positive predictive value
- RT-PCR
- Reverse transcriptase polymerase chain reaction
- RdRp
- RNA-dependent RNA polymerase
- RoB
- Risk of bias
- SOB
- Shortness of breath
- UK
- United Kingdom
References




